

Abstract
Tau is a microtubule associated protein that is localized to the axon in neurons. During pathological conditions including frontotemporal dementia (FTD) a shift in tau isoforms occurs that leads to enhanced expression of a form of tau with four (rather than 3) microtubule binding repeats; this has been postulated to alter microtubule structure. Second harmonic generation (SHG) is a technique that allows the visualization of intact microtubules in axons of living neurons without the need for labeling or fixing. We examined how the presence of exogenous tau influences SHG in living neurons. Results show the presence of tau significantly enhances SHG, specifically in neuronal axons, despite the presence of tau throughout the entire cell. Data also suggest that the presence or absence of the fourth microtubule binding repeat does not significantly alter tau’s ability to enhance SHG. These results provide evidence that SHG is a useful, noninvasive tool to study tau-microtubule interactions in axons; further, it appears that tau overexpression, rather than specific isoforms, is the major contributor to tau-induced changes in axonal microtubule SHG signal.
Introduction
Microtubules are dynamic, labile structures that undergo tight regulation by numerous proteins. Microtubules provide important functions for most cell types including structural support, intracellular transport, and chromosome separation during cell division. Perhaps the most well studied microtubule associated protein to date is tau protein, since changes in tau splicing and tau mutations lead to a neurodegenerative disease called frontotemporal dementia (FTD) with parkinsonism linked to chromosome 17 (FTDP-17), a clinical syndrome leading to neuronal loss and dementia. Tau is also a key component of one of the pathological hallmarks of Alzheimer’s disease (AD), the neurofibrillary tangle (NFT). Tau is uniquely localized in neurons, and is associated with microtubules only in axons where the microtubules adopt an asymmetric conformation. Understanding the way in which tau and microtubules interact in living cells is crucial to understanding how disruption of this process might lead to cell dysfunction or death.
Approximately 50 tau gene mutations are associated with FTDP-17. Many of the mutations are within the introns adjacent to exon 10, and have been shown to alter tau splicing to favor inclusion of exon 10 which encodes an additional microtubule binding domain. Tau isoforms containing exon 10 are referred to as 4 repeat (4R) isoforms, whereas those lacking exon 10 are referred to as 3 repeat (3R) isoforms. Alterations of exon 10 splicing are also seen in nonfamilial cases of FTD (1-8), supranuclear palsy (8-10), and in some instances of AD (3,8,11,12). We have also recently shown that alternative splicing of an amino terminal domain (exon 2) as well as exon 10 occur differentially in AD and control brain tissue (12). Other FTDP-17 tau mutations lead instead to amino acid changes, often centered near the microtubule bindng domains and these are also believed to alter tau-microtubule dynamics. One of the most common of these mutations is the P301L mutation (4,13-15).
These studies have provided valuable information about the marked effect tau splicing has in terms of association with neurodegeneration, but have lacked a functional readout for the effect tau alterations have on intact microtubules within neurons. In isolated tau/tubulin preparations, the presence of the fourth microtubule binding repeat in tau appears to stabilize microtubules, compared to isoforms containing only three repeats (16,17). By contrast, and somewhat contrary to expectations based on these in vitro data, recent fluorescence recovery after photobleaching (FRAP) data suggest that 3R tau and 4R tau have very similar properties with regard to tau/tubulin association in intact retinal ganglion cells (18).
We have now explored this issue using an alternative approach to examine the effect of tau splice forms on microctubules in intact neurons. Second harmonic generation (SHG) is a nonlinear optical microscopy method that has inherent properties that make it suitable for visualizing microtubule structure in living cells. In SHG, light is generated by a structure at exactly one-half the wavelength of incident light; unlike fluorescence, the SHG signal can be elicited by a broad range of incident light and the emitted photons “track” at exactly one-half the wavelength of incident light. Moreover, the process is nonabsorptive, so that there is no photobleaching or free radical generation (for review see (19)). SHG arises only from inherently asymmetric chemical structures including some biological molecules. A number of proteins have been shown to generate second harmonic signals, including collagen (20,21), myosin (22-24), and axonal microtubules (19,25,26), with axonal microtubules being the weakest source of SHG.
In this study we take advantage of the observation that axonal microtubules give rise to an SHG signal due to their asymmetric structure (26). Disruption of axonal microtubules causes loss of SHG signal (26). We now utilize SHG as a functional readout for microtubule structure in living cultured neurons and examine the effect of the presence of exogenous tau in transfected neurons. Results clearly demonstrate that the presence of exogenous tau significantly enhances SHG signal. Surprisingly, and in contrast to extrapolation from biophysical measures of isolated tau/microtubule interaction assays, our results suggest that the three repeat and four repeat isoforms of tau, and even a P310L FTDP-17-associated mutant form of tau, all seem to alter microtubules/SHG to a similar extent.
Experimental Procedures
Cell Culture and Western Blot Analysis
Primary cortical neurons were prepared from cerebral cortices of CD-1 strain mouse embryos (day 15-17 of gestation). Cortices were dissociated by trypsinization for 5 min at RT and cells were resuspended in Neurobasal (NB) medium supplemented with 10% FBS, 2 mmol/L Gln, 100 U/ml penicillin and 100 μg/ml streptomycin and centrifuged at 100 g for 10 min and resuspended in NB/B27 (NB medium containing 2% (v/v) B-27 supplement), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mmol/L Gln and plated at 1 × 106 on 42 mm round coverslips (Hemogenix, Colorado Springs, CO), coated with Poly-D-Lysine (20 μg/ml), in 60 mm cell culture dishes (Corning, Lowell, MA). Neurons were grown for 7-10 days in vitro and transiently transfected with the indicated constructs utilizing Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol.
