

Abstract
Technological advances in genotyping have given rise to hypothesis-based association studies of increasing scope. As a result, the scientific hypotheses addressed by these studies have become more complex and more difficult to address using existing analytic methodologies. Obstacles to analysis include inference in the face of multiple comparisons, complications arising from correlations among the SNPs (single nucleotide polymorphisms), choice of their genetic parametrization and missing data. In this paper we present an efficient Bayesian model search strategy that searches over the space of genetic markers and their genetic parametrization. The resulting method for Multilevel Inference of SNP Associations, MISA, allows computation of multilevel posterior probabilities and Bayes factors at the global, gene and SNP level, with the prior distribution on SNP inclusion in the model providing an intrinsic multiplicity correction. We use simulated data sets to characterize MISA’s statistical power, and show that MISA has higher power to detect association than standard procedures. Using data from the North Carolina Ovarian Cancer Study (NCOCS), MISA identifies variants that were not identified by standard methods and have been externally “validated” in independent studies. We examine sensitivity of the NCOCS results to prior choice and method for imputing missing data. MISA is available in an R package on CRAN.
Key words and phrases: AIC, Bayes factor, Bayesian model averaging, BIC, Evolutionary Monte Carlo, false discovery, genetic models, lasso, model uncertainty, single nucleotide polymorphism, variable selection
1. Introduction
Recent advances in genotyping technology have resulted in a dramatic change in the way hypothesis-based genetic association studies are conducted. While previously investigators were limited by costs to investigating only a handful of variants within the most interesting genes, researchers may now conduct candidate-gene and candidate-pathway studies that encompass many hundreds or thousands of genetic variants, often single nucleotide polymorphisms (SNPs). For example, the North Carolina Ovarian Cancer Study (NCOCS) [Schildkraut et al. (2008)], an ongoing population-based case-control study, genotyped 2129 women at 1536 SNPS in 170 genes on 8 pathways, where “pathway” is defined as a set of genes thought to be simultaneously active in certain circumstances.
The analytic procedure most commonly applied to association studies of this scale is to fit a separate model of association for each SNP that adjusts for design and confounder variables. As false discoveries due to multiple testing are often a concern, the level of significance for each marginal test of association is adjusted using Bonferroni or other forms of false discovery correction [Storey (2002); Wacholder (2004); Balding (2006)]. While these methods have been shown to be effective in controlling the number of false discoveries reported, correlations between the markers may limit the power to detect true associations [Efron (2007)]. The NCOCS study provides a case in point. When simple marginal methods are applied to the NCOCS data, no SNPs are identified as notable.
Marginal SNP-at-a-time methods do not address directly many of the scientific questions in candidate pathway studies, such as “Is there an overall association between a pathway and the outcome of interest?” and “Which genes are most likely to be driving this association?” The Multilevel Inference for SNP Association (MISA) method we describe here is designed to simultaneously address these questions of association at the level of SNP, gene and pathway.
MISA, in contrast to the marginal methods, identifies ten SNPs of interest in the NCOCS study. To date, one of these (ranked tenth by MISA) has been validated in external data by a large multi-center consortium [Schildkraut et al. (2009)]; additional testing is underway for other top SNPs discovered by MISA. To buttress this empirical evidence, we demonstrate using simulation studies (Section 4) that MISA has higher power to detect associations than other simpler procedures, with a modest increase in the false discovery rate (Figure 1).
Fig. 1.

True and false positive rates of MISA versus alternative methods.
In the next section we describe the Bayesian hierarchical model behind MISA and highlight how it addresses many of the key issues in analysis of SNP association studies: identification of associated SNPs and genetic models, missing data, inference for multi-level hypotheses and control of the false discovery rate. Like stepwise logistic regression [Balding (2006)], lasso [Park and Hastie (2008); Shi, Lee and Wahba (2007); Wu et al. (2009)] and logic regression [Ruczinski, Kooperberg and LeBlanc (2003); Kooperberg and Ruczinski (2004); Schwender and Ickstadt (2007)], MISA improves upon marginal, SNP-at-a-time methods by modeling the outcome variable as a function of a multivariate genetic profile, which provides measures of association that are adjusted for the remaining markers. MISA uses Bayesian Model Averaging [Hoeting et al. (1999)] to combine information from multiple models of association to address the degree to which the data support an association at the level of individual SNPs, genes and pathways, while taking into account uncertainty regarding the best genetic parametrization. By using model averaging, MISA improves upon methods that select a single model, which may miss important SNPs because of LD structure. We show how the prior distribution on SNP inclusion provides a built-in multiplicity correction. Because missing data are a common phenomenon in association studies, we discuss two options for handling this problem.
In Section 3 we present an Evolutionary Monte Carlo algorithm to efficiently sample models of association according to their posterior probabilities. In Section 4 we apply our method to simulated data sets and demonstrate that MISA outperforms less complex and more commonly used alternatives for detecting associations in modestly powered candidate-gene case-control studies. The simulation approach may also be used to guide selection of the prior hyperparameters given the study design. In Section 5 we return to the NCOCS study and present results from the analysis of a single pathway from that study. We examine the sensitivity of results to prior hyperparameter choice and methods for imputing missing data. We conclude in Section 6 with recommendations and a discussion of future extensions.
2. Models of association
We consider SNP association models with a binary phenotype, such as presence or absence of a disease as in case-control designs. For i = 1, …, n, let Di indicate the disease status of individual i, where Di = 1 represents a disease case and Di = 0 represents a control. For each individual, we have S SNP measurements, where SNP s is either homozygous common (As As), heterozygous (as As or As as), homozygous rare (as as), or missing and is coded as 0, 1, 2, representing the number of rare alleles, or NA if the SNP is missing for that individual. We will discuss methods for imputing missing SNP data in Section 2.3. In addition to the SNP data, for each individual we have a q-dimensional vector of design and potential confounding variables that will be included in all models, henceforth referred to as “design” variables.
