

Abstract
Using a mouse model, we tested the hypotheses that severe burn trauma causes metabolic disturbances in skeletal muscle, and that these can be measured and repeatedly followed by in vivo electron paramagnetic resonance (EPR). We used a 1.2-GHz (L-band) EPR spectrometer to measure partial pressure of oxygen (pO2) levels, redox status and oxidative stress following a non-lethal burn trauma model to the left hind limbs of mice. Results obtained in the burned mouse gastrocnemius muscle indicated a significant decrease in tissue pO2 immediately (P=0.032) and at 6 h post burn (P=0.004), compared to the gastrocnemius of the unburned hind limb. The redox status of the skeletal muscle also peaked at 6 h post burn (P=0.027) in burned mice. In addition, there was an increase in the EPR signal of the nitroxide produced by oxidation of the hydroxylamine (CP-H) probe at 12 h post burn injury, indicating a burn-induced increase in mitochondrial reactive oxygen species (ROS). The nitroxide signal continued to increase between 12 and 24 h, suggesting a further increase in ROS generation post burn. These results confirm genomic results, which indicate a downregulation of antioxidant genes and therefore strongly suggest the dysfunction of the mitochondrial oxidative system. We believe that the direct measurement of tissue parameters such as pO2, redox and ROS by EPR may be used to complement measurements by nuclear magnetic resonance (NMR) in order to assess tissue damage and the therapeutic effectiveness of antioxidant agents in severe burn trauma.
Keywords: electron paramagnetic resonance, oximetry, partial pressure of oxygen, redox, skeletal muscle, burn, trauma, mitochondria, reactive oxygen species, nuclear magnetic resonance, genomics
Introduction
Electron paramagnetic resonance (EPR) is a magnetic resonance-based technique that detects species with unpaired electrons, such as free radicals (organic or inorganic) and transition metal ions. The development of low frequency (1200 MHz and below) EPR spectrometers has led to the in vivo application of the technique in a variety of animal models. The most extensive applications of in vivo EPR have been repeated non-invasive measurements of oxygen, nitric oxide, free radicals, pH and tissue redox status (1–9). EPR in vivo has significantly contributed to the understanding of various pathological changes, and has the potential to become an important clinical tool (4,9). Often, it may also be complementary to nuclear magnetic resonance (NMR) (1).
NMR is a useful method for measuring biomarkers in vivo, and has already shown relevance in the study of the metabolism and bioenergetics of skeletal muscle (10–25). In addition to NMR spectroscopy, the development of high-throughput microarray systems, capable of simultaneously measuring the expression of thousands of genes, has also greatly advanced our ability to detect and measure biomarkers. By exploring the expression of certain organ-specific candidate genes, the biological relevance of NMR data can be validated in microarray experiments examining the same specimen (22,23,25). For assessing oxidative damage after burn trauma, in vivo EPR using nitroxides is complementary to NMR since NMR cannot measure redox status and reactive oxygen species (ROS), while EPR can.
Here, for the first time, we report tissue partial pressure of oxygen (pO2), redox status and ROS measurement by in vivo EPR in intact proximal skeletal muscle tissue following burn trauma in mice. The potential significance of our findings include the in vivo non-invasive nature of the EPR measurements, which can serve to follow tissue pathology and to monitor the effectiveness of antioxidant agents in order to alleviate the symptoms of severe burn trauma. With the development and application of in vivo EPR oximetry in clinical settings (4,9), its potential application in the clinical management of burn injury, alongside NMR, might also prove to be very useful.
Materials and methods
Study design
Due to the in vivo repetitive nature of EPR measurements, the EPR experiments followed a longitudinal random-effects study design. This is powerful since each mouse contributed information at each of the time points, and the clustered data were modeled by a compound symmetry correlation structure (26). The genomics followed a cross-sectional study design.
Experimental animals
Male 6-week-old CD1 mice weighing 20–25 g were purchased from the Charles River Laboratory (Boston, MA). The animals were maintained on a regular light-dark cycle (lights on from 8:00 to 20:00 h) at an ambient temperature of 22±1°C and had free access to food and water. All animal experiments were approved by the Subcommittee on Research Animal Care of the Massachusetts General Hospital, Boston, MA, and by the Institutional Animal Care and Use Committee of Dartmouth Medical School, Hanover, NH.