H4 human neuroglioma cells were maintained in Opti-MEM (Gibco, Carlsbad CA) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Carlsbad CA). Where indicated, cells were transiently transfected with the identified constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol.
Tau constructs were subcloned into either mRFP-N1 or eCFP-N1 vectors (Clontech, Mountainview, CA). Using Tau3R(-2-3), Tau3R(+2+3), Tau4R(-2-3), or Tau4R(+2+3) in pcDNA3.1(+), all generous gifts from Dr. Michael Hutton, as templates, PCR was performed using the following primers, 5’-CTCGAGATGGCTGAGCCCCGCCAGGAGTTCGAAG-3’ (forward) and 5’-GGATCCAAACCCTGCTTGGCCAGGGAGGCAGAC-3’ (reverse), digested with Xho1 and BamH1 and ligated into appropriate color vectors. The integrity of each construct was verified using DNA sequencing analysis.
H4 human neuroglioma cells were transiently transfected with Lipofectamine 2000 according to the manufacturer’s protocol, and collected 48 hr posttransfection in 100 μl 2X SDS lysis buffer (0.25 M Tris-Cl, pH 7.5, 2% SDS, 5 mM EDTA, 5 mM EGTA, 10 % glycerol supplemented with a protease inhibitor pellet (Gibco, Carlsbad, CA). Cell lysates were then sonicated for 10 sec and boiled for 10 min. Samples were then centrifuged at room temp for 10 min at 15,000 g. Sample concentrations were determined using the bicinchoninic acid assay (Pierce). Samples were diluted to 1 μg/μl in 2X SDS running buffer and loaded onto precast 4-12% Tris-Glycine gradient polyacrylamide gels (Invitrogen, Carlsbad, CA), electrophoresed and transferred to PVDF membranes. Membranes were blocked for 1 hr at room temp in TBST (20 mM Tris-Cl pH 7.6, 137 mM NaCl, 0.05% Tween 20) and 5% milk and incubated in TBST and milk at 4°C overnight in indicated primary antibody. Membranes were then washed 3× 10 min in TBST at room temp and incubated for 1 hr with appropriate HRP-conjugated secondary antibody in TBST and milk. Membranes were washed 3× 10 min in TBST at room temp and visualized with enhanced chemiluminescence (Amersham).
Live Cell Imaging and SHG
Primary neurons on coverslips were fitted in POC (Hemogenix, Colorado Springs, CO) closed chamber with 1 mm silicone gasket between glass coverslips, containing Hank’s balanced salt solution (HBSS) prewarmed to 37°C. The chamber was affixed in the heated stage of a Zeiss Axiovert 200 inverted microscope. All images were acquired on the Zeiss LSM-510 META confocal microscope fitted with He/Ne 543 nm laser for RFP fluorescence and Coherent (Santa Clara, CA) Chameleon Mode-Locked Ti/Saph tunable (720-930nm) laser with ~140 fs pulse width, ~ 20-30 mW average power with linear polarization at the sample, for CFP and SHG imaging. All images were acquired using a Zeiss 25X 0.8 NA Plan-NEOFLUAR water/oil immersion lens for epifluorescence and an infinity corrected Zeiss 20X 0.75 N/A PlanAPOCHROMAT dry lens affixed to a custom condenser tube (fabricated by Zeiss, Jana Germany) affixed to the condenser housing, which projected the forward propagating signal onto a photomultiplier tube (PMT, Hamamatsu). A 400 ± 25 nm bandpass filter was fitted between the condenser housing and PMT. This aided in both reducing background fluorescence at the SHG PMT and also verifying the SHG nature of the signal observed, as 800 nm light was used to induce SHG signal and the resulting 400 nm signal should pass through the filter unobstructed, while producing an SHG signal using 900 nm should result in a loss of the SHG signal as the 450 nm signal will be blocked by the bandpass filter. The bandpass filter also ensured that no fluorescent signal from the fluorophores used pass through to the PMT, as CFP emits around 450 nm and RFP emits around 590 nm, and both are blocked by the bandpass filter. To further ensure the signal obtained was from SHG and not autofluorescence, a polarizer was placed in front of the SHG PMT and the polarizer was rotated to demonstrate the polarized nature of the SHG signal. Since the SHG signal is a polarized signal, rotating the polarizer perpendicular with respect to the laser polarization should result in a loss of SHG signal, in contrast to fluorescence emissions.
Images were collected using Zeiss LSM 3.5 software and exported as JPEG images for analysis in Image J. The Neuron J macro plugin for Image J was utilized to trace and quantify SHG signal in all sample images and the resulting intensity data was analyzed using Graphpad Prism 4 (San Diego, CA) to carry out t tests or One Way ANOVAs. Data are presented as mean ± SEM for each condition and results were considered significant if p < 0.05.
Results
Second Harmonic Generation Signal is Detectable in Single Neuronal Processes
Recent work has shown that SHG signal arises mainly from axonal processes in neurons, and not from the cell body or dendrites (26), in acute hippocampal slices. Therefore, we sought to determine whether it was possible to visualize an SHG signal from a single neuronal process in a cultured neuron that would allow us to visualize intact axonal microtubules in a living neuron. Figure 1 shows the morphology of a single neuron transfected with RFP and visualized with the red fluorescence signal. RFP was excited with a 543 nm He/Ne laser and the emitted light at 590 nm was directed to the PMT through a 570-610 nm bandpass filter. Similarly the SHG signal was obtained with 800 nm excitation and collected in the forward direction after passing the signal through a 375-425 nm bandpass filter. Weak SHG signal was observed in single processes extending from each neuron. Several characteristics suggest that this is SHG: the 400 nm signal is seen only in the axon (as expected given the asymmetric microtubule structure unique to axons), whereas tau and autofluorescent moieties are most strongly seen in the cell body. This signal is lost with treatment with colchicine, arguing that it is microtubule dependent. We inserted a polarizer in the light path between the sample and the PMT. Rotating the polarizer between 0 and 90 degrees should either allow a polarized signal to pass or block a polarized signal, depending on the orientation, while fluorescent signals should be unaffected by the orientation of the polarizer. As expected, when the polarizer is in the 0 degree position, the SHG signal is strong. However, when the polarizer is shifted to the 90 degree position, the SHG signal is completely lost. As another test, the excitation wavelength was changed to 900 nm, the SHG signal observed in a 375-425 emission filter was lost (as expected for SHG) (Fig 2). Finally, we demonstrated that, if the optics were reconfigured to detect the back propagating signal, the signal was undetectable, again as expected for SHG which is predominantly forward propagating, unlike a fluorescent signal.