We use logistic regression models to relate disease status to the design variables and subsets of SNPs. We denote the collection of all possible models by 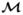 . An individual model, denoted by
. An individual model, denoted by 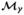 , is specified by the S dimensional vector γ, where γs indicates the inclusion and SNP-specific genetic parametrization of SNP s in model : γs = 0 if SNPs ∉ , γs = 1 if SNPs ∈ with a log-additive parametrization, γs = 2 if SNPs ∈ with a dominant parametrization, and γs = 3 if SNPs ∈ with a recessive parametrization. When no homozygous rare cases or controls are observed, we fix the genetic parametrization to be log-additive. Under each of these genetic parametrizations, SNP s may be encoded using one degree of freedom. In particular, for the log-additive model, the design variable representing SNP s is a numeric variable equal to the number of copies of the risk allele as. For the dominant model, we use an indicator variable of whether allele as is present (homozygous rare or heterozygous) and for the recessive model, an indicator variable of whether SNP s has the homozygous rare genotype. For each individual, the logistic regression under model assuming complete data is given by
, is specified by the S dimensional vector γ, where γs indicates the inclusion and SNP-specific genetic parametrization of SNP s in model : γs = 0 if SNPs ∉ , γs = 1 if SNPs ∈ with a log-additive parametrization, γs = 2 if SNPs ∈ with a dominant parametrization, and γs = 3 if SNPs ∈ with a recessive parametrization. When no homozygous rare cases or controls are observed, we fix the genetic parametrization to be log-additive. Under each of these genetic parametrizations, SNP s may be encoded using one degree of freedom. In particular, for the log-additive model, the design variable representing SNP s is a numeric variable equal to the number of copies of the risk allele as. For the dominant model, we use an indicator variable of whether allele as is present (homozygous rare or heterozygous) and for the recessive model, an indicator variable of whether SNP s has the homozygous rare genotype. For each individual, the logistic regression under model assuming complete data is given by
| (2.1) |
where xγi represents the coding of SNPs in model and θγ is the vector of model specific parameters (α0, αT,
), with intercept α0, vector of design variable coefficients α and log-odds ratios βγ. Prospective models for disease outcome given multivariate genetic marker data as in equation (2.1) provide measures of association that are adjusted for other markers which can increase the power to detect associations [Balding (2006)], however, one is faced with an extremely large collection of possible models. While stepwise selection methods may be used to select a single model [Cordell and Clayton (2002)], this leads to difficulty in interpreting the significance of SNPs in the selected model. Bayesian model averaging is an alternative to stepwise selection methods and is an effective approach for identifying subsets of likely associated variables, for prioritizing them and for measuring overall association in the presence of model uncertainty [see the review articles by Hoeting et al. (1999) and Clyde and George (2004) and the references therein].
2.1. Posterior inference
Posterior model probabilities measure the degree to which the data support each model in a set of competing models. The posterior model probability of any model in the space of models is expressed as
where p(D|) is the (marginal) likelihood of model obtained after integrating out model-specific parameters θγ with respect to their prior distribution, and p() is the prior probability of .
While posterior probabilities provide a measure of evidence for hypotheses or models, it is often difficult to judge them in isolation, as individual model probabilities may be “diluted” as the space of models grows [Clyde (1999); George (1999); Clyde and George (2004)]. Bayes factors (BF) [Kass and Raftery (1995)] compare the posterior odds of any two models (or hypotheses) to their prior odds
and measures the change in evidence (on the log scale) provided by data for one model, 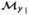 , to another,
, to another, 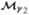 , or for pairs of hypotheses. Goodman (1999) and Stephens and Balding (2009) provide a discussion on the usefulness of Bayes factors in the medical context and Wakefield (2007) illustrates their use in controlling false discoveries in genetic epidemiology studies. Below we define Bayes factors for quantifying association at multiple levels (global, gene and SNP) and assessing the most likely SNP-specific genetic parametrization.
, or for pairs of hypotheses. Goodman (1999) and Stephens and Balding (2009) provide a discussion on the usefulness of Bayes factors in the medical context and Wakefield (2007) illustrates their use in controlling false discoveries in genetic epidemiology studies. Below we define Bayes factors for quantifying association at multiple levels (global, gene and SNP) and assessing the most likely SNP-specific genetic parametrization.
2.1.1. Global Bayes factor
The Bayes factor in favor of HA, the alternative hypothesis that there is at least one SNP associated with disease, to H0, the null hypothesis that there is no association between the SNPs under consideration and disease, measures the relative weight of evidence of HA to H0. The null model corresponding to H0 is the model which includes only design variables and no SNPs, and is denoted 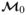 . The alternative hypothesis is represented by all of the remaining models in . Because the space of models is large, the null model (or any single model in general) may receive small probability (both prior and posterior), even when it is the highest posterior probability model (this illustrates the dilution effect of large model spaces); Bayes factors allow one to judge how the posterior odds compare to one’s prior odds.
. The alternative hypothesis is represented by all of the remaining models in . Because the space of models is large, the null model (or any single model in general) may receive small probability (both prior and posterior), even when it is the highest posterior probability model (this illustrates the dilution effect of large model spaces); Bayes factors allow one to judge how the posterior odds compare to one’s prior odds.
The Global Bayes factor for comparing HA to H0 may be simplified to
| (2.2) |
which is the weighted average of the individual Bayes factors BF(: ) for comparing each model in HA to the null model with weights given by the prior probability of conditional on being in HA, p(|HA). Because the alternative is a composite hypothesis, the resulting Global Bayes factor is not independent of the prior distribution on the models that comprise the alternative, thus, the prior distribution on models will play an important role in controlling the (relative) weights that models of different sizes receive. For a large number of SNPs, it is impossible to enumerate the space of models and posterior summaries are often based on models sampled from the posterior distribution. In equation (2.2), if we replace the average over all models in HA with the average over the models in 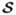 (the collection of unique models sampled from the posterior distribution), the result
(the collection of unique models sampled from the posterior distribution), the result
is a lower bound for the Bayes factor for testing global association. If the lower bound indicates evidence of an association, then we can be confident that this evidence will only increase as we include more models.
2.1.2. SNP Bayes factors
While it is of interest to quantify association at the global level, interest is primarily in identifying the gene(s) and variant(s) within those genes that drive the association. We begin by defining SNP inclusion probabilities and associated Bayes factors. These marginal summaries are adjusted for the other potentially important SNPs and confounding variables and provide a measure of the strength of association at the level of individual SNPs. Given each sampled model ∈ and the model specification vectors γ = (γ1, γ2, …, γS) previously defined in Section 2, the inclusion probability for SNP s is estimated as
| (2.3) |
where p(|D, ) is the posterior probability of a model re-normalized over the sampled model space. The SNP Bayes factor is the ratio of the posterior odds of the SNP being associated to the prior odds of the same, and is defined as
where p(γs ≠ 0) is the prior probability of SNP s being associated. Estimates of the SNP Bayes factor may be obtained using the estimated SNP inclusion probabilities from (2.3).
2.1.3. Gene Bayes factor
In cases where there are SNPs in Linkage Disequilibrium (LD), SNP inclusion probabilities may underestimate the significance of an association at a given locus. This occurs because SNPs in LD may provide competing explanations for the association, thereby diluting or distributing the probability over several markers. Since the amount of correlation between markers across different genes is typically negligible, calculating inclusion probabilities and Bayes factors at the gene level will not be as sensitive to this dilution. A gene is defined to be associated if one or more of the SNPs within the given gene are associated. Hence, we define the gene inclusion probability as
where Γg = 1 if at least one SNP in gene g is in model and is zero otherwise. The gene Bayes factor is defined as
where p(Γg = 1) is the prior probability of one or more SNPs in gene g being associated.
2.1.4. Interpreting evidence
Jeffreys [(1961), page 432], presents a descriptive classification of Bayes factors into “grades of evidence” (reproduced in Table 1) to assist in their interpretation [see Kass and Raftery (1995)]. In the context in which he presents the grades, he defined the Bayes factor assuming equal prior odds, making it equivalent to posterior odds and enabling a meaningful interpretation in terms of probabilities. It is not clear whether he intended his descriptive grades to be used more broadly for interpreting Bayes factors or for interpreting posterior probabilities.
Table 1.