Hind limb burn model
Mice were anesthetized by intraperitoneal (i.p.) injection of 40 mg/kg pentobarbital sodium and were randomized into burn or control groups. The left hind limb of all mice in both groups (control and burn) was shaved, and each mouse in the burn group was subjected to a non-lethal scald injury of 3–5% total body surface area (TBSA) by the immersion of its left hind limb in 90°C water for 3 sec, as previously described (27). The TBSA of each mouse was calculated using Meeh's formula: A = k × W2/3, where A is the surface area in cm2, k is a proportionality constant of 12.3, and W is the weight of the mouse in grams. Mice were resuscitated with 2 ml of 0.9% saline i.p. The gastrocnemius muscle was excised from the hind limbs of both control and treated mice and was immersed in 1 ml TRIzol® (GibcoBRL, Invitrogen, Carlsbad, CA) for RNA extraction at 6 h, 12 h, 1 day and 3 days post burn, with n=3 measurements at each time point. After injury, the animals were given analgesia in the form of buprenorphine 0.05–0.1 mg/kg SQ, as needed.
Non-invasive in vivo EPR
EPR measurements were carried out with a 1.2-GHz EPR spectrometer equipped with a microwave bridge and external loop resonator specially designed for in vivo experiments. The optimal spectrometer parameters were: incident microwave power, 10 mW; magnetic field center, 400 gauss; modulation frequency, 27 kHz. Modulation amplitude was one-third of the EPR line width, with a scan time of 10 sec. The methods described here are well established and routinely used in the EPR Center for Viable Systems at Dartmouth Medical School, Hanover, NH (1–9).
Measurement of pO2 using in vivo EPR
Repeated non-invasive measurements of the tissue pO2 of the gastrocnemius muscle of 4 control mice and 4 burned mice were carried out using in vivo EPR oximetry. Approximately 80–100 μg of sterilized EMS char (charcoal powder) was injected in the gastrocnemius muscle of the left hind limb of each mouse using an 18-gauge needle and plunger. The mice were allowed to recover for one week after char implantation. Once implanted, the paramagnetic material remains at the site and allows repeated tissue pO2 measurements without further invasion (3,4,28).
For in vivo EPR oximetry, mice were anesthetized by 1.5% isoflurane with 30% O2 and were gently placed in the magnet of the 1.2-GHz EPR spectrometer for pO2 measurements. Rectal temperature was continuously monitored and maintained at 37±1°C using a warm air blower and warm water pad. After a 30-min baseline measurement (pre-burn), the left hind limb of the mouse was exposed to hot water (90°C) for 3 sec. For control experiments, the left hind limbs of the mice were exposed to 37°C water for 3 sec. The mice were immediately transferred back to the EPR magnet, and the tissue pO2 was measured for another 30 min (post-burn pO2) and at time points as indicated in the results.
Redox status measurements using nitroxides by in vivo EPR
Nitroxides exist in biological systems as a redox pair, namely the nitroxide free radical form and the diamagnetic hydroxylamine (one-electron reduction product of the nitroxide free radical). Nitroxides are reduced to the corresponding hydroxylamine in cell suspensions as well as in vivo by cellular reducing equivalents. Consequently, the pharmacology of nitroxides reports the redox status of the tissue (7,29,30). Mice were anesthetized using 1.5% isoflurane with 30% O2 delivered through a nose cone. The animals were placed on the warm water pad, and the tail vein was cannulated with a heparin-filled 30-gauge catheter for infusion of the nitroxide probe (3-carbamoyl-2,2,5,5-tetramethylpyrrolidin-1-yloxyl free radical, Sigma-Aldrich). The nitroxide (150 mg/kg) was injected slowly (over 1 min) through the tail vein cannula. The left hind limb of the mice was immediately exposed to 90°C water for 3 sec, and the mice were quickly moved to the EPR magnet and positioned for the measurement of the signal intensity of the injected nitroxide in the gastrocnemius muscle of the left hind limb over time (Fig. 2A). The time difference between the infusion of the nitroxide and the start of EPR acquisition was 2 min. This time was maintained in all the experiments. The change in signal intensity of the middle component of the nitroxide was followed over time (Fig. 2B), and data were fitted using exponential decay kinetics (Fig. 3) to determine the rate constants. For control experiments, the hind limb was exposed to 37°C water for 3 sec, and the measurements were repeated at identical time points. The same procedure was followed to measure the decay kinetics of the injected nitroxide at various time points, which indicated the redox status of the muscle.
Figure 2.

(A) Typical in vivo EPR spectra of the CPA (3-carbamoyl-2,2,5,5-tetramethylpyrrolidin-1-yloxyl free radical, 150 mg/kg) nitroxide injected into mice through the tail vein. The spectra were collected over the gastrocnemius muscle of the mice using a 1.2-GHz EPR spectrometer equipped with an external loop resonator. (B) The middle component of the EPR spectrum was used to follow the reduction of the nitroxide over time to determine tissue redox status.