Fig. 1. Weak SHG Signal is Obtainable from Non-transfected Neurons.
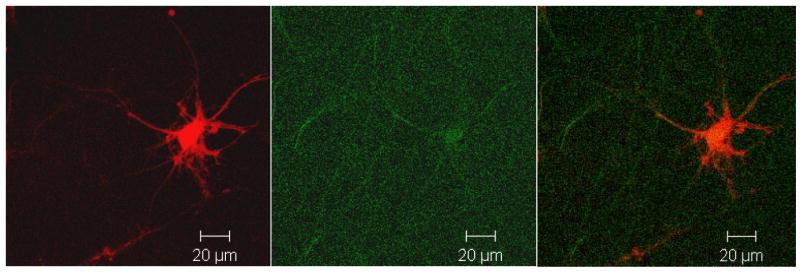
Photomicrograph showing a neuron transfected with RFP alone and visualized simultaneously in the 543/590 nm fluorescent excitation/emission signal (Red) and 800/400 nm SHG excitation/emission (green). Notice that there is very little SHG present in cells that have endogenous levels of mouse tau expressed and that express RFP, but that there is signal detectable. Micrograph is representative of at least 3 neurons in each transfection from 3 separate mice.
Fig. 2. SHG Signal, but Not GFP Signal is Abolished by Blocking Polarized Light With a Polarizer.
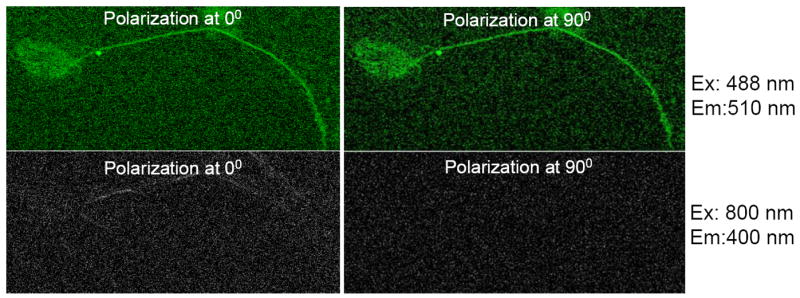
To conclusively demonstrate the SHG nature of the signal obtained in the SHG channel, a polarizer was placed in the lightpath of the SHG signal and the orientation of the polarizer was positioned at either 0° or 90°, and both the GFP and SHG signals were obtained from GFPTau4R(+2+3)-transfected neurons. As expected, when the polarizer was changed and the GFP signal of tau-transfected neurons was obtained, there was no difference in the GFP signal. Conversely, the SHG signal is present only in the 0° position of the polarizer and is abolished when it is set to 90°, indicating the polarized nature of the SHG signal. This also rules out autofluorescent signals as contaminating the SHG signal, as they would not be affected by the position of the polarizer. Micrograph is representative of three neurons transfected with tau.
Overexpression of tau increases the SHG signal
We next examined whether altering tau levels would enhance SHG signal. As a first step, we characterized tau expression in H4 cells. Figure 3 shows a western blot analysis of various tau constructs expressed in H4 cells to verify the biochemical properties of each construct. The different isoforms’ molecular weights correspond to the presence or absence of the fourth microbutubule binding repeat (4R, representing exon 10) or the 2 amino-terminal inserts (±2±3). All constructs migrated to the expected molecular weight, indicating that the proteins were expressed and processed appropriately in cells.
Fig. 3. Biochemical Properties of Tagged Tau Constructs are Normal.
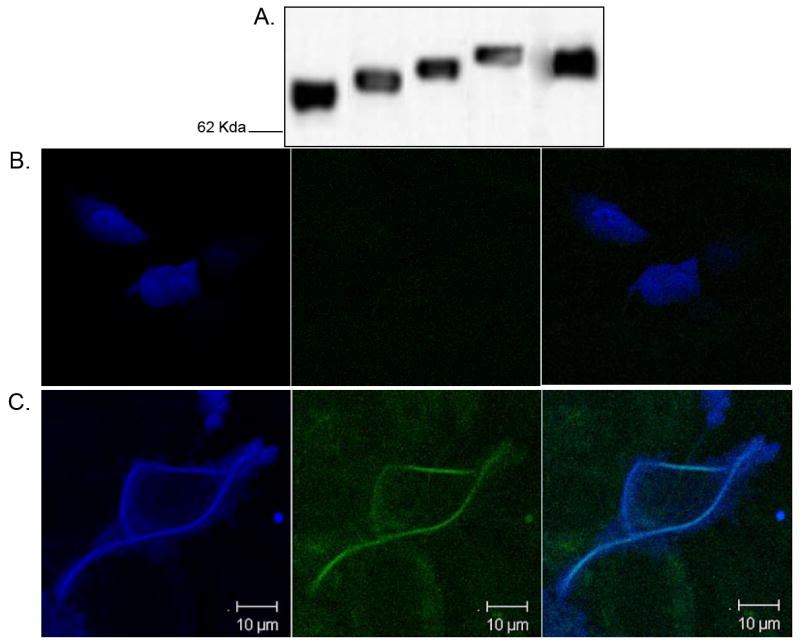
Representative blot showing various tagged tau constructs were transiently transfected into H4 human neuroglioma cells and (A) cell lysates were immunoblotted and probed with a pan tau antibody (AB-3). Western blot shows that all expressed tau migrates to the expected molecular weight and that the presence of a C-terminal fluorescent protein (CFP or RFP) does not significantly alter tau processing. All constructs were transfected utilizing 1 μg of DNA per condition, and total lysate protein concentration was normalized for each sample. (B) Expression of CFP in H4 neuroglioma cells leads to robust expression of CFP throughout the entire cell and fluorescence throughout the entire cell, with no SHG signal detectable in transfected cells. Imaging of fluorescently-tagged tau (C) shows that tau is localized throughout entire cell. SHG signal however comes from filamentous structures that contain both tau and tubulin that encircle the nucleus and continue into processes. Notice specifically that the microtubule network seems to reorganize into circular structures in transfected H4 cells and that robust SHG signal is present in more than one process. Photomicrographs are representative of at least three cells from each of at least three separate preparations.