Jeffrey’s grades of evidence [Jeffreys (1961), page 432]
| Grade | BF (HA: H0) | Evidence against H0 |
|---|---|---|
| 1 | 1 – 3.2 | Indeterminate |
| 2 | 3.2 – 10 | Positive |
| 3 | 10 – 31.6 | Strong |
| 4 | 31.6 – 100 | Very strong |
| 5 | >100 | Decisive |
Jeffreys was well aware of the issues that arise with testing several simple alternative hypotheses against a null hypothesis [Jeffreys (1961), Section 5.04], noting that if one were to test several hypotheses separately, that by chance one might find one of the Bayes factors to be less than one even if all null hypotheses were true. He suggested that, in this context, the Bayes factors needed to be “corrected for selection of hypotheses” by multiplying by the prior odds.
Experience has shown that detectable SNP associations are relatively infrequent, hence, the prior odds of any given SNP being marginally associated in the typical genetic association study should be small. For this reason, Stephens and Balding (2009) suggest that marginal Bayes factors calculated assuming equal prior odds be interpreted in light of a prior odds more appropriate to the study at hand. Our approach to the problem of exploring multiple hypotheses is to embed each of the potential submodels (corresponding to a subset of SNPs) into a single hierarchical model. Unlike the marginal (one-at-a-time) Bayes factors in Stephens and Balding (2009) that are independent of the prior odds on the hypotheses, our SNP Bayes factors are based on comparing composite hypotheses and hence do depend on the prior distribution over models, which implicitly adjusts for the selection of hypotheses.
While Bayes factors do not provide a measure of absolute support for or against a hypothesis (except with even prior odds), the log Bayes factor does provide a coherent measure of how much the data change the support for the hypothesis (relative to the prior) [Lavine and Schervish (1997)]. Applying Jeffreys grades to Bayes factors using priors distributions that account for competing hypotheses provides an idea of the impact of the data on changing prior beliefs, but ultimately posterior odds provide a more informative measure of evidence and model uncertainty.
2.2. Prior distributions, Laplace approximations and marginal likelihoods
We assume normal prior distributions for the coefficients θγ with a covariance matrix that is given by a constant 1/k times the inverse Fisher Information matrix. For logistic regression models, analytic expressions for p(D|) are not available and Laplace approximations or the Bayes Information Criterion are commonly used to approximate the marginal likelihood [Raftery (1986); Wakefield (2007); Burton et al. (2007)]. Using a Laplace approximation with the normal prior distribution [Wilson et al. (2010)], the posterior probability of model takes the form of a penalized likelihood
| (2.4) |
where dev(; D) = −2 log(p(D|θ̂γ, )) is the model deviance, and the penalty term pen() encompasses a penalty on model size induced by the choice of k in the prior distribution on coefficients θγ and the prior distribution over models. Because we expect that effect sizes will be small, we calibrate the choice of k based on the Akaike information criterion [Wilson et al. (2010)], leading to
2.3. Missing data
The expression in (2.4) assumes complete data on all SNPs. Missing SNP data, unfortunately, are the norm rather than the exception in association studies. Removing all subjects with any missing SNP genotype data will typically result in an unnecessary loss of information and potential bias of estimated effects if the missing data are nonignorable. It is possible, however, to exploit patterns in LD to efficiently impute the missing genotypes given observed data [Balding (2006)]. We use fastPHASE [Stephens, Smith and Donnelly (2001); Servin and Stephens (2007)] to sample haplotypes and missing genotypes (Gm) given the observed unphased genotypes (Go). This assumes that the pattern of missing data is independent of case-control status, which, if not true, may lead to serious biases [Clayton et al. (2005)]. This assumption may be examined by using indicator variables of missingness as predictors in MISA.
The posterior probabilities of models given the data are obtained by averaging the marginal likelihood of a model over imputed genotype data:
| (2.5) |
where I is the number of imputed data sets, dev(; D, Go, Gm) is the deviance based on the completed data, and Ψ() is an estimate of the un-normalized posterior model probability for model . We have found that the number of imputed sets must be on the order of I = 100 to provide accurate estimates of posterior quantities. This has a significant computational impact in the model search algorithm described in Section 3. As a simple alternative, we approximate (2.5) by a modal approximation, where the missing genotypes are imputed with the mode of the sampled genotypes using fastPHASE. While it is well known that plugging in a single estimate for the missing data under-estimates uncertainty, the modal approximation provides dramatic computational savings. In Section 5 we examine the sensitivity of results to the method of imputing missing data and find that the modal approximation gives comparable results for SNP BFs.
2.4. Choice of prior distribution on models
The prior distribution on the space of models , p(), completes our model specification. The frequentist approach for SNP association studies usually involves some form of adjustment for multiple-testing, which can, in effect, penalize the researcher who looks beyond single-SNP models of association to multiple SNP models or models of interactions. Under the Bayesian approach, posterior evidence in the data is judged against the prior odds of an association using Bayes factors, which should not be affected by the number of tests that an investigator chooses to carry out [Balding (2006)].
While it has been common practice to adopt a “noninformative” uniform distribution over the space of models for association (this is after marginalizing over the possible genetic models for each SNP), this choice has the potentially undesirable “informative” implication that of the SNPs are expected to be associated a priori, and the prior odds of at least one SNP being included (which is used in the global Bayes factor) depends on the number of tests (2S) (Table 2).
Table 2.
General prior characteristics and limiting behavior (in parentheses) of the Bin(S, 1/2), BetaBinomial(1, 1) and BetaBinomial(1, λS) distribution on model size
| Binomial (S, 1/2) | Beta-Binomial (1, 1) | Beta-Binomial (1, λS) | ||||
|---|---|---|---|---|---|---|
| Expected model size |
|
|
|
|||
| Global prior odds of an association |
|
S(∞) |
|
|||
| Marginal prior odds of an association | 1 | 1 |
|
|||
| Prior odds of adding a variable | 1 |
|
|
A recommended alternative is the Beta-Binomial distribution on the model size, which provides over-dispersion, added robustness to prior misspecification and multiplicity corrections as a function of the number of variables [Ley and Steel (2009); Scott and Berger (2010); Cui and George (2008)]. We construct a hierarchical prior distribution over the space of models defined by subsets of SNPs and their genetic parametrizations as follows. For any SNP included in the model, we assign a uniform distribution over the possible genetic parametrizations. The prior distribution on the model size sγ is Bin(S, ρ) conditional on ρ, and for the last stage, ρ is assigned a Beta (a, b) distribution. Integrating over the distribution on ρ leads to the BetaBinomial (a, b) distribution on model size,
| (2.6) |
and the following distribution on models,
| (2.7) |
where B(·, ·) is the beta function and the factor of 1/3 accounts for the distribution over genetic parametrizations.
2.4.1. Default hyperparameter choice
Following Ley and Steel (2009) and Scott and Berger (2010), we recommend a = 1 as a default, so that the prior distribution on model size is nonincreasing in sγ. The hyperparameter b can then be chosen to reflect the expected model size, the global prior probability of at least one association or the marginal prior odds that any SNP is associated (Table 2). A default choice is to set b = 1, leading to a uniform distribution on model size [Ley and Steel (2009); Scott and Berger (2010)]. Like the binomial distribution, the BetaBinomial(1, 1) distribution results in an expected model size of (Table 2), although the BetaBinomial(1, 1) distribution has a larger variance than the Bin(S, 1/2). Alternatively, if b is proportional to S, b = λS, the expected model size approaches a limit of as S approaches infinity.
