

Abstract
Neurotropic herpesviruses depend on long-distance axon transport for the initial establishment of latency in peripheral ganglia (retrograde transport) and for viral spread in axons to exposed body surfaces following reactivation (anterograde transport). Images of neurons infected with herpes simplex virus type 1 (HSV-1), acquired using electron microscopy, have led to a debate regarding why different types of viral structures are seen in axons and which of these particles are relevant to the axon transport process. In this study, we applied time-lapse fluorescence microscopy to image HSV-1 virion components actively translocating to distal axons in primary neurons and neuronal cell lines. Key to these findings, only a small fraction of viral particles were engaged in anterograde transport during the egress phase of infection at any given time. By selective analysis of the composition of the subpopulation of actively transporting capsids, a link between transport of fully assembled HSV-1 virions and the neuronal secretory pathway was identified. Last, we have evaluated the seemingly opposing findings made in previous studies of HSV-1 axon transport in fixed cells and demonstrate a limitation to assessing the composition of individual HSV-1 particles using antibody detection methods.
Several members of the alphaherpesvirus subfamily infect and establish latency within the peripheral nervous system of their natural hosts. The human pathogen herpes simplex virus type 1 (HSV-1) and the veterinary pathogen pseudorabies virus (PRV) are commonly studied models of herpesvirus neural infection. These viruses initially infect exposed skin or mucosal tissue and spread to innervating sensory or sympathetic nerve endings. Neurotropic herpesviruses enter nerve terminals by fusion of the envelope with the plasma membrane (47), resulting in release of the capsid and tegument into the cytosol (36, 37). Following fusion, capsids together with a subset of tegument proteins engage the host microtubule transport machinery and move intracellularly to the neuronal cell body (4, 35, 58), where the DNA genomes are deposited in nuclei and latency is established. HSV-1 and PRV are highly conserved both in postentry capsid transport dynamics and in the composition of the moving particle in axons during this early stage of infection (4, 35). Upon reactivation, progeny viral particles sort into axons from the neuronal cell body and move anterograde back toward nerve terminals, where infection spreads to exposed tissue and ultimately to new hosts. Reactivated spread of HSV-1 can result in several pathologies that range from mild (herpes labialis) to severe (keratitis). The ability of neurotropic herpesviruses to undergo long-distance directed movement in axons is critical both for the establishment of latency and for recurrent disease resulting from reactivation.
Understanding the process by which HSV-1 traffics in axons is fundamental to deciphering the molecular mechanisms of microtubule-based transport and how the virus disseminates in animals and causes disease. Several laboratories have used transmission electron microscopy (TEM) to examine the composition of progeny intracellular HSV particles in axons at late time points postinfection. Collectively the TEM observations are ambiguous, with some research groups reporting the presence of enveloped capsids in vesicles (13, 33, 37), others the presence of non-membrane-associated capsids (32, 50), and some noting the presence of both forms in axons (26, 31, 46a, 54). Immunofluorescence microscopy of a neuroblastoma cell line infected with HSV-1 detected only a minority of capsids in neurites for which a corresponding envelope signal could be detected (60-62). In contrast, TEM studies of the closely related neuroinvasive herpesvirus PRV have consistently shown that the majority of viral particles within axons are enveloped and resident in the lumen of vesicles during the egress phase of infection (8, 10, 15, 21, 38, 39). The PRV findings are further substantiated by time-lapse microscopy of fluorescently tagged recombinant viruses, which demonstrates that progeny capsids actively undergoing anterograde transport to distal axons are associated with membrane components (5, 21).
These differences have led to one hypothesis that the human pathogen HSV-1 uses a mechanism that is distinct from that of PRV to disseminate from neuron cell bodies to the distal axon (reviewed in references 14 and 16). Whether these two neuroinvasive herpesviruses have evolved multiple mechanisms to disseminate within the nervous system or share a conserved transport process has important ramifications for both the molecular mechanisms of virus intracellular transport and pathogenesis. To date, all interpretations of HSV-1 anterograde axon transport are based on images captured from fixed cells. Both to better understand the dynamics of HSV-1 axon transport and to specifically examine the composition of actively trafficking HSV-1 particles in axons, we have provided here a time-lapse study of HSV-1 composition during anterograde axon transport in neurons. The results support a model of HSV-1 transport by way of the neuronal secretory pathway and that HSV-1 and PRV transport occurs by a fundamentally conserved process. Differences between HSV-1 and PRV that may have contributed to the disparate historical accounting of how these neuroinvasive herpesviruses traffic in axons are discussed. Finally, because these findings oppose the interpretations of previous immunofluorescence studies of HSV-1, additional experiments are included that may help to explain this apparent dichotomy.