We examined whether microtubules in H4 neuroglioma cells, which morphologically are flat with several long processes, would show SHG signal. No SHG could be detected from H4 cells transfected with CFP (Fig 3B). By contrast, imaging of H4 cells transfected with tau showed robust tau expression in the cell body and processes, but interestingly the SHG signal arose primarily from perinuclear compartments where tau levels were highest, where the microtubule network appears to reorganize within the perikeria (Fig 3C).
We next examined whether transfection of tau would enhance SHG in neurons, and if so, in which cellular compartment. Given that tau is a microtubule binding protein that promotes the assembly and stability of microtubules in axons (reviewed in (27)), it might follow that expression of tau would increase axonal SHG signal. To further test this hypothesis, and also to determine if the presence of a fluorescent tag on tau would change SHG, neurons were transfected with different isoforms of either TauRFP, untagged tau cotransfected with RFP, or RFP alone. SHG signal was measured by tracing SHG positive neurites using the Neuron J macro for Image J and analyzing the intensity across the entire length of the process.
Since transfection efficiency is very low in cultured neurons with our transfection method, it is possible to visualize the processes of a single neuron and follow them for many hundreds of microns, without interference from the processes of neighboring, untransfected neurons. When TauRFP-transfected neurons were examined, the RFP fluorescence and SHG signals were distinct (Fig 4A): SHG signal was present in only one process, whereas the TauRFP fluorescence signal was present in multiple processes and the cell body. To verify that the signal obtained in the SHG channel was indeed SHG and not contaminating fluorescence, the wavelength of the excitation laser was changed from 800 nm to 900 nm. As expected the SHG signal was abolished, since the bandpass filter in front of the SHG detector blocked 450nm emission (Fig 4B). As a further control, we added a 450 nm bandpass filter to the turret, and could switch between the 400 nm and 450 nm filters. Reconfiguring the detectors demonstrated that the signal was substantially forward propagating, and could not be detected in the back propagating configuration (data not sown). As expected, using 800 nm light and the 400 nm filter, there was detectable SHG signal. To test the hypothesis that the SHG signal observed is due to microtubule structures, sister cultures of Tau-RFP transfected neurons were treated with 100 μM Colchicine for 30 min and imaged as above. Since colchicine completely disrupts microtubule stability, any SHG signal arising from microtubules should be attenuated. As expected, treatment of cells with colchicine led to the complete loss of SHG signal (Fig 4C). These results clearly demonstrate that SHG signal is obtainable from a single neuronal process that is presumably the axon, and that the source of SHG signal within that process is the microtubule network.
Fig. 4. Tau Enhances SHG Signal in Transfected Neurons.
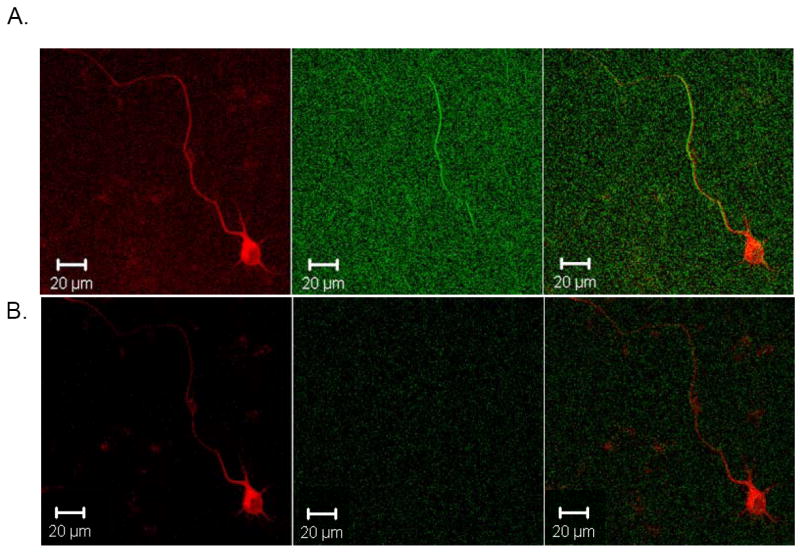

Embryonic day 15 mouse primary neurons were transfected with TauRFP for 48 hr and individual transfected cells were examined for SHG signal. A. Transfected neuron showing both TauRFP expression throughout neuron and SHG signal at 800 nm in single process, which is presumably the axon, final panel shows merged image. B. Same cell as in A, showing TauRFP signal in first panel and SHG signal when 900 nm excitation wavelength is used in second panel, showing that SHG signal is not detectable when 900 nm excitation is used, as expected, as the bandpass filter before SHG detecting PMT cuts out all light above 425 nm. Third panel shows merged image. Micrographs are representative of at least 3 neurons per transfection from at least three separate mouse primary neuronal culture preparations. C. Sister cultures of primary neurons as in A and B that were transfected with TauRFP and also treated with 100 μM colchicine for 30 min prior to imaging, which completely depolymerizes microtubules. First panel shows TauRFP signal in cell and a similar amount of TauRFP expression as in A and B. Second panel shows SHG signal which is completely abolished, as expected, indicating the microtubule nature of the SHG signal. Third panel shows merged image. Micrographs are representative of at least three transfected neurons for three separate mouse primary culture preparations.