The choices for hyperparameters have implications for the global Bayes factor. The BetaBinomial(1, 1) has a global prior odds of association equal to the number of SNPs, S, and would be appropriate for the case where increasing the number of SNPs under consideration reflects increased prior certainty that an overall (global) association can be detected. Under the BetaBinomial(1, λS), the global prior odds are constant, 1/λ, reflecting a prior odds for overall association that is independent of the number of genes/SNPs tagged. Also, with both Beta–Binomial prior distributions, the prior odds of incorporating an additional SNP in any model decreases with model size sγ and approaches 0 in the limiting case as the number of SNPs, S, increases. This provides an implicit multiple testing correction in the number of SNPs (rather than tests) that are included in the study of interest. The BetaBinomial(1, λS) achieves this by keeping the global (pathway) prior odds of an association constant while decreasing the marginal prior odds of any one of the SNPs being associated as the number of SNPs increases. As a skeptical “default” prior, we suggest the hyperparameters a = 1 and b = S which leads to the global prior odds of there being at least one association of 1 and the marginal prior odds of any single SNP being associated of 1/S.
3. Stochastic search for SNPs
Given the number of SNPs under consideration, enumeration of all models for S greater than 25–30 is intractable. While it is possible to enumerate all single variable SNP models, the number of models with 2 or 3 SNPs allowing for multiple genetic parametrizations is in the millions or more for a typical modern hypothesis-oriented study. Stochastic variable selection algorithms [see Clyde and George (2004), for a review] provide a more robust search procedure than stepwise methods, but also permit calculation of posterior probabilities and Bayes factors based on a sample of the most likely candidate models from the posterior distribution.
MISA makes use of a stochastic search algorithm based on the Evolutionary Monte Carlo (EMC) algorithm of Liang and Wong (2000). EMC is a combination of parallel tempering [Geyer (1991)] and a genetic algorithm [Holland (1975)] and samples models based on their “fitness.” While originally designed to find optimal models based on AIC, in our application the fitness of the models is given by ψ(),
where Ψ() is defined in equation (2.5) and is equal to the log of the un-normalized posterior model probability. This results in models being generated according to their posterior probability.
The EMC algorithm requires that we specify the number of parallel chains that are run and the associated temperature for each chain that determines the degree of annealing. If the temperatures are too spread out for the number of chains, then the algorithm may exhibit poor mixing and slow convergence. Liang and Wong (2000) show that even with all chains run at a temperature of 1 (no annealing), EMC outperforms alternative sampling methods such as Gibbs sampling and Reversible Jump MCMC in problems where strong correlations among the predictor variables lead to problems with exploring multiple modes in the posterior distribution. We have found that a constant temperature ladder with 5 parallel chains provides good mixing and finds more unique models than using a custom temperature ladder based on the prescription in Liang and Wong (2000), and recommend the constant temperature ladder as a default. To assess convergence, we take two independent EMC runs using randomly chosen starting points and examine trace plots of the fitness function. We use the marginal likelihoods from the set of unique models in the sample for inference and compute estimates of marginal posterior inclusion probabilities for each run. We continue running the two instances of the EMC algorithm until the posterior probabilities derived from each are sufficiently close. This leads to longer running times than those suggested by conventional convergence diagnostic such as Gelman–Rubin [Gelman and Rubin (1992)].
Efficiency of stochastic algorithms often diminishes as the total number of models increases. For this reason, we have found it useful to reduce the number of SNPs included in the EMC search using a screen when S is large. Such a screen will typically be fairly permissive, leaving only the weakest candidates out of the stochastic search. The screen should be quick to calculate, adjust for the same design variables and consider the same genetic parametrizations as in the full analysis. In our analyses, we calculated marginal (i.e., SNP-at-a-time) Bayes factors for each of the log-additive, dominant and recessive models of association against the model of no association. We ordered SNPs according to the maximum of the three marginal Bayes factors and retained those with a maximum marginal BF greater than or equal to one. More details are available in Wilson et al. (2010).
4. Simulation comparison
We used the 124 simulated case-control data sets [details of the simulation can be found in Wilson et al. (2010)] to estimate true and false positive rates for MISA and seven other alternative procedures:
Bonferroni
We fit a logistic regression model for each SNP under the log-additive parametrization and calculate the p-value for testing association using a Chi-Squared test. We use a Bonferroni corrected level α = 0.05 test to declare a SNP associated.
Adjusted Bonferroni
We fit a logistic regression model for each SNP under the log-additive parametrization and calculate the p-value for testing association using a Chi-Squared test. We use a Bonferroni corrected level α test to declare a SNP associated where α is chosen so that the proportion of false positives detected is the same as in MISA using the default BetaBinomial(1, S) prior.
Benjamini–Hochberg
We fit the same SNP-at a time logistic regression as above, but declare a SNP to be associated if it has a Benjamini–Hochberg false discovery rate of less than 0.05.
Marginal BF
This also utilizes the single SNP at a time logistic regression, but calculates a BF for association under each of the three genetic models. If the maximum BF over the three genetic models is greater than 3.2, we declare the SNP associated. See Wilson et al. (2010) for more detail.
Stepwise LR (AIC)
We use a stepwise multiple logistic regression procedure to select SNPs based on AIC. Each SNP is coded using 2 degrees of freedom to select among the three genetic models. SNPs in the final model are called associated.
Stepwise LR (BIC)
Same as above but using BIC to select models.
Lasso
We use the Lasso2 package in R [Lokhorst et al. (2009)] that is based on the algorithm developed by Osborne, Presnell and Turlach (2000) to select SNPs based on the least absolute shrinkage and selection operator. Each SNP is coded using 2 degrees of freedom to represent the three genetic models and all SNPs in the final model with coefficients greater than zero are called associated.
MISA
We reduced the number of SNPs using the marginal Bayes factor method above to eliminate SNPs with a marginal BF ≥ 1. We ran MISA using the default BetaBinomial(1, S) and the BetaBinomial(1/8, S) prior distributions on the models using two runs of 400,000 iterations based on convergence of the marginal inclusion probabilities. SNPs are called associated if their MISA SNP BF is greater than 3.2. All SNPs that did not pass the marginal screen step in MISA were declared not associated.
The first four are single SNP methods, while the last three are multi-SNP methods that take into account the genetic parametrization for each SNP.