Figure 3.
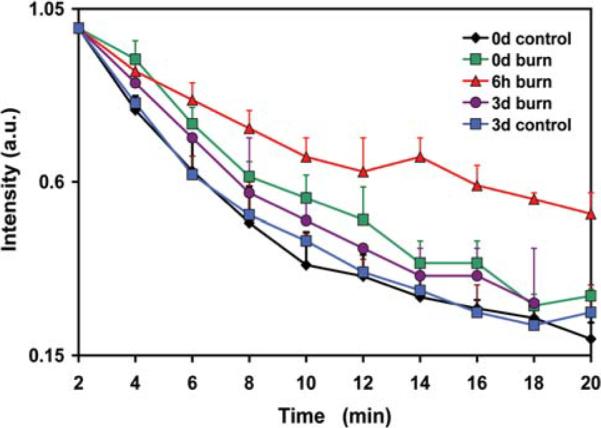
The decay kinetics of the nitroxide (CPA) in the gastrocnemius muscle of the mice in control and burn groups. The redox status of the control group was measured at day 0 (0d) and day 3 (3d). The redox status of the burn group was measured at day 0, 6 h and day 3. Values are means ± SE; n=4.
ROS measurement using nitroxides by in vivo EPR spectroscopy
We measured ROS using EPR spectroscopy with a spin trapping technique that has previously been used to detect ROS production in vivo (31). The upregulation or down-regulation of ROS was measured using a cyclic 1-hydroxy-3-carboxy-2,2,5,5-tetramethylpyrrolidine probe (CPH, Alexis Biochemicals). This EPR inactive probe is oxidized to the corresponding nitroxide (CP•) by ROS. The increase in the EPR signal intensity of the nitroxide provides an indirect measure of the ROS in a tissue. Optimized spectrometer parameters and procedures as described above were used for these experiments. The CPH probe (50 mg/kg) was injected intravenously and the formation of the CP• was followed over time by the placement of the EPR resonator over the gastrocnemius muscle. The increase in the EPR signal intensity of the oxidized probe over time indicated the formation of ROS in the gastrocnemius muscle of the control and burn mice.
RNA extraction
At 6 h, 1 day and 3 days post burn, 3 burned and 3 control mice were anesthetized at each time point by i.p. injection of 40 mg/kg pentobarbital, and the gastrocnemius was excised. All mice were then administered a lethal dose of pentobarbital (200 mg/kg i.p.), while the gastrocnemius muscle was isolated and immediately immersed in 1 ml TRIzol for RNA isolation. The muscle was homogenized for 60 sec using a Brinkmann Polytron PT 3000 homogenizer (Brinkmann Instruments, Westbury, NY) before the extraction of total RNA. Chloroform (200 μl) was added to the homogenized muscle and mixed by inverting the tube for 15 sec. After centrifugation at 12,000 × g for 15 min, the upper aqueous phase was collected and precipitated by adding 500 μl isopropanol. Further centrifugation at 12,000 × g for 10 min separated the RNA pellet, which was then washed with 500 μl 70% ethanol and centrifuged at 7,500 × g for 5 min prior to air-drying. The pellet was re-suspended in 100 μl DEPC-H20. An RNeasy Kit (Qiagen, Valencia, CA) was used to purify the RNA according to the manufacturer's protocol. Purified RNA was quantified by UV absorbance at 260 and 280 nm and stored at −70° for DNA microarray analysis.
Microarray hybridization
Biotinylated cRNA was generated with 10 μg of total cellular RNA according to the protocol outlined by Affymetrix Inc. (Santa Clara, CA). The cRNA was hybridized onto MOE430A oligonucleotide arrays (Affymetrix), stained, washed and scanned according to Affymetrix protocol.
Genomic data analysis
The scanned images of cRNA hybridization were converted to cell intensity files (CEL files) with the Microarray suite 5.0 (MAS, Affymetrix). Data were scaled to a target intensity of 500, and for each time point all possible pair-wise array comparisons of the replicates to control mice were carried out (i.e., four combinations when the two arrays from each time point were compared with the two arrays from control mice) using a MAS 5.0 change call algorithm. Probe sets that had a signal value difference ≥100 and in which both samples were present were scored as differentially modulated when i) the number of change calls in the same direction were at least 3, 4 and 6 when the number of comparisons were 4, 6 and 9, respectively, and ii) the other comparisons were unchanged. Such scoring was to partially compensate for biological stochasticity and technical variation. Based on the ratios of 100 genes determined to be invariant in most of the conditions tested (Affymetrix) in the hind limb of burn and control animals, an additional constraint of a minimum ratio of 1.65 was applied to control the known false positives at 5% hind limb.