MATERIALS AND METHODS
Plasmid construction.
PCR template plasmids, pEP-EGFP-in and pEP-mRFP1-in, were a gift from Nikolaus Osterrieder and were used to insert the monomeric fluorescent proteins GFP (green fluorescent protein) and mRFP1 (monomeric red fluorescent protein) into herpesvirus bacterial artificial chromosome (BAC) clones using a two-step BAC recombination protocol (7, 66). A pEP-CMV>GFP-in template plasmid was made by modifying a cytomegalovirus (CMV)-driven GFP expression construct that duplicated an internal portion of the CMV promoter and inserted the aphAI gene (encoding kanamycin resistance) and an I-SceI cleavage site between the duplicated sequences. This was achieved by amplifying the last 393 nucleotides (nt) of the CMV immediate-early (IE) promoter along with an endogenous downstream NheI restriction site with the primers 5′ GGGATATCGGATCCGGTAAACTGCCCACTTGG (EcoRV and BamHI sites underlined) and 5′ GCGCTAGCGGATCTGAC (NheI site underlined). The PCR product was digested with NheI and EcoRV and cloned into a CMV-EGFP expression construct using a unique SnaBI site in the CMV promoter. This produced a 143-nt duplication with a BamHI site at the center. The aphAI gene and I-SceI cleavage site cassette from pEP-EGFP-in were cloned into the BamHI site by PCR amplification using primers encoding 5′ BglII sites. pGS2435 is a derivative of pEP-CMV>GFP-in that additionally encodes two PRV US4 (gG) homology sequences flanking the CMV IE promoter repeats and downstream poly(A) sequence to allow for homologous recombination into the US4 locus of the PRV pBecker3 infectious clone using two-step BAC recombination (66). The US4 homology sequences were obtained from pGS202, which contains the SalI fragment of the US4 open reading frame (ORF) from pALM104 (30), which was provided by Lynn Enquist. A 384-nt portion of the US4 gene was amplified from pGS202 using the primers 5′ GCCAGCTGCCACCGGGACGGACTACTCTCG (PvuII site is underlined) and 5′ CGGCATGCATCGTGAGCCCGTGCTTCATGC (SphI site is underlined), and the PCR product was cloned upstream of the CMV IE promoter in pEP-CMV>GFP-in at PvuII and SphI (pGS2434). A 735-nt portion of the US4 gene was amplified off pGS202 using the primers 5′ GCGAATTCGCTTCCCCTCGACCGACGAG (EcoRI site is underlined) and 5′ GCACATGTTAGATCGAGGTGCGGATGTCG (AflIII site is underlined), and the PCR product was cloned into pGS2434 downstream of the poly(A) tail at EcoRI and AflIII to create pGS2435. Plasmid constructs encoding the neuropeptide Y signal sequence fused to GFP (peNPYss-noMetGFP) and the Vamp2 open reading frame fused to GFP (peVamp2-GFP) were a generous gift from Gary Banker and were used to create derivatives of pGS2435 that additionally encoded for NPYss-GFP or Vamp2-GFP expression under the control of the CMV IE promoter. The peNPYss-noMetGFP and peVamp2-GFP plasmids were digested with NheI and BsrGI, and the coding sequences of the fusion constructs were cloned into pGS2435 at NheI and BsrGI (partial digestion). This resulted in the pEP-CMV>NPYss-GFP-in and pEP-CMV>Vamp2-GFP-in plasmids.
Virus construction.