Figure 1 shows the proportion of SNPs detected by each of the methods as a function of the assumed true odds ratio. Thus, at an odds ratio of 1.00 we plot the proportion of SNPs that were falsely declared associated by each of the methods. While both Bonferroni and Benjamini–Hochberg have the smallest false positive rates, they have much lower power to detect true associations than any of the other methods; the marginal BF has the highest power out of the three marginal methods, and is comparable to lasso, a multi-SNP method. Stepwise model selection using BIC has the lowest power of the multiple SNP model selection procedures. Stepwise logistic regression using AIC to select a model, on the other hand, has high power to detect associations, but an unacceptably high false positive rate (44%). With the exception of stepwise/AIC, the MISA methods have higher power than the alternatives at all odds ratios (ORs) in the simulation, with the gain in power most noticeable for the smaller ORs, those encompassing the range 1.25–1.75 typically seen in practice [Flint and Mackay (2009)]. This increase in power comes at the cost of only a slight increase in the false positive rate. Overall, MISA using the default BetaBinomial(1, S) prior distribution is able to detect 9% as many associations at the SNP level and 13% as many at the gene level than the marginal BF method used alone. In addition, MISA is able to detect 19% as many true associations at the SNP level and 27% as many at the gene level as the calibrated Bonferroni method (the two methods have the same Type I error rate).
4.1. Sensitivity to hyperparameters
We examined a range of parameters (a and b) for the Beta-Binomial prior distribution on model size (Table 3) to assess sensitivity of true positive and false positive rates. In practice, this may be done by reweighting the MCMC output using the new prior distribution, without resorting to additional MCMC runs, as long as high posterior probability models receive adequate support under both prior distributions.
Table 3.
Estimated overall false and true positive rates with standard errors and prior odds (PO) of association at the gene and SNP levels. The values in bold characterize the method selected for use in the analysis of the NCOCS ovarian cancer example
| Method: | True positive |
False positive |
PO of assoc. |
|||||
|---|---|---|---|---|---|---|---|---|
| Gene (se) | SNP (se) | Gene (se) | SNP (se) | Global | SNP | |||
| n: | 1020 | 1020 | 5546 | 54980 | ||||
| MISA | ||||||||
| a | b | |||||||
| 1 |
|
0.77 (0.006) | 0.669 (0.007) | 0.128 (0.001) | 0.025 (0.0001) | 2.00 | 0.04 | |
| 1/2 | · | 0.809 (0.005) | 0.704 (0.007) | 0.166 (0.001) | 0.031 (0.0001) | 0.74 | 0.020 | |
| 1/4 | · | 0.846 (0.004) | 0.729 (0.006) | 0.189 (0.001) | 0.041 (0.0002) | 0.32 | 0.009 | |
| 1/8 | · | 0.874 (0.003) | 0.739 (0.006) | 0.259 (0.001) | 0.048 (0.0002) | 0.15 | 0.005 | |
| 1/16 | · | 0.896 (0.003) | 0.746 (0.006) | 0.341 (0.001) | 0.065 (0.0003) | 0.07 | 0.002 | |
| 1/32 | · | 0.904 (0.003) | 0.746 (0.006) | 0.437 (0.001) | 0.090 (0.0003) | 0.04 | 0.001 | |
| 1 | S | 0.784 (0.005) | 0.685 (0.007) | 0.150 (0.001) | 0.027 (0.0001) | 1.00 | 0.020 | |
| 1/2 | · | 0.821 (0.005) | 0.716 (0.006) | 0.185 (0.001) | 0.035 (0.0001) | 0.42 | 0.009 | |
| 1/4 | · | 0.855 (0.004) | 0.736 (0.006) | 0.207 (0.001) | 0.044 (0.0002) | 0.19 | 0.005 | |
| 1/8 | · | 0.877 (0.003) | 0.743 (0.006) | 0.280 (0.001) | 0.053 (0.0002) | 0.09 | 0.002 | |
| 1/16 | · | 0.899 (0.003) | 0.746 (0.006) | 0.368 (0.001) | 0.073 (0.0003) | 0.04 | 0.001 | |
| 1/32 | · | 0.904 (0.003) | 0.746 (0.006) | 0.465 (0.001) | 0.098 (0.0004) | 0.02 | 0.001 | |
| 1 |
|
0.791 (0.005) | 0.696 (0.007) | 0.169 (0.001) | 0.029 (0.0001) | 0.67 | 0.01 | |
| 1/2 | · | 0.825 (0.005) | 0.722 (0.006) | 0.190 (0.001) | 0.037 (0.0002) | 0.29 | 0.006 | |
| 1/4 | · | 0.855 (0.004) | 0.735 (0.006) | 0.222 (0.001) | 0.048 (0.0002) | 0.14 | 0.003 | |
| 1/8 | · | 0.878 (0.003) | 0.744 (0.006) | 0.291 (0.001) | 0.057 (0.0002) | 0.07 | 0.002 | |
| 1/16 | · | 0.898 (0.003) | 0.746 (0.006) | 0.377 (0.001) | 0.075 (0.0003) | 0.03 | 0.001 | |
| 1/32 | · | 0.902 (0.003) | 0.746 (0.006) | 0.474 (0.001) | 0.099 (0.0004) | 0.02 | 0.0004 | |
| Marg. BF | 0.695 (0.007) | 0.627 (0.007) | 0.171 (0.001) | 0.041 (0.0002) | – | 1.00 | ||
| lasso | 0.708 (0.007) | 0.607 (0.008) | 0.158(0.001) | 0.022 (0.0001) | – | – | ||
| Step. AIC | 0.993 (0.000) | 0.794 (0.005) | 0.969 (0.0001) | 0.445 (0.001) | – | – | ||
| Step. BIC | 0.680 (0.007) | 0.547 (0.008) | 0.122(0.001) | 0.015 (0.0001) | – | – | ||
| BH | 0.439 (0.008) | 0.419 (0.008) | 0.013 (0.0001) | 0.011 (0.0001) | – | – | ||
| Bonf. | 0.337 (0.007) | 0.330 (0.008) | 0.003 (0.00001) | 0.006 (0.00002) | – | – | ||
| Adj. Bonf. 1 | 0.618 (0.007) | 0.574 (0.008) | 0.069 (0.0003) | 0.027 (0.0001) | – | – | ||
| Adj. Bonf. 2 | 0.708 (0.007) | 0.644 (0.007) | 0.184 (0.001) | 0.053 (0.0002) | – | – | ||
Over the range of values for (a, b), MISA has a higher gene and SNP true positive rate than any of the other simpler procedures, with the exception of Stepwise AIC. In general, decreasing a leads to higher true positive rates, but at the expense of higher false positive rates. The SNP false positive rate is modest, ranging from 0.025 to 0.099, providing effective control of the experiment wide error rate. While these rates are higher than the false positive rates under Bonferroni or Benjamini–Hochberg, eliminating a SNP from consideration that truly is associated has a higher scientific cost than continuing to collect data to confirm that a SNP is really a null finding. Because the NCOCS will follow up apparent associations, a higher true positive rate with a modest increase in false positives was preferable.
The hyperparameters a = 1/8 and b = S, highlighted in bold in Table 3, were selected for comparison with the default choice (a = 1, b = S) in the analysis of the NCOCS data presented in the next section. MISA using the BetaBinomial(1/8, S) is able to detect 19% as many true associations at the SNP level and 26% as many at the gene level as the marginal BF method used alone. In addition, MISA with the BetaBinomial(1/8, S) prior is able to detect 14% as many true associations at the SNP level and 24% as many at the gene level as a calibrated Bonferroni method (the two methods have the same Type I error rate).