Statistical analysis
The Kolmogorov-Smirnov and Levene's tests were used to assess the normality and homogeneity of variances, respectively. Differences between control and burn groups were evaluated at each time point using two-way repeated-measures ANOVA (32) with time and group as factors for pO2, and by the Student's t-test for the rest. Statistical analysis was conducted using SPSS, version 15.0 (SPSS, Chicago, IL), and a two-tailed α level of 0.05 was used as the criterion for the statistical significance of all comparisons.
Results
Tissue pO2 alterations after burn
The changes in tissue pO2 observed at different time points post burn are shown in Fig. 1. The decrease in tissue pO2 was significant immediately (P=0.033) and at 6 h (P=0.004) as compared to the control hind limbs, and returned to the initial tissue pO2 after 3 days (n=4 mice per group). At day 3 and thereafter, the tissue pO2 of the burned group did not differ from the controls. The tissue pO2 of the control hind limb was constant throughout the experiments.
Figure 1.
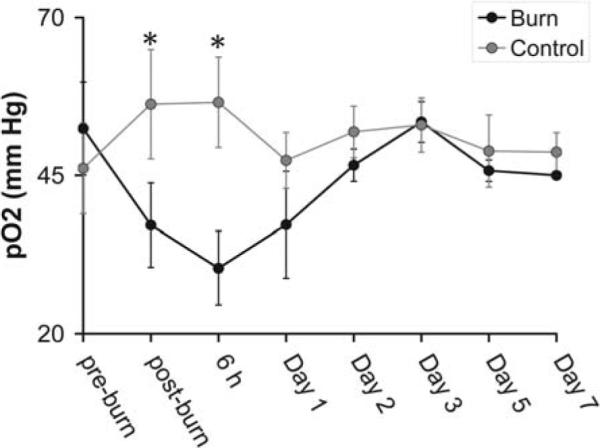
Repeated in vivo tissue pO2 measurements of the gastrocnemius skeletal muscle of the mice using EPR oximetry pre and post burn trauma at different time points. The post-burn data were acquired immediately after the burn followed by measurements at 6 h and on days 1–7. Values are the means ± SE; *P<0.05, n=4.
Burn injury affects the tissue redox status as measured by EPR
The results obtained for redox measurements are shown in Fig. 3. The control mice had the same skeletal muscle redox status on day 0 and 3. Also, no significant difference was found in the decay rates of the nitroxide at day 0 (immediately post burn) and day 3 between the control and burn groups. The decay rate of the nitroxide in the burn group at 6 h (0.000604±0.00008 sec−1) differed significantly from the decay rate in the control group at day 0 (0.00165±0.00024 sec−1, P=0.027) and from the burn group at time 0 (0.00143±0.00151 sec−1, P=0.018, t-test).
ROS production after burn
We measured ROS production with EPR in vivo using of the oxidation of hydroxylamine (CP-H). Before burn (baseline), a low-intensity EPR signal was detectable from the mouse hind limb. Twelve hours after burn, the oxidized CP-H signal increased, indicating an increase in ROS generation by the burn. The oxidized CP-H signal increased between 12 and 24 h, suggesting a further increase in ROS formation (Fig. 4). No such increase in the EPR signal of the nitroxide was observed in the control group at all time points.
Figure 4.
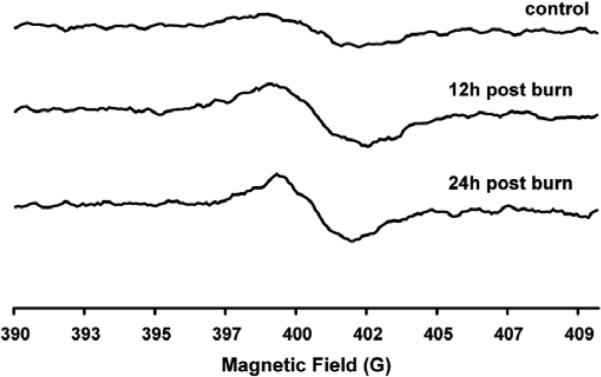
Typical in vivo EPR spectra of the oxidized CPH spin trap injected via the tail vein, and the spectra collected over the gastrocnemius tissue of the mice pre-, 12 h and 24 h post burn trauma. The oxidation of the hydroxylamine to the nitroxide occurs due to its oxidation by ROS in the tissue.