As an additional control we inserted a Berek compensator (New Focus) into the excitation light path (to circularly polarize the laser source) and examined the SHG signal from the processes. Figure 5 shows that, as expected, even when the excitation beam is circularly polarized only one process shows a strong SHG signal.
Fig. 5. Using Circularly Polarized Light Does Not Alter Pattern of SHG in Transfected Neurons.
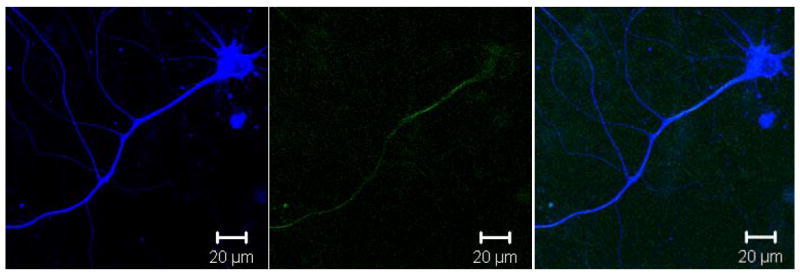
Representative photomicrograph showing a single mouse neuron transfected with Tau4R(+2+3)CFP and visualized with 800 nm two photon excitation. Left panel shows TauCFP localization throughout the entire cell, including all processes and cell body. Middle panel shows the forward propagating SHG signal after circularly polarizing the excitation source which should eliminate the dependence of the angle of orientation seen with linearly polarized light. Note that only a single process is still visible in the SHG channel, confirming that the primary neurite that is visible with SHG is the axon. Right panel shows merge of first two images and colocalization of the SHG signal with tau in the axon, again indicating that TauCFP enhances SHG in only the axon. Micrographs are representative of at least 3 separate neurons from at least 3 separate transfections, all showing that circularly polarized light does not significantly affect the pattern of SHG signal.
Expression of either tagged or untagged tau led to a significant and equal increase in SHG signal (Fig 5), over that of RFP alone (RFP = 15.0 ± 1.5, TauRFP = 32.1 ± 4.6, and Tau + RFP =31.7 ± 2.6; p < 0.001 for both RFP vs. TauRFP and RFP vs. Tau + RFP). The presence of either RFP or CFP fusion constructs on the C-terminus of tau did not further enhance or decrease the SHG signal from microtubules, when compared to tranfection with untagged tau (p > 0.05).
One possibility that could account for increase SHG signal from tau-transfected axons, is that expression of tau significantly increases the overall diameter of the axon, and therefore increases the signal by giving rise to a signal from a larger axonal cross section. To rule this out, images of axons of both RFP- or TauRFP-transfected neurons were examined in Image J and the axonal diameter was measured at least 20 μm distal to the axon hillock and before the first major bifurcation of the axon. Three separate measurements per axon were averaged and a two-tailed t-test revealed that there was no significant difference between RFP- and TauRFP-transfected axons. Therefore, tau expression does not increase SHG simply by increasing the size of the axon (data not shown).
Different tau isoforms do not significantly differ in enhancing SHG
In in vitro assays, 4 repeat tau binds microtubules with a greater affinity and polymerizes the formation of microtubules to a greater extent than 3 repeat tau (16,28)(reviewed in (29)). Having shown that tau enhances SHG signal in neurons, we next sought to determine whether different tau isoforms would differentially affect SHG signal. Tau-CFP constructs were generated for ease of planned co-transfection experiments and in all instances were equivalent to the transfection protocol utilized for Tau-mRFP constructs used above. Neuronal cultures were transfected with one of the following constructs: CFP, Tau3R(-2-3)CFP, Tau3R(+2+3)CFP, Tau4R(-2-3)CFP, Tau4R(+2+3)CFP. SHG signal was determined for each condition. As expected, expression of any isoform of tau enhanced the observed SHG signal, compared to empty vector or CFP transfected neurons (Fig 7). Surprisingly, however, there was no significant difference between exon 10 containing or exon 10 skipping constructs, indicating that the presence of 3 or 4 microtubule binding repeats does not significantly affect tau’s ability to enhance microtubule SHG signal in living neurons. No morphological changes in axon shape were noted qualitatively and measuring axon diameter revealed no difference between tau-transfected and control neurons.
Fig. 7. Different Tau Isoforms Do Not Significantly Differ in Their Ability to Enhance SHG.