5. Ovarian cancer association analysis
In this section we describe a MISA candidate pathway analysis of data from the ongoing NCOCS ovarian cancer case-control association study. The NCOCS is a population based study that covers a 48 county region of North Carolina [Schildkraut et al. (2008)]. Cases are between 20 and 74 years of age and were diagnosed with primary invasive or borderline epithelial ovarian cancer after January 1, 1999. Controls are frequency matched to the cases by age and race and have no previous diagnosis of ovarian cancer. In the analysis we present, we focus on self-reported Caucasians and a specific histological subtype of the cancer, leaving us a total of 397 cases and 787 controls. Because the ovarian cancer results have not yet been published, we have anonomyzed the pathway, the genes chosen to represent it and the IDs of the SNPs tagging variation in those genes. The pathway is comprised of 53 genes tagged by 508 tag SNPs.
All models fit in the screen and by MISA included the patient’s age as a design variable. We used the modal approximation to fill in missing SNP data. We screened 508 SNPs using marginal Bayes factors, retaining S = 70 SNPs that exceeded the threshold of 1 in favor of an association. Using the default hyperparameters a = 1 and b = S, we ran two independent runs of the algorithm from independent starting points for a total of 1.2 million iterations—the point at which the SNP marginal inclusion probabilities from the two independent runs were determined to be in sufficiently close agreement.
On the basis of this analysis, we estimate a lower bound on the pathway-wide Bayes factor for asspociation to be BF(HA: H0) = 7.67 (which is also the posterior odds for this prior). This constitutes “positive” evidence in favor of an association between the pathway and ovarian cancer based on Jeffreys’ grades of evidence and corresponds to a posterior probability that the pathway is associated of roughly 0.89. Figure 2 summarizes the associations of the ten SNPs that had a SNP BF greater than 3.2, while Figure 3 illustrates the nine genes that contained these SNPs and two others that received comparable support. SNPs and genes in the pathway are denoted by a two-level name (e.g., S1 and G1) where the number represents the rank of the SNP or gene by its respective Bayes factor. These plots provide a graphical illustration of the top 100 models ∈ selected on the basis of their posterior model probabilities. Models are ordered on the x-axis in descending probability and the width of the column associated with a model is proportional to that probability. SNPs (Figure 2) or genes (Figure 3) are represented on the y-axis. The presence of a SNP or gene in a model is indicated by a colored block at the intersection of the model’s column and the SNP’s or gene’s row. In Figure 2 the color of the block indicates the parametrization of the SNP: purple for log-additive, blue for recessive and red for dominant. The “checkerboard” pattern (as opposed to the presences of more vertical bars) suggests substantial model uncertainty.
Fig. 2.

Image plot of the SNP inclusion indicators for the SNPs with marginal Bayes factors greater than 3.2 and the top 100 Models. The color of the inclusion block corresponds to the genetic parametrization of the SNP in that model. Purple corresponds to a log-additive parametrization, red to a dominant parametrization and blue to a recessive parametrization. SNPs are ordered on the basis of their marginal SNP Bayes factors which are plotted on the right axis across from the SNP of interest. Width of the column associated with a model is proportional to its estimated model probability.
Fig. 3.

Image plot of the gene inclusion indicators for the top 100 Models. Genes are ordered based on their marginal gene Bayes factors which are plotted on the right axis. Columns correspond to models and have width proportional to the estimated model probability, models are plotted in descending order of posterior support. The color is chosen to be neutral since the genetic parametrizations are not defined at the gene level.
The top five models depicted in Figure 2 include only a single SNP in addition to age at diagnosis (the design variable is omitted in the figure as it is included in all models). The top model includes SNP S1 in gene G1 under the log-additive genetic parametrization, which is estimated to have an odds ratio (OR) of approximately 1.42 (the posterior mode). The second ranked model includes only SNP S2 in gene G1 under the log-additive genetic parametrization with an estimated OR of 1.37. Note that the study has relatively low power to detect effects of this magnitude (Figure 1).
Figure 2 also illustrates that many of the top models beyond the first five include multiple SNPs. This suggests that if we were to restrict our attention to single SNP models, we would potentially lose substantial information regarding their joint effects. For example, model six is comprised of both SNP S3 from gene G5 and SNP S1 from gene G1, while model 12 is comprised of both SNP S3 from gene G5 and SNP S2 from gene G2. In both cases, SNP S3 is included in models with a SNP from gene G1. This may indicate that not only are SNPs S1, S2 and S3 important as single effects in the top four models, but that their combined effects may be of interest. Note that, in cases where the disease variant is unmeasured but “tagged,” several tagged SNPs may be required to explain variation at that locus.
The SNP Bayes factors of S1 (BF = 42.2) and S2 (BF = 17.8) provide “strong evidence” of changes in prior beliefs, however, the marginal posterior probabilities of association with ovarian cancer are 0.38 and 0.20, respectively. Figure 2 illustrates that when one of SNP S1 or S2 is included in a model, the other is often not (at least in the top 50 models). This trade off often arises when SNPs are correlated (i.e., in high linkage disequilibrium). In this case, R2 is 0.5 suggesting fairly strong LD between SNPs S1 and S2, in which case the joint inclusion probabilities are more meaningful than marginal probabilities. Both SNP 1 and SNP 2 are in gene G1 which has a gene Bayes factor of 31.95 (Figure 3) and posterior probability of association of 0.58. These probabilities need to be interpreted in the context of model uncertainty; conditional on the pathway being associated with ovarian cancer, the probability that gene G1 is driving the association is 0.58/0.89 = 0.65. However, there remains substantial uncertainty regarding which genes and SNPs may explain it, as the posterior mass is spread over competing models/hypotheses. The positive support for an association suggests the continuation of data accrual to refine these posterior probabilities.
Gene G1 and other genes in Figure 3 highlight a caution regarding the interpretation of Bayes factors as a measure of absolute support with composite hypotheses. The gene Bayes factor for G1 is 31.95, which is smaller than the SNP Bayes factors for S1 (42.2). The posterior probability that gene G1 is associated is based on summing the probabilities of all models that include at least one SNP from that gene (S1, S2 and S51), hence, the posterior probability for gene inclusion is always greater than or equal to the probability that any one SNP is included (i.e., posterior probabilities observe a monotonicity property with composite hypotheses). Bayes factors (and p-values) for composite hypotheses do not share this monotonicity property [Lavine and Schervish (1997)]. Bayes factors for comparing composite hypotheses may be expressed as the ratio of the weighted average (with respect to the prior distribution) of marginal likelihoods conditional on the hypotheses, which may decrease the evidence in favor of a composite hypothesis when a subset of the individual hypotheses have low likelihood. As mentioned in Section 2.1.4, while Bayes factors do not provide a coherent measure of absolute support because of their nonmonotonicity property, Lavine and Schervish (1997) show that the log Bayes factor does provide a coherent measure of how much the data change the support for the hypothesis (relative to the prior). Hence, they do provide useful summaries of changes in prior beliefs of association in large association studies with many competing models/hypotheses.