Expression of antioxidant genes after burn
In our transcriptome studies, we compared the expression of all genes that might lead to metabolic dysfunction in skeletal muscle after burn. We found the differential expression of three metabolic genes at 3 time points following a localized, but severe, hind limb burn (Table I). Slc25a4 or Ant1 gene expression was downregulated at 1 and 3 days post burn, Sod2 expression was downregulated at day 1, and the expression of Gpx1 was moderately upregulated at 3 days post burn.
Table I.
Differential expression of selected antioxidant genes at 6 h, day 1 and day 3 post burn trauma.
| GenBank accession no. | Product | Gene symbol | 6h | Day 1 | Day 3 |
|---|---|---|---|---|---|
| AV327862 | Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 4 | Slc25a4 or Ant1 | / | −1.41 | −1.61 |
| AW701319 | Superoxide dismutase 2, mitochondrial | Sod2 | / | −1.72 | / |
| BI219063 | Glutathione peroxidase 1 | Gpx1 | / | / | +1.81 |
Values represent the relative expression intensity in burn animals versus normal control animals. (/) not differentially expressed compared to controls.
Discussion
In vivo EPR was successfully used to investigate changes in tissue pO2, redox state and ROS generation after burn injury. The downregulation of the antioxidant genes Ant1 and Sod2 in skeletal muscle, which parallels the rise of mitochondrial ROS as detected by in vivo EPR, suggests that insufficient oxidative defense following local burn trauma may lead to mitochondrial dysfunction via mtDNA oxidative damage due to ROS toxicity. Our findings suggest that tissue pO2, redox state and ROS by EPR in skeletal muscle could serve as markers of metabolic changes for following the pathophysiology of burn trauma and associated oxidative damage.
The principal findings of the present study are complementary to findings by NMR, suggesting that a major factor in the progression of mitochondrial skeletal muscle dysfunction in burns results from defects in oxidative phosphorylation (OXPHOS) (22–24,33,34). Indeed, findings with the same mouse `local' burn model (affecting the hind limb and representing 3–5% of total body surface area) demonstrated that burn injury causes a significant dysregulation in OXPHOS and a decrease in the ATP synthesis rate (23). Taken together, NMR and the present EPR oximetry data suggest that the decrease in ATP synthesis rate at day 3 previously observed following burn trauma (23) might be preceded by a significant reduction in tissue pO2 at 6 h, as observed in this study. Of note, the ATP synthesis rate remains significantly low, even after tissue pO2 have normalized, which indicates that the dysregulation of OXPHOS in burn trauma is not tissue O2-dependent. On the other hand, the dysregulation of OXPHOS in burn trauma at 24 h and thereafter is preceded by the downregulation of a key transcription factor, the peroxisome proliferator-activated receptor γ coactivator 1, or PBC-1ß (23). These are hypothesized to play a central role in regulating energy homeostasis and metabolism (35) at 6 h post burn (23), which coincides with significant changes in EPR-derived parameters at the same time point as shown in this study. This downregulation of PBC-1ß by 6 h post burn probably causes the downregulation of the genes involved in OXPHOS (23) and in detoxification, mainly Ant1 and Sod2 (Table I), since the expression of such genes is downregulated by 24 h post burn. As a result of this dysregulation, the balance of detoxification and ROS generation, which has been detected as accumulating as early as 12 h post burn, is prevented. To this end, the present study confirms the notion that certain components of the ROS scavenging pathway are linked by the PBC-1ß to mitochondrial OXPHOS, apparently enabling or disabling cells to maintain normal redox status in response to changing oxidative capacity (36).
Another major finding of this study is the effect of the burn on skeletal muscle redox status, which coincided with decreased expression in two major antioxidant systems; that is, a decrease in Ant1 and Sod2 gene expression, and a moderate increase in Gpx1 gene expression. The mitochondria are one of the major sources of endogenous ROS in the cell, including superoxide anions (O2•-), hydrogen peroxide (H2O2), and hydroxyl radicals (OH• ) (37). Since OH• is largely limited by diffusion, it is more likely to cause damage directly to the mitochondria, especially to the inner membrane where they are generated. Additionally, ROS can initiate damage to nucleic acids, proteins and lipids (38,39). Since mitochondrial DNA (mtDNA) is located closer to the site of ROS generation, lacks protective histones and has more limited base excision repair mechanisms than the nucleus, it is more vulnerable to oxidative damage than nuclear DNA (nDNA) (40,41). Moreover, due to its proximity to OXPHOS in the mitochondrial inner membrane, mtDNA has been hypothesized to be a major target for ROS damage. Hence, inhibition of OXPHOS is predicted to result in the progressive destruction of mtDNA. Since OXPHOS is significantly inhibited in skeletal muscle post burn (23,24), we hypothesized that the mitochondria should produce higher levels of ROS post burn. This has been confirmed in this study. The increased ROS should induce antioxidant defenses and prevent damage to mitochondrial macromolecules; in the absence of such defenses oxidative damage ensues. Based on these findings, we suggest that increased ROS results in oxidative damage of mitochondrial macromolecules, possibly including mtDNA. Indeed, this is what happens in Ant1 knockout mice, the mitochondria of which produce increased levels of ROS and show a striking increase in the accumulation of mtDNA rearrangements (42). In agreement with this, increased mitochondrial oxidative stress is reported in mice lacking the glutathione peroxidase-1 gene (43).