PRV-GS1236 is a dual-fluorescence virus that encodes mRFP1 fused to the N terminus of the VP26 capsid protein and GFP fused to the C terminus of the gD envelope protein, and it was previously described (5). PRV-GS1236 is referred to as “PRV RFP-cap/GFP-env” throughout this report. All recombinants constructed for this study were created using a two-step BAC recombination protocol (66) performed with the Escherichia coli strain GS1783, which encodes inducible Red and I-SceI activities. All HSV-1 recombinant viruses were derived from the pYEbac102 infectious clone of HSV-1 strain F (65), and PRV recombinant viruses were derived from the pBecker3 infectious clone of PRV strain Becker (56).
HSV1-YEbac102 is referred to as “wild type” throughout this report. HSV1-GS2822 is a monofluorescent red-capsid-tagged derivative of HSV-1 harboring the mRFP1 coding sequence fused to the 5′ end of the UL35 gene (encodes the VP26 capsid protein) and is referred to as “HSV-1 RFP-cap” throughout this report. Primers to insert the mRFP1 ORF as a translational fusion to the 5′ end of the HSV-1 UL35 ORF were 5′ CCGACACCCCCATATCGCTTCCCGACCTCCGGTCCCGATGGCCTCCTCCGAGGACG and 5′ CGGTGGTAACGGTGCTGGGGCGGTGAAATTGCGGGACGGCGGCGCCGGTGGAGTGG (the UL35 start methionine codon is in bold, and sequence with homology to the pEP-mRFP1-in template is underlined). HSV1-GS2971 encodes a monofluorescent envelope-tagged derivative that contains egfp fused to the UL27 gene (encodes the gB envelope glycoprotein) and is based on a previous design in which GFP was inserted following the signal peptide sequence at amino acid 43 (63). Primers to insert the GFP ORF as a translational fusion to amino acid 43 of UL27 were 5′ GGCGGCTCCGAGTTCCCCCGGCACGCCTGGGGTCGCGGCCGTGAGCAAGGGCGAGGAG and 5′ GCGCCGGAGTGGCAGGGCCCCCGTTCGCCGCCTGGGTCGCCTTGTACAGCTCGTCCATGC (sequence with homology to the pEP-GFP-in template is underlined). HSV1-GS2971 is referred to as “HSV-1 GFP-env” throughout this report. HSV1-GS2843 is a dual-fluorescence strain that encodes both mRFP1-VP26 and GFP-gB and was isolated by recombining egfp into the pGS2822 infectious clone. HSV1-GS2843 is referred to as “HSV-1 RFP-cap/GFP-env” throughout this report.
HSV1-GS3353 and HSV1-GS3561 are derivatives of HSV-1 RFP-cap in which the CMV IE promoter followed by either the neuropeptide Y signal sequence fused to GFP (HSV1-GS3353) or the Vamp2 ORF fused to GFP (HSV1-GS3561) were recombined into the US5 (gJ) locus of the pGS2822 infectious clone using homology encoded in the 5′ end of PCR primers. Primers used to insert CMV IE-NPYss-GFP-polyA and CMV IE-Vamp2-GFP-polyA into the HSV-1 US5 locus were 5′ AGAAACAGCACACGACTTGGCGTTCTGTGTGTCGCGATGTAGTTATTAATAGTAATCAATTACGGGGTCATTAG and 5′ TTATACGACAACTGGGTCCATGTAGGGATGGTAACGCCCAGTTTGGACAAAGGACAACTAGAATGC (the US5 start methionine codon is in bold, and sequence with homology to pEP-CMV>NPYss-GFP-in and pEP-CMV>Vamp2GFP-in is underlined). HSV1-GS3353 is referred to as “HSV-1 RFP-cap/NPYss-GFP” and HSV1-GS3561 is referred to as “HSV-1 RFP-cap/Vamp2-GFP” throughout this report. PRV-GS2727 and PRV-GS2795 were made by releasing the US4-flanked sequence from pEP-CMV>NPYss-GFP-in and pEP-CMV>Vamp2GFP-in (respectively) by AflIII and PvuII digestion followed by two-step recombination into the US4 locus of the pGS847 infectious clone, which was previously described (58). PRV-GS2727 is referred to as “PRV RFP-cap/NPYss-GFP” and PRV-GS2795 is referred to as “PRV RFP-cap/Vamp2-GFP” throughout this report. The recombinant mono- and dual-fluorescence viruses are summarized in Table 1.
TABLE 1.