5.1. Sensitivity analysis
In this section we consider sensitivity of the results in the NCOCS study to the prior distribution on the models and to the method of imputation. The simulation study suggests that priors with smaller values of a may identify more associated SNPs. We estimated that the BetaBinomial(1/8, S) prior distribution on model size has a false positive rate comparable to the marginal BF method, but a much higher true positive rate, in the scenarios we considered. Full data imputation, achieved by averaging over the distribution of missing SNPs, is probabilistically correct, but computationally expensive. Thus, if the use of modal imputation provides an accurate approximation to BF calculated using full imputation, the computational efficiency of MISA can be greatly improved at small cost.
For purposes of this analysis, we used the set of unique models identified by the EMC search with modal imputations and a = 1 and calculated 3 additional sets of BFs. First, we obtained marginal likelihoods for each of these models using 100 imputed data sets with missing SNPs filled in based on their estimated distribution. Second, we calculated BFs using the BetaBinomial(1/8, S) and BetaBinomial(1, S) prior distributions using the marginal likelihoods under the full and modal imputations. We applied ANOVA to these four sets of BFs to compare the effects of prior hyperparameters and imputation methods after adjusting for SNP using the ranked SNP BFs.4
Table 4 shows that the method of imputation has no significant effect on the ranking of SNP BFs. This suggests that, for purposes of model search and calculation of BFs, we may use the modal imputed genotypes in place of full imputation, with significant computational savings. For purposes of parameter estimation, we suggest that the use of full imputation using a subset of the top models and top SNPs as using a plug-in approach for imputation is known to underestimate uncertainty.
Table 4.
Analysis of variance for the ranked SNP Bayes factors contrasting the prior hyperparameters (default a = 1 versus a = 1/8) and method of imputation (full imputation with 100 data sets versus a modal estimate of the missing genotypes) for the 70 SNPs in the NCOCS pathway that passed the marginal screen
| d.f. | Sum Sq. | Mean Sq. | F-value | Pr(> F) | |
|---|---|---|---|---|---|
| SNP | 69 | 1635891.00 | 23708.57 | 208.04 | <2 × 10−16 |
| Prior | 1 | 169641.66 | 169641.66 | 1488.60 | 0.0000 |
| Impute | 1 | 134.41 | 134.41 | 1.18 | 0.28 |
| Prior:impute | 1 | 53.16 | 53.16 | 0.47 | 0.50 |
| Residuals | 207 | 23589.77 | 113.96 |
We anticipated that the prior distribution would have a significant effect based on the higher true positive and false positive rates estimated from the simulation study and by considering differences in the prior odds. While Table 4 suggests that overall the rankings are different between the two prior distributions, the top 20 SNPs have the same rank under each of the four methods, leading to no qualitative differences in our conclusions about the top SNPs. The prior odds for any given SNP’s inclusion in a model are 8 times lower under the BetaBinomial(1/8, S) prior distribution than under to the BetaBinomial(1, S) prior distribution; the resulting SNP BFs are 2.8 times higher under the BetaBinomial(1/8, S) prior distribution than those under the BetaBinomial(1, S) prior distribution. As a result, eight more SNPs are above the 3.2 threshold used by the NCOCS to determine SNPs worthy of additional study.
5.2. External validation and comparison
To provide a basis of comparison, we applied the methods described in the simulation study (Section 4) to the NCOCS data. We omitted stepwise logistic regression using AIC because of its poor operating characteristics. The marginal FDR methods of Bonferroni and Benjamini–Hochberg failed to identify any significant SNPs. Lasso, which accounts for correlation among SNPS, also failed to identify any SNPS. Stepwise logistic regression using BIC selected a model with three of the top four SNPs identified by MISA—S1.G1, S3.G5 and S4.G4—but failed to identify S2.G1, which has correlation 0.71 with SNP S1.G1. This highlights a problem with selection methods that ignore model uncertainty.
The NCOCS proposed two SNPs—S10 and S14 in G9—for external validation by the Ovarian Cancer Association Consortium (OCAC), a large international multi-center consortium of ovarian cancer case-control studies. The decision to focus on these variants was made on the basis of results from an earlier version of the NCOCS data set and on the basis of the strong prior interest NCOCS researchers had in the gene (and not on the basis of the analysis described above). Under the default BetaBinomial(1, S) prior distribution, only SNP S10 in G9 exceeds the 3.2 threshold and the G9 BF is only 2.28. In contrast, under the BetaBinomial(1/8, S) prior distribution, both SNPs S10 and S14 (LD 0.62) in G9 have SNP BFs greater than 3.2 (8.70 and 5.99, respectively) and the gene BF is 6.18. An additional three SNPs in the same gene were proposed by another member of the consortium on the basis of uncorrected p-values. Of the five SNPs proposed for validation, only SNPs S10 and S14 were confirmed to be associated with serous invasive ovarian cancer by OCAC [Schildkraut et al. (2009)].
6. Discussion
In this paper we describe MISA, a natural framework for multi-level inference with an implicit multiple comparisons correction for hypothesis based association studies. MISA allows one to quantify evidence of association at three levels: global (e.g., pathway-wide), gene and SNP, while also allowing for uncertainty in the genetic parametrization of the markers. We have evaluated MISA against established, simple to implement and more commonly used methods and demonstrated that our methodology does have higher power than these methods in detecting associations in modestly powered candidate pathway case-control studies. The improvement in power is most noticeable for odds ratios of modest (real world) magnitude and comes at the cost of only a minimal increase in the false positive rate. Like stepwise logistic regression, lasso and logic regression, MISA improves upon marginal, SNP-at-a-time methods by considering multivariate adjusted associations. By using model averaging, MISA improves upon these multivariate methods that select a single model, which may miss important SNPs because of LD structure. These improvements have concrete implications for data analysis: MISA identified SNPs in the NCOCS data that were subsequently externally validated; none of the less complex methods considered here highlighted these SNPs to be of interest. Currently, other top ranked SNPs in genes identified by MISA are undergoing external validation. Finally, we note that while MISA was developed for binary outcomes in case-control studies, MISA is readily adaptable to accommodate other forms of outcome variables (e.g., quantitative traits or survival) that are naturally modeled within a GLM framework.
Supplementary Material
Footnotes
Ranks that were used as residuals on the log scale still exhibited strong departures from normality.
Web resources. The URL for the software for the methodology and simulations presented in this paper is as follows: http://www.isds.duke.edu/gbye/packages.html.
Bayesian model search and multilevel inference for SNP association studies: Supplementary materials (DOI: 10.1214/09-AOAS322SUPP; .pdf). In this supplement we provide details for: (1) Derivation of the implied prior distribution on the regression coefficients when AIC is used to approximate the marginal likelihood in logistic regression, (2) Description of the marginal Bayes factor screen used to reduce the number of SNPs in the MISA analysis, (3) Details of how the simulated genetic data sets used in the power analysis of MISA were created and information on the statistical software we developed for this purpose, and (4) Location of the freely available software resources referred to in this and the parent document.
Contributor Information
Melanie A. Wilson, Email: maw27@stat.duke.edu, Department of Statistical Science, Duke University, Durham, North Carolina 27708-0251, USA.
Edwin S. Iversen, Email: iversen@stat.duke.edu, Department of Statistical Science, Duke University, Durham, North Carolina 27708-0251, USA.
Merlise A. Clyde, Email: clyde@stat.duke.edu, Department of Statistical Science, Duke University, Durham, North Carolina 27708-0251, USA.