In aging, where we also have mitochondrial dysfunction, reports have indicated that somatic mtDNA mutations cause OXPHOS dysfunction without affecting antioxidant systems and ROS production, and that OXPHOS dysfunction is the primary inducer of premature aging in mtDNA-mutated mice (44). Others, however, have reported that electron leakage from the electron transport chain causes specific damage to their subunits and increased ROS generation as oxidative damage accumulates (45). This leads to further mitochondrial dysfunction in a cyclical process, which underlies the progressive physiological decline of the aged mouse heart (45). In another study, the DNA damage induced by acrolein in HepG2 cells was related to oxidative stress produced as a result of increased ROS and the depletion of glutathione (GSH) (45,46). Studies in heart muscle suggest that antioxidant vitamin therapy following burn trauma provides at least partial cardio-protection by inhibiting translocation of the transcription factor NF-κB and by interrupting cardiac inflammatory cytokine secretion (47). Additional studies by the same research group support the hypothesis that oxidative stress critically influences the heart following burn injury, suggesting that antioxidant strategies designed to either inhibit free radical formation or to scavenge free radicals may provide organ protection in patients with burn injury (48). To date, there have been no studies that stimulated mitochondrial production of ROS or measured the resulting ROS and/or bioenergetic and oxidative damage in the skeletal muscle.
Finally, our results indicate that EPR is a promising tool for making an in vivo assessment of the consequences of burn injury in well-established burn injury models. We also propose that it is complementary to NMR, which has already shown the consequences of bioenergetic and mitochondrial dysfunction in vivo (22–25). Thus, our results lead us to conclude that bioenergetic dysfunction and insufficient oxidative defense following local burn trauma may lead to oxidative damage. In our future multidisciplinary studies, we plan to test this hypothesis, to potentially assign physiological functions to key regulatory genes, and to demonstrate whether these genes are involved in burn injury. These insights will lead to new therapeutic strategies for the prevention of potentially fatal complications in burn patients. We also believe that our findings may be clinically relevant in molecular medicine.
Acknowledgements
This work was supported in part by a Shriners Hospital for Children research grant no. 8893 to A. Aria Tzika, a National Institutes of Health (NIH) Center Grant P50GM021700 (Ronald G. Tompkins, PI; A. Aria Tzika, Director of NMR Core), and by a National Institute of Diabetes and Digest and Kidney (NIDDK) research grant no. DK072112 to Nadeem Khan. We thank Dr Huagang Hou, Dartmouth Medical School, for advice on the procedures for EPR measurements. We also thank Drs Katie Edmondson and Ann Power Smith of Write Science Right for editorial assistance.