Virus strains
| Virus strain | Description | Capsid fusion | Envelope fusion | Vesicle label | Titer (PFU/ml) |
|---|---|---|---|---|---|
| HSV 1-YEbac102 | Wild type | 7.5 × 108 | |||
| HSV 1-GS2822 | RFP-cap | mRFP1-VP26 | 2.8 × 108 | ||
| HSV 1-GS2971 | GFP-env | GFP-gB | 1.0 × 108 | ||
| HSV 1-GS2843 | RFP-cap/GFP-env | mRFP1-VP26 | GFP-gB | 7.7 × 107 | |
| HSV 1-GS3353 | RFP-cap/NPYssGFP | mRFP1-VP26 | NPYssGFP | 1.0 × 108 | |
| HSV 1-GS3561 | RFP-cap/Vamp2-GFP | mRFP1-VP26 | Vamp2-GFP | 4.5 × 108 | |
| PRV-GS1236 | RFP-cap/GFP-env | mRFP1-VP26 | gD-GFP | 3.0 × 108 | |
| PRV-GS2727 | RFP-cap/NPYssGFP | mRFP1-VP26 | NPYssGFP | 3.5 × 108 | |
| PRV-GS2795 | RFP-cap/Vamp2-GFP | mRFP1-VP26 | Vamp2-GFP | 7.3 × 107 |
Cells and virus propagation.
HSV-1 recombinants were isolated following transfection of BAC infectious clones into Vero cells expressing Cre recombinase using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and were subsequently passaged on Vero cells as previously described (4). PRV recombinants were isolated following electroporation of BAC infectious clones into PK15 cells and were subsequently passaged on PK15 cells (35). Viral propagation kinetics were assessed by single-step growth curve analysis, and viral titers were measured by plaque assay as previously described with the exception that Vero cells were used in place of PK15 cells (56). In the case of HSV1-GS2843 (HSV-1 RFP-cap/GFP-env), spontaneous syncytial activity sporadically appeared with serial passage. Stocks displaying syncytial activity were discarded.
Neuronal culture.
Dissociated dorsal root ganglia (DRG) sensory neurons were purchased from Lonza (Walkersville, MD) (rat DRG) or isolated from embryonic chickens (E8-E11) and cultured as previously described (57). DRG neurons were cultured for 2 to 3 days before infection. A derivative of a mouse catecholaminergic central nervous system cell line, Cath.a derived (CAD), was maintained in Dulbecco's modified Eagle medium (DMEM) F12 supplemented with 8% fetal bovine serum (FBS) and differentiated in culture by serum starvation for 4 days before infection (52).
Virus purification.
HSV-1 wild type, RFP-cap, GFP-env, and RFP-cap/GFP-env were each used to infect one 850-cm2 roller bottle (Corning) of 80 to 90% confluent Vero cells at a multiplicity of infection (MOI) of 3. Viral particles were purified from infected cell supernatants at 20 h postinfection (hpi) as previously described with the exception that viral particles were initially pelleted through a 10% Nycodenz cushion (5).
Nucleocapsid isolation.
Two 22.5-cm dishes of confluent Vero cells were infected with HSV-1 RFP-cap at an MOI of 10. Infected cells were harvested 1 day postinfection (dpi), spun at 1,000 × g for 10 min at 4°C, and washed once with phosphate-buffered saline (PBS). Infected cell pellets were resuspended in 50 ml of 1% NP-40 lysis buffer (0.15 M NaCl, 0.01 M Tris-HCl [pH 7.2], 2 mM MgCl2, and 1% NP-40) supplemented with 5 mM dithiothreitol (DTT) and protease inhibitor cocktail (Sigma) and incubated on ice for 30 min. Nuclei were pelleted at 1,000 × g for 10 min at 4°C, and pellets were separated from cytoplasmic material, washed, and resuspended in 3 ml of NP-40 lysis buffer. Resuspended nuclei were then passed several times through a series of syringes (18-, 21-, and 25-gauge) and supplemented with 100 U of DNase I (MP Biomedicals) before being brought to a final volume of 5 ml in NP-40 lysis buffer. The sample was passed two additional times through a 25-gauge needle. Debris was removed by centrifugation at 3,000 × g for 10 min at 4°C, and the cleared supernatant was overlaid on 35% sucrose in TNE (500 mM NaCl, 1 mM EDTA, 20 mM Tris [pH 7.6]) and centrifuged at 25,000 rpm in a Beckman SW41 rotor for 1 h at 4°C. The resulting pellet was resuspended in 250 μl of TNE.