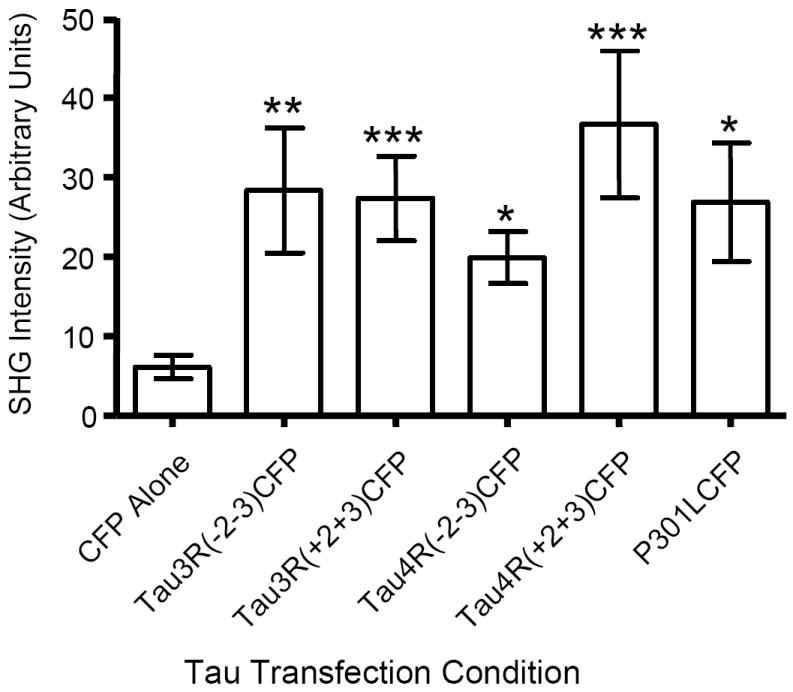
Primary mouse neurons were transiently transfected with the indicated tau isoform fused to CFP or CFP alone and SHG signal was measured. Note that CFP alone was different from all groups, and that there were no intergroup or differences that were the result of tau isoform, or even the mutated P301L form of tau. CFP mean ± SEM = 6.12 ± 1.48, Tau3R(-2-3)CFP = 28.35 ± 7.85, Tau3R(+2+3)CFP = 27.34 ± 5.31, Tau4R(-2-3)CFP = 19.88 ± 3.28, Tau4R(+2+3)CFP = 36.65 ± 9.26, TauP301L-CFP = 26.85 ± 7.45, n = 20-30 neurons/condition.
Mutations in the tau gene found in FTDP-17 have been shown to decrease the in vitro association of tau with microtubules and rate of microtubule polymerization (13,17,30-34). To determine if tau mutations alter axon-derived SHG in living neurons, we transfected neurons with P301LTau4R(+2+3)CFP and examined the effect on SHG signal. Surprisingly, the mutant tau enhances SHG signal observed in transfected neurons (Fig 8), to essentially the same extent as other tau isoforms.
Fig. 8. FTDP-17 Mutant Tau Does Not Differentially Impact SHG Generation.
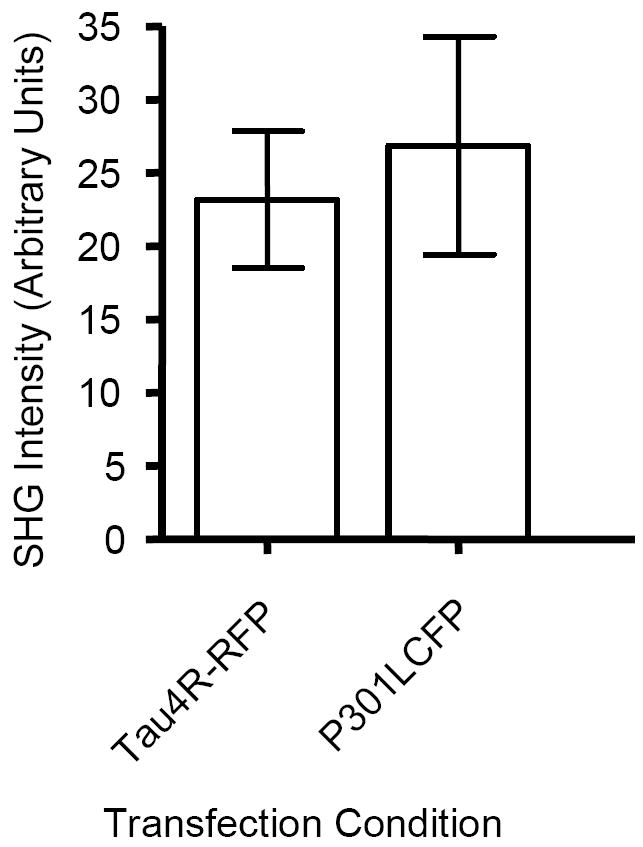
Primary mouse neurons were transiently transfected with indicated Tau construct. Surprisingly, expression of either TauRFP or P301L-Tau4RCFP results in a high level of SHG signal with mean ± SEM being 33.5 ± 4.7 and 37.2 ± 7.4, respectively, n = 20-30 neurons per condition.
Discussion
Our earlier studies suggested that axonal microtubule structures gave rise to SHG, as detected in bundles of axons in slice preparations (26), and we proposed that the signal was due to a unique structure of axonal microtubules. Our current experiments demonstrate that the unique SHG signal can be detected in single axons. The signal is lost with the addition of pharmacological disruption of microtubules by colchicine, and enhanced (only in axons) by transfection with the axonal microtubule binding protein tau, suggesting that tau-microtubule interactions gave rise to the enhanced signal. These data are consistent with the idea that the signal seen in axon bundles originates from the axonal microtubule structure itself, rather than from extracellular compartments such as collagen or basement membrane. This observation further supports the idea that the source of SHG signal in axons is the asymmetric polarity of axonal microtubules.
Interestingly, H4 neuroglioma cells have very little or no SHG signal present, even in the many small neurites that encircle the cells. However, when tau is transfected into H4 cells, the microtubule network, along with tau, rearranges into circular filamentous structures that give rise to significant SHG signals. There are also multiple processes that can be observed with SHG signal transfected H4 cells. These data suggest that H4 cells differ significantly from neurons in their processing of tau and organization of microtubules within processes, and highlight the importance of studying tau-microtubule interactions in axonal compartments.