References
- 1.Gallez B, Swartz HM. In vivo EPR: when, how and why? NMR Biomed. 2004;17:223–225. doi: 10.1002/nbm.913. [DOI] [PubMed] [Google Scholar]
- 2.Swartz HM, Khan N, Buckey J, et al. Clinical applications of EPR: overview and perspectives. NMR Biomed. 2004;17:335–351. doi: 10.1002/nbm.911. [DOI] [PubMed] [Google Scholar]
- 3.Helisch A, Wagner S, Khan N, et al. Impact of mouse strain differences in innate hindlimb collateral vasculature. Arterioscler Thromb Vasc Biol. 2006;26:520–526. doi: 10.1161/01.ATV.0000202677.55012.a0. [DOI] [PubMed] [Google Scholar]
- 4.Khan N, Williams BB, Hou H, Li H, Swartz HM. Repetitive tissue pO2 measurements by electron paramagnetic resonance oximetry: current status and future potential for experimental and clinical studies. Antioxid Redox Signal. 2007;9:1169–1182. doi: 10.1089/ars.2007.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Towner RA, Sturgeon SA, Khan N, Hou H, Swartz HM. In vivo assessment of nodularin-induced hepatotoxicity in the rat using magnetic resonance techniques (MRI, MRS and EPR oximetry) Chem Biol Interact. 2002;139:231–250. doi: 10.1016/s0009-2797(02)00002-9. [DOI] [PubMed] [Google Scholar]
- 6.James PE, Bacic G, Grinberg OY, et al. Endotoxin-induced changes in intrarenal pO2, measured by in vivo electron paramagnetic resonance oximetry and magnetic resonance imaging. Free Radic Biol Med. 1996;21:25–34. doi: 10.1016/0891-5849(95)02221-x. [DOI] [PubMed] [Google Scholar]
- 7.Swartz HM, Khan N, Khramtsov VV. Use of electron paramagnetic resonance spectroscopy to evaluate the redox state in vivo. Antioxid Redox Signal. 2007;9:1757–1771. doi: 10.1089/ars.2007.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan N, Swartz H. Measurements in vivo of parameters pertinent to ROS/RNS using EPR spectroscopy. Mol Cell Biochem. 2002;234:341–357. [PubMed] [Google Scholar]
- 9.Khan N, Williams B, Swartz H. Clinical applications of in vivo EPR: rationale and initial results. Appl Magn Reson. 2006;30:185–199. [Google Scholar]
- 10.Szczepaniak LS, Babcock EE, Schick F, et al. Measurement of intracellular triglyceride stores by 1H spectroscopy: validation in vivo. Am J Physiol. 1999;276:E977–E989. doi: 10.1152/ajpendo.1999.276.5.E977. [DOI] [PubMed] [Google Scholar]
- 11.Kuhlmann J, Neumann-Haefelin C, Belz U, Kramer W, Juretschke HP, Herling AW. Correlation between insulin resistance and intramyocellular lipid levels in rats. Magn Reson Med. 2005;53:1275–1282. doi: 10.1002/mrm.20501. [DOI] [PubMed] [Google Scholar]
- 12.Boesch C, Slotboom J, Hoppeler H, Kreis R. In vivo determination of intra-myocellular lipids in human muscle by means of localized 1H-MR-spectroscopy. Magn Reson Med. 1997;37:484–493. doi: 10.1002/mrm.1910370403. [DOI] [PubMed] [Google Scholar]
- 13.Schick F, Eismann B, Jung WI, Bongers H, Bunse M, Lutz O. Comparison of localized proton NMR signals of skeletal muscle and fat tissue in vivo: two lipid compartments in muscle tissue. Magn Reson Med. 1993;29:158–167. doi: 10.1002/mrm.1910290203. [DOI] [PubMed] [Google Scholar]
- 14.Neumann-Haefelin C, Kuhlmann J, Belz U, et al. Determinants of intramyocellular lipid concentrations in rat hindleg muscle. Magn Reson Med. 2003;50:242–248. doi: 10.1002/mrm.10518. [DOI] [PubMed] [Google Scholar]
- 15.Hwang JH, Pan JW, Heydari S, Hetherington HP, Stein DT. Regional differences in intramyocellular lipids in humans observed by in vivo 1H-MR spectroscopic imaging. J Appl Physiol. 2001;90:1267–1274. doi: 10.1152/jappl.2001.90.4.1267. [DOI] [PubMed] [Google Scholar]
- 16.Krssak M, Petersen KF, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 17.Krssak M, Petersen KF, Bergeron R, et al. Intramuscular glycogen and intramyocellular lipid utilization during prolonged exercise and recovery in man: a 13C and 1H nuclear magnetic resonance spectroscopy study. J Clin Endocrinol Metab. 2000;85:748–754. doi: 10.1210/jcem.85.2.6354. [DOI] [PubMed] [Google Scholar]
- 18.Jacob S, Machann J, Rett K, et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–1119. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 19.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001;86:5755–5761. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 20.Anderwald C, Bernroider E, Krssak M, et al. Effects of insulin treatment in type 2 diabetic patients on intracellular lipid content in liver and skeletal muscle. Diabetes. 2002;51:3025–3032. doi: 10.2337/diabetes.51.10.3025. [DOI] [PubMed] [Google Scholar]
- 21.Sinha R, Dufour S, Petersen KF, et al. Assessment of skeletal muscle triglyceride content by (1)H nuclear magnetic resonance spectroscopy in lean and obese adolescents: relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes. 2002;51:1022–1027. doi: 10.2337/diabetes.51.4.1022. [DOI] [PubMed] [Google Scholar]