Western blot analysis.
HSV-1 wild type, GFP-env, and RFP-cap/GFP-env were each used to infect confluent Vero cells in a 6-well tray at an MOI of 2. Lysates of infected and uninfected cells were harvested in 500 μl of 2× final sample buffer (6.25 mM Tris [pH 6.8], 10% glycerol, 0.01% bromophenol blue, 2% sodium dodecyl sulfate, and 10% β-mercaptoethanol) at 1 dpi. Lysate samples were boiled for 5 min, and 10 μl of each was electrophoresed through an 8% sodium dodecyl sulfate polyacrylamide gel followed by transfer to a polyvinylidene fluoride membrane (Pall). For detection of GFP, the membrane was incubated with mouse anti-GFP antibody B-2 (Santa Cruz Biotechnology) diluted 1:1,000, followed by incubation with horseradish peroxidase-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) diluted 1:10,000. For detection of HSV-1 gB, the membrane was incubated with a 1:5,000 dilution of rabbit polyclonal antiserum R74 (provided by Patricia Spear) followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch) diluted 1:50,000. For detection of GFP in purified virions, HSV-1 GFP-env virions were mixed with equal volumes of 2× final sample buffer, while HSV-1 wild-type and RFP-cap/GFP-env virions were mixed with 2× final sample buffer at a 2:1 volume ratio. Samples were boiled for 5 min, and 15 μl of each was electrophoresed through a 4 to 20% sodium dodecyl sulfate polyacrylamide gel (Bio-Rad) followed by transfer to a polyvinylidene fluoride membrane (Pall). The membrane was incubated with mouse anti-GFP antibody as described above. For detection of gB and VP5 antigens in purified virions, equal volumes of virions were mixed with 2× final sample buffer and boiled for 5 min. A 7.5-μl volume of the wild-type sample and 20 μl of the HSV-1 GFP-env and RFP-cap/GFP-env samples were electrophoresed through a 4 to 20% sodium dodecyl sulfate polyacrylamide gel (Bio-Rad), followed by transfer to a polyvinylidene fluoride membrane (Pall). The membrane was incubated with rabbit polyclonal antiserum specific for HSV-1 gB as described above. The HSV-1 VP5 major capsid protein was detected with mouse anti-VP5 H1.4 (Biodesign International, Sato, ME) diluted 1:3,000, followed by incubation with horseradish peroxidase-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch) diluted 1:10,000. For all Western blots, horseradish peroxidase signal was detected by incubation with luminol-coumeric acid H202 chemiluminescence solution, and exposed film was digitized with an EDAS 290 documentation system (Kodak). Relative intensities of gB and VP5 protein bands were quantitated using the ImageJ software program, and gB expression was normalized to VP5 protein levels (1).
Immunofluorescence analysis.
Isolated nucleocapsids and purified virions were diluted in PBS, adhered to polyornithine-treated coverslips immediately following isolation, and maintained at 37°C for 2 h before fixation. Vero cells were seeded 1:10 on coverslips and infected the following day with HSV-1 RFP-cap at 1 × 106 PFU per coverslip. Samples were either fixed at 24 hpi for Vero cell analysis or at 48 hpi for analysis of viral particles released from Vero cells onto bare regions of the coverslip. Dissociated chick sensory DRG neurons were seeded on polyornithine-treated coverslips at low density for 2 days before infection with HSV-1 RFP-cap at 1 × 106 PFU per coverslip. For these experiments, the MOI was not calculated for either Vero or DRG cultures due to the sparse cell densities. Infected DRG neurons were fixed at 24 hpi. All samples were processed for immunofluorescence identically to the methods described by Snyder et al. (62). Briefly, samples were fixed with 4% paraformaldehyde for 30 min, permeabilized in 0.2% Triton X-100 for 15 min, and blocked with 2% goat serum for 1 h. For detection of VP5, two mouse monoclonal antibodies, 3B6 (Virusys, North Berwick, ME) and H1.4 (Biodesign International, Sato, ME), were mixed at a 1:1 ratio and diluted 1:500. The IL-73-1 mouse monoclonal antibody (provided by Patricia Spear) was diluted 1:500 for detection of gC. Alexa 488-conjugated goat anti-mouse IgG (Molecular Probes, Eugene, OR) secondary antibodies were diluted 1:2,000.