Our results, following tau transfections, are unexpected from several perspectives: overexpressed tau appears in the soma, and all processes of a neuron, and localizes with tubulin in all cellular compartments. However, SHG signal was observed only in single processes. These data suggest that tau-tubulin interactions in the soma or dendritic processes do not stabilize microtubules into the same conformation in all cell compartments and instead that a unique conformation occurs in axons. The enhancement of SHG signal in these axonal compartments is consistent with the idea that excess tau (in the axon) can stabilize microtubules or enhance microtubule bundling; if so, these data using an in vivo biophysical measure in neocortical neurons complement and extend observations in retinal ganglion cells (RGCs) in which tau overexpression leads to functional impairments in organelle transport (33,35-39). Consistent with this idea, it was demonstrated by Mandelkow et al. (40) that overexpressed tau leads to an increased density of microtubule bundles in axons and we postulate that this bundling underlies enhanced SHG signal. Although it is not clear how this concept-of being an improved SHG source compares to microtubule stability measured in vitro, our data demonstrate that overexpressed tau changes the biophysical properties of the axon in a striking fashion. One possibility that could account for increased SHG signal from tau-transfected axons is that expression of tau significantly increases the overall diameter of the axon, and therefore increases the signal from a larger axonal cross-section. To rule this out, images of axons of both RFP- or TauRFP-transfected neurons were examined in image J and the axonal diameter was measured at least 20 μm distal to the axon hillock and before the first major bifurcation of the axon. Three separate measurements per axon were averaged and a two-tailed T-test revealed that there was no significant difference between RFP- and TauRFP-axons. Therefore, tau expression does not increase SHG by increasing the size of the axon (Data not shown).
We hypothesized that four repeat tau would enhance SHG to a greater extent than three repeat tau. Our results suggest, however, that the presence of the fourth microtubule binding repeat had very little effect on microtubule SHG signal, implying that within the microenvironment of an intact axon, the presence of the fourth microtubule binding repeat does not significantly change the tau-induced microtubule conformation. This is consistent with the findings of Konzack et al (41), who demonstrated using both FRAP and FCS, that the dwell time of a single molecule of tau on microtubules is very short, and that there was no significant difference between three repeat and four repeat tau, with respect to how rapidly it diffused through the RGC axon or moved off and on the microtubules. These data suggest that within the physiological environment of an axon, tau isoform has little effect on microtubule-tau interactions.
The picture that emerges from our data is that tau interacts with microtubules in a specific asymmetric conformation in axons to generate a unique conformation that supports SHG. Using this assay of tau-microtubule interactions, it appears that the presence of the fourth microtubule binding repeat may exert less effect on the tau-microtubule interaction, in axons, than previously thought. Thus, although changes in tau splicing are clearly important in FTDP-17 and related conditions, our current data as well as that of Konzack et al. (18) suggest that the elevation of 4R tau may have additional pathogenic mechanisms independent of differential 3R vs. 4R effects on axonal tau-microtubule interactions.
Fig. 6. Expression of Either a Tagged Tau or Untagged Tau Results in Increased SHG.
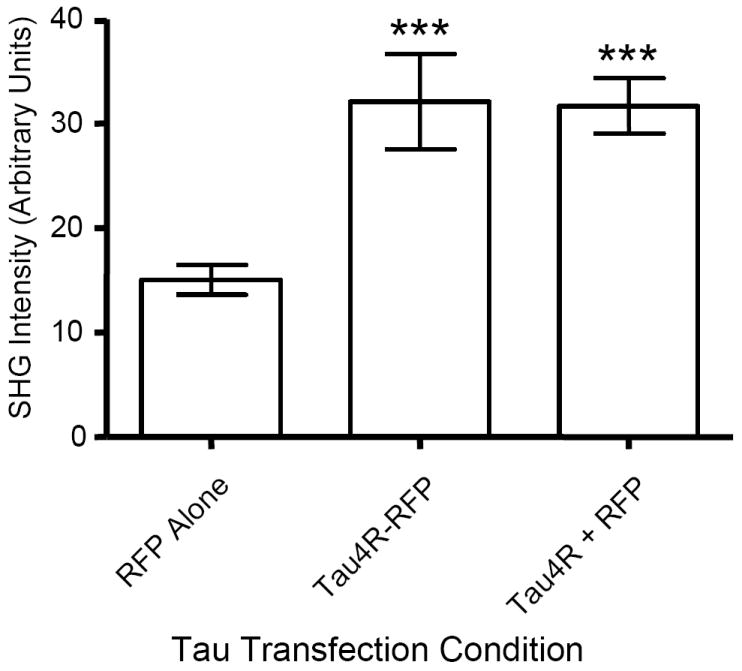
Mouse primary neurons were transiently transfected with either RFP alone, TauRFP, or Tau + RFP and SHG signal was quantitated for each condition. Expression of RFP alone results in a modest amount of SHG signal (mean ± SEM = 15 ± 1.5) while expression of either TauRFP (32.1 ± 4.6) or Tau + RFP (31.7 ± 2.6) result in a statistically significant increase in SHG signal, p < 0.001 for each when compared to RFP alone (n=15 for each condition), n = 25 neurons/condition. Interestingly, TauRFP and Tau + RFP are not significantly different.
Acknowledgments
The authors would like to thank Dr. Jennifer M. Gass and Dr. Michael L. Hutton of the Mayo Clinic for their generous gifts of Tau3R(-2-3), Tau3R(+2+3), Tau4R(-2-3), and Tau4R(+2+3) isoforms. We would also like to thank Brad Reynolds of Zeiss microimaging for assistance with modifications of the LSM 510 instrumentation. Supported by NIH AG 026249, EB000768, and P50 AG 05134.