Scott C. Schmidler, Email: scs@stat.duke.edu, Department of Statistical Science, Duke University, Durham, North Carolina 27708-0251, USA
Joellen M. Schildkraut, Email: schil001@mc.duke.edu, Department of Community and Family Medicine, Duke University, Durham, North Carolina 27713, USA.
References
- Balding DJ. A tutorial on statistical methods for population association studies. Nature. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- Clayton DG, Walker NM, Smyth DJ, Pask R. Population structure differential bias and genomic control in a large-scale casecontrol association study. Nature Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- Clyde M. Bayesian Statistics 6—Proceedings of the Sixth Valencia International Meeting. Oxford Univ. Press; New York: 1999. Bayesian model averaging and model search strategies (with discussion) pp. 157–185. [Google Scholar]
- Clyde M, George EI. Model uncertainty. Statist Sci. 2004;19:81–94. [Google Scholar]
- Cordell HJ, Clayton DG. A unified stepwise regression procedure for evaluating the relative effects of polymorphisms within a gene using case/control or family data: Application to HLA in type 1 diabetes. AJHG. 2002;70:124–141. doi: 10.1086/338007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, George EI. Empirical Bayes vs. fully Bayes variable selection. J Statist Plan Inference. 2008;138:888–900. [Google Scholar]
- Efron B. Correlation and large-scale simultaneous significance testing. J Amer Statist Assoc. 2007;102:93–103. [Google Scholar]
- Flint J, Mackay TFC. Genetic architecture of quantitative traits in mice, flies and humans. Genome Research. 2009;19:723–733. doi: 10.1101/gr.086660.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Rubin D. Inference from iterative simulation using multiple sequences. Statist Sci. 1992;7:457–472. [Google Scholar]
- George E. Discussion of “Model averaging and model search strategies”. In: Clyde M, Bernardo JM, Berger JO, Dawid P, Smith AFM, editors. Bayesian Statistics 6—Proceedings of the Sixth Valencia International Meeting. Oxford Univ. Press; Oxford: 1999. pp. 157–185. [Google Scholar]
- Geyer CJ. Markov chain Monte Carlo maximum likelihood. Proc. 23rd Symp. Interface. Computing Science and Statistics; 1991. pp. 156–163. [Google Scholar]
- Goodman SN. Toward evidence-based medical statistics. 2: The Bayes factor. Annal Intern Med. 1999;130:1005–1013. doi: 10.7326/0003-4819-130-12-199906150-00019. [DOI] [PubMed] [Google Scholar]
- Hoeting JA, Madigan D, Raftery AE, Volinsky CT. Bayesian model averaging: A tutorial (with discussion) Statist Sci. 1999;14:382–401. Corrected version available at http://www.stat.washington.edu/www/research/online/hoeting1999.pdf.
- Holland JH. Adaptation in Natural and Artificial Systems. Univ. Michigan Press; Ann Arbor, MI: 1975. [Google Scholar]
- Jeffreys H. Theory of Probability. 3. Oxford Univ. Press; 1961. [Google Scholar]
- Kass RE, Raftery AE. Bayes factors. J Amer Statist Assoc. 1995;90:773–795. [Google Scholar]
- Kooperberg C, Ruczinski I. Identifying interacting SNPs using Monte Carlo logic regression. Genetic Epidemiology. 2004;28:157–170. doi: 10.1002/gepi.20042. [DOI] [PubMed] [Google Scholar]
- Lavine M, Schervish MJ. Bayes factors: What they are and what they are not. Amer Statist. 1997;53:119–122. [Google Scholar]
- Ley E, Steel MF. On the effect of prior assumptions in Bayesian model averaging with applications to growth regression. Appl Econometrics. 2009;24:651–674. [Google Scholar]
- Liang F, Wong WH. Evolutionary Monte Carlo: Applications to cp model sampling and change point problem. Statist Sinica. 2000;10:317–342. [Google Scholar]
- Lokhorst J, Venables B. lasso2: L1 constrained estimation aka “lasso”. R package version 1.2–10 2009 [Google Scholar]
- Osborne MR, Presnell B, Turlach BA. On the LASSO and its dual. J Comp Graph Statist. 2000;9:319–337. [Google Scholar]
- Park MY, Hastie T. Penalized logistic regression for detecting gene interactions. Bioinformatics. 2008;9:30–50. doi: 10.1093/biostatistics/kxm010. [DOI] [PubMed] [Google Scholar]
- Raftery AE. A note on Bayes factors for log-linear contingency table models with vague prior information. J Roy Statist Soc Ser B. 1986;48:249–250. [Google Scholar]
- Ruczinski I, Kooperberg C, LeBlanc M. Logic regression. J Computat Graph Statist. 2003;12:475–511. [Google Scholar]
- Schildkraut JM, Moorman PG, Bland AE, Halabi S. Cyclin E over-expression in epithelial ovarian cancer characterizes an etiologic subgroup. Cancer Epidemiology Biomarkers and Prevention. 2008;17:585–593. doi: 10.1158/1055-9965.EPI-07-0596. [DOI] [PubMed] [Google Scholar]
- Schildkraut JM, Goode EL, Clyde MA, Iversen ES. Single nucleotide polymorphisms in the TP53 region and susceptibility to invasive epithelial ovarian cancer. Cancer Research. 2009;69:2349–2357. doi: 10.1158/0008-5472.CAN-08-2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender H, Ickstadt K. Identification of SNP interactions using logic regression. Biostatistics. 2007;9:187–198. doi: 10.1093/biostatistics/kxm024. [DOI] [PubMed] [Google Scholar]
- Scott JG, Berger JO. Bayes and empirical-Bayes multiplicity adjustment in the variable-selection problem. Ann Statist. 2010;38:2587–2619. [Google Scholar]
- Servin B, Stephens M. Imputation-based analysis of association studies: Candidate regions and quantitative traits. PLOS Genetics. 2007:3. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Lee K, Wahba G. Detecting disease-causing genes by lasso-patternsearch algorithm. BMC Proceedings. 2007;1(Suppl 1):S60. doi: 10.1186/1753-6561-1-s1-s60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Balding DJ. Bayesian statistical methods for genetic association studies. Nature Genet. 2009;10:681–690. doi: 10.1038/nrg2615. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith N, Donnelly P. A new statistical method for haplotype reconstruction from population data. The American Journal of Human Genetics. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. A direct approach to false discovery rates. J Roy Statist Soc. 2002;64:479–498. [Google Scholar]
- Wacholder S. Assessing the probability that a positive report is false: An approach for molecular epidemiology studies. Journal of the National Cancer Institute. 2004;96:434–442. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. The American Journal of Human Genetics. 2007;81:208–227. doi: 10.1086/519024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust . Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Iversen ES, Clyde MA, Schmidler SC, Schildkraut JM. Supplement to “Bayesian Model Search and Multilevel Inference for SNP Association Studies”. 2010 doi: 10.1214/09-AOAS322SUPP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Chen YF, Hastie T, Sobel E, Lange K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics. 2009;25:714–721. doi: 10.1093/bioinformatics/btp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.