- 22.Astrakas LG, Goljer I, Yasuhara S, et al. Proton NMR spectroscopy shows lipids accumulate in skeletal muscle in response to burn trauma-induced apoptosis. FASEB J. 2005;19:1431–1440. doi: 10.1096/fj.04-2005com. [DOI] [PubMed] [Google Scholar]
- 23.Padfield KE, Astrakas LG, Zhang Q, et al. Burn injury causes mitochondrial dysfunction in skeletal muscle. Proc Natl Acad Sci USA. 2005;102:5368–5373. doi: 10.1073/pnas.0501211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tzika AA, Mintzopoulos D, Padfield K, et al. Reduced rate of adenosine triphosphate synthesis by in vivo 31P nuclear magnetic resonance spectroscopy and downregulation of PGC-1ß in distal skeletal muscle following burn. Int J Mol Med. 2008;21:201–208. [PubMed] [Google Scholar]
- 25.Tzika AA, Astrakas LG, Cao H, et al. Murine intramyocellular lipids quantified by NMR act as metabolic biomarkers in burn trauma. Int J Mol M ed. 2008;21:825–832. [PubMed] [Google Scholar]
- 26.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 27.Tomera JF, Martyn J. Systemic effects of single hindlimb burn injury on skeletal muscle function and cyclic nucleotide levels in the murine model. Burns Incl Therm Inj. 1988;14:210–219. doi: 10.1016/0305-4179(88)90040-x. [DOI] [PubMed] [Google Scholar]
- 28.Dunn J, Swartz H. In vivo electron paramagnetic resonance oximetry with particulate materials. Methods. 2003;30:159–166. doi: 10.1016/s1046-2023(03)00077-x. [DOI] [PubMed] [Google Scholar]
- 29.He G, Kutala VK, Kuppusamy P, Zweier JL. In vivo measurement and mapping of skin redox stress induced by ultraviolet light exposure. Free Radic Biol Med. 2004;36:665–672. doi: 10.1016/j.freeradbiomed.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 30.Ilangovan G, Li H, Zweier JL, Kuppusamy P. In vivo measurement of tumor redox environment using EPR spectroscopy. Mol Cell Biochem. 2002;234:393–398. [PubMed] [Google Scholar]
- 31.Liu S, Timmins GS, Shi H, Gasparovic CM, Liu KJ. Application of in vivo EPR in brain research: monitoring tissue oxygenation, blood flow, and oxidative stress. NMR Biomed. 2004;17:327–334. doi: 10.1002/nbm.899. [DOI] [PubMed] [Google Scholar]
- 32.Vittinghoff E, Glidden D, Shiboski S, McCulloch C. Linear, Logistic, Survival, and Repeated Measures Models. Springer; New York: 2005. Regression Methods in Biostatistics. [Google Scholar]
- 33.Padfield KE, Zhang Q, Gopalan S, et al. Local and distant burn injury alter immunoinflammatory gene expression in skeletal muscle. J Trauma. 2006;61:280–292. doi: 10.1097/01.ta.0000230567.56797.6c. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Cao H, Astrakas LG, et al. Uncoupling protein 3 expression and intramyocellular lipid accumulation by NMR following local burn trauma. Int J Mol Med. 2006;18:1223–1229. [PubMed] [Google Scholar]
- 35.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 36.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Wallace DC, Melov S. Radicals r'aging. Nat Genet. 1998;19:105–106. doi: 10.1038/448. [DOI] [PubMed] [Google Scholar]
- 38.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 39.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAAPS J. 2006;8:E521–E531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 41.Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci USA. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esposito LA, Kokoszka JE, Waymire KG, Cottrell B, MacGregor GR, Wallace DC. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic Biol Med. 2000;28:754–766. doi: 10.1016/s0891-5849(00)00161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trifunovic A, Hansson A, Wredenberg A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choksi KB, Papaconstantinou J. Age-related alterations in oxidatively damaged proteins of mouse heart mitochondrial electron transport chain complexes. Free Radic Biol Med. 2008;44:1795–1805. doi: 10.1016/j.freeradbiomed.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Jiang L, Geng C, Cao J, Zhong L. The role of oxidative stress in acrolein-induced DNA damage in HepG2 cells. Free Radic Res. 2008;42:354–361. doi: 10.1080/10715760802008114. [DOI] [PubMed] [Google Scholar]
- 47.Horton JW, White DJ, Maass DL, Hybki DP, Haudek S, Giroir B. Antioxidant vitamin therapy alters burn trauma-mediated cardiac NF-kappaB activation and cardiomyocyte cytokine secretion. J Trauma. 2001;50:397–407. doi: 10.1097/00005373-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Horton JW. Free radicals and lipid peroxidation mediated injury in burn trauma: the role of antioxidant therapy. Toxicology. 2003;189:75–88. doi: 10.1016/s0300-483x(03)00154-9. [DOI] [PubMed] [Google Scholar]