References
- 1.Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. J Biol Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 2.Ishizawa K, Ksiezak-Reding H, Davies P, Delacourte A, Tiseo P, Yen SH, Dickson DW. Acta Neuropathol (Berl) 2000;100:235–244. doi: 10.1007/s004019900177. [DOI] [PubMed] [Google Scholar]
- 3.Ksiezak-Reding H, Shafit-Zagardo B, Yen SH. J Neurosci Res. 1995;41:583–593. doi: 10.1002/jnr.490410504. [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Am J Pathol. 1998;153:1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y. Ann Neurol. 1998;43:193–204. doi: 10.1002/ana.410430209. [DOI] [PubMed] [Google Scholar]
- 7.Sergeant N, David JP, Lefranc D, Vermersch P, Wattez A, Delacourte A. FEBS Lett. 1997;412:578–582. doi: 10.1016/s0014-5793(97)00859-4. [DOI] [PubMed] [Google Scholar]
- 8.Ingelsson M, Ramasamy K, Russ C, Freeman SH, Orne J, Raju S, Matsui T, Growdon JH, Frosch MP, Ghetti B, Brown RH, Irizarry MC, Hyman BT. Acta Neuropathol (Berl) 2007;114:471–479. doi: 10.1007/s00401-007-0280-z. [DOI] [PubMed] [Google Scholar]
- 9.Sergeant N, Wattez A, Delacourte A. J Neurochem. 1999;72:1243–1249. doi: 10.1046/j.1471-4159.1999.0721243.x. [DOI] [PubMed] [Google Scholar]
- 10.Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR. Brain. 2000;123(Pt 5):880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- 11.Yasojima K, McGeer EG, McGeer PL. Brain Res. 1999;831:301–305. doi: 10.1016/s0006-8993(99)01486-9. [DOI] [PubMed] [Google Scholar]
- 12.Conrad C, Zhu J, Conrad C, Schoenfeld D, Fang Z, Ingelsson M, Stamm S, Church G, Hyman BT. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04857.x. [DOI] [PubMed] [Google Scholar]
- 13.Nagiec EW, Sampson KE, Abraham I. J Neurosci Res. 2001;63:268–275. doi: 10.1002/1097-4547(20010201)63:3<268::AID-JNR1020>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 14.Fischer D, Mukrasch MD, von Bergen M, Klos-Witkowska A, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Biochemistry. 2007;46:2574–2582. doi: 10.1021/bi061318s. [DOI] [PubMed] [Google Scholar]
- 15.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 16.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Embo J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott CW, Klika AB, Lo MM, Norris TE, Caputo CB. J Neurosci Res. 1992;33:19–29. doi: 10.1002/jnr.490330104. [DOI] [PubMed] [Google Scholar]
- 18.Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E. J Neurosci. 2007;27:9916–9927. doi: 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohler W, Millard AC, Campagnola PJ. Methods. 2003;29:97–109. doi: 10.1016/s1046-2023(02)00292-x. [DOI] [PubMed] [Google Scholar]
- 20.Freund I, Deutsch M, Sprecher A. Biophys J. 1986;50:693–712. doi: 10.1016/S0006-3495(86)83510-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim BM, Eichler J, Reiser KM, Rubenchik AM, Da Silva LB. Lasers Surg Med. 2000;27:329–335. doi: 10.1002/1096-9101(2000)27:4<329::aid-lsm5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 22.Both M, Vogel M, Friedrich O, von Wegner F, Kunsting T, Fink RH, Uttenweiler D. J Biomed Opt. 2004;9:882–892. doi: 10.1117/1.1783354. [DOI] [PubMed] [Google Scholar]
- 23.Plotnikov S, Juneja V, Isaacson AB, Mohler WA, Campagnola PJ. Biophys J. 2006;90:328–339. doi: 10.1529/biophysj.105.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plotnikov SV, Millard AC, Campagnola PJ, Mohler WA. Biophys J. 2006;90:693–703. doi: 10.1529/biophysj.105.071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debarre D, Pena AM, Supatto W, Boulesteix T, Strupler M, Sauviat MP, Martin JL, Schanne-Klein MC, Beaurepaire E. Med Sci (Paris) 2006;22:845–850. doi: 10.1051/medsci/20062210845. [DOI] [PubMed] [Google Scholar]
- 26.Dombeck DA, Kasischke KA, Vishwasrao HD, Ingelsson M, Hyman BT, Webb WW. Proc Natl Acad Sci U S A. 2003;100:7081–7086. doi: 10.1073/pnas.0731953100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoothoff WH, Johnson GV. Biochim Biophys Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Brandt R, Lee G. J Biol Chem. 1993;268:3414–3419. [PubMed] [Google Scholar]
- 29.Goedert M, Klug A, Crowther RA. J Alzheimers Dis. 2006;9:195–207. doi: 10.3233/jad-2006-9s323. [DOI] [PubMed] [Google Scholar]
- 30.Cho JH, Johnson GV. J Neurochem. 2004;88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 31.Ross JL, Santangelo CD, Makrides V, Fygenson DK. Proc Natl Acad Sci U S A. 2004;101:12910–12915. doi: 10.1073/pnas.0402928101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samsonov A, Yu JZ, Rasenick M, Popov SV. J Cell Sci. 2004;117:6129–6141. doi: 10.1242/jcs.01531. [DOI] [PubMed] [Google Scholar]
- 33.Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Embo J. 2002;21:4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Arch Biochem Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- 35.Thies E, Mandelkow EM. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. J Cell Sci. 1999;112(Pt 14):2355–2367. doi: 10.1242/jcs.112.14.2355. [DOI] [PubMed] [Google Scholar]
- 39.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandelkow EM. 2008 [Google Scholar]
- 41.Konzack S, Thies E, Marx A, Mandelkow E-M, Mandelkow E. J Neurosci. 2007;27:9916–9927. doi: 10.1523/JNEUROSCI.0927-07.2007. %R 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]