Fluorescence microscopy.
All images were acquired with an inverted wide-field Nikon Eclipse TE2000-U microscope using automated fluorescence filter wheels (Sutter Instruments, Novato, CA), a 60× 1.4 numerical aperture oil objective (Nikon), and a Cascade:650 camera (Photometrics, Roper Scientific). The microscope was housed in an environmental box maintained at 37°C (In Vivo Scientific). The Metamorph software package was used for image acquisition and processing (Molecular Devices, Downington, PA).
Immunofluorescence imaging of nucleocapsids, gradient-purified extracellular virions, and nascent viral particles released from Vero cells onto the coverslip was accomplished by acquiring static images with 1-s RFP and 4-s GFP sequential exposures. To image GFP-gB incorporation into extracellular virions, Vero cells were seeded 1:10 on coverslips and infected the following day with either HSV-1 RFP-cap or RFP-cap/GFP-env at 1 × 105 PFU per coverslip, and images were captured 2 to 3 days postinfection. The presence of GFP or Alexa 488 fluorescent emissions from individual capsid-containing particles was scored using a custom automated image processing algorithm that detected particles based on RFP emissions and determined the presence or absence of corresponding GFP emissions from the same diffraction-limited emission source (11). Typically, between 10 and 100 punctae were scored per image, with multiple images analyzed for each sample.
Living primary sensory neurons and differentiated CAD cells were imaged in sealed chambers as previously described (57). To assess the frequency of egress transport, dissociated sensory chick DRG neurons were infected with 1 × 106 PFU per coverslip and imaged at 9, 12, 15, 18, and 21 hpi. Static images of one proximal and one distal field from seven infected neurons were captured at each time point with 1-s RFP and 4-s GFP sequential exposures. HSV-1 egress transport was imaged in chick or rat dissociated DRG neurons and differentiated CAD cells from 18 to 24 hpi. PRV egress transport was imaged in chick dissociated DRG neurons from 10 to 13 hpi. For all egress transport experiments, neurons were infected with approximately 1 × 106 PFU per coverslip. Time-lapse imaging of mRFP1 and GFP emissions was accomplished by continuous sequential imaging using automated excitation-and-emission filter wheels (Sutter Instruments, Novato, CA) with 100-ms exposures for each channel.
RESULTS
Isolation and characterization of fluorescent viruses.
A monofluorescent mRFP1-VP26 virus (referred to as “HSV-1 RFP-cap”) was used to track HSV-1 capsids in living cells (4). Four HSV-1 glycoproteins were initially modified as GFP fusions for use as an envelope marker: glycoproteins B, C, D, and H (gB, gC, gD, and gH). HSV-1 encoding a GFP fusion to gC produced bright fluorescence in the axon membrane that overwhelmed emissions from individual viral particles, gD-GFP and GFP-gH fusions resulted in poor fluorescence during infection, and a gH-GFP fusion impaired virus spread (data not shown). These problems were largely absent when the coding sequence for GFP was inserted behind the codons for the gB signal sequence, resulting in a translational fusion of GFP to the N-terminal ectodomain of gB (referred to as “HSV-1 GFP-env”), similar to previous approaches (51, 63). To simultaneously image capsids and the gB envelope protein, a dual-fluorescence derivative of HSV-1 encoding both markers (referred to as “HSV-1 RFP-cap/GFP-env”) was isolated. All viruses were derived from the HSV-1 (strain F) pYEbac102 infectious clone and are summarized in Table 1 (65).
Both mono- and dual-fluorescence recombinants of HSV-1 propagated slower than unmodified HSV-1 in Vero cells, which is consistent with a previously characterized recombinant strain of HSV-1 encoding similar fluorescent fusions (Fig. 1) (63). Although the ability of GFP-env viruses to replicate within a log of the wild-type replication indicates that the essential function of gB in viral fusion and entry was not disrupted (6, 68), the reporter viruses are mutants since they did not fully recapitulate the kinetics of HSV-1 propagation. In contrast, the kinetics of retrograde transport in axons following entry was not impaired by placing GFP on gB, and retrograde transporting particles were never associated with GFP-gB fluorescence emissions, indicating that viral fusion and entry were not disrupted (i.e., the viral particles were not in endosomes; data not shown) (36, 47). Expression of the GFP-gB fusion protein was confirmed by Western blot analysis (Fig. 2) and was observed predominantly in the nuclear membrane and endoplasmic reticulum of Vero cells infected with HSV-1 RFP-cap/GFP-env, consistent with previous fluorescence microscopy reports (data not shown) (3, 20, 28, 53, 70).
FIG. 1.
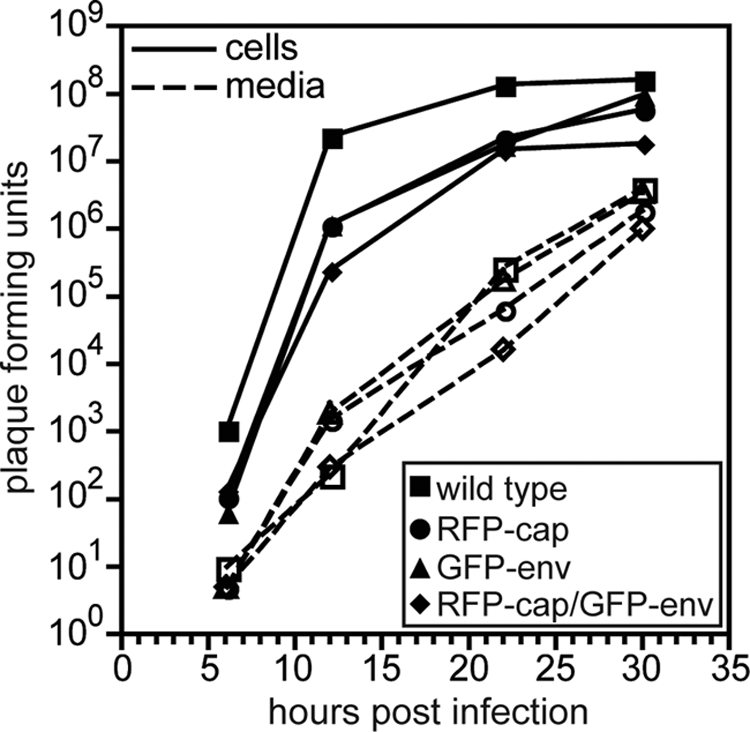
Propagation kinetics of fluorescent viruses. Single-step growth curves of unmodified HSV-1 strain F (wild type) and three recombinant derivatives (RFP-cap, GFP-env, and RFP-cap/GFP-env) are shown. Infected cell supernatants (dashed lines, open symbols) and adherent Vero cells (solid lines, filled symbols) were harvested at the indicated times, and titers were quantitated by plaque assay.
FIG. 2.
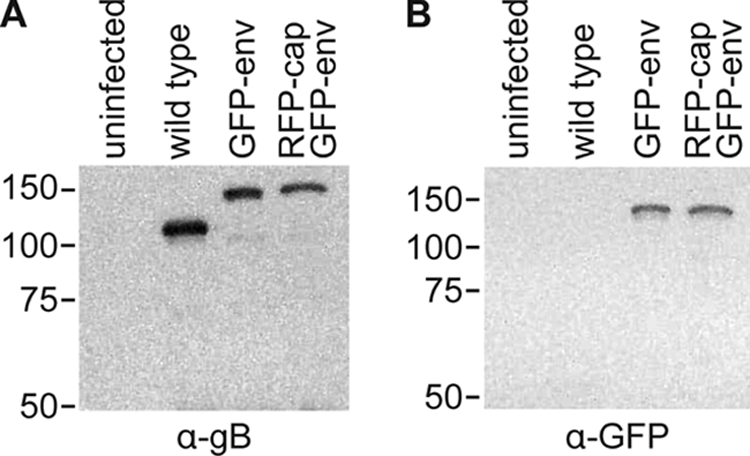
Fluorescent fusion protein expression. Western blot of lysates from Vero cells that were either uninfected or infected with the indicated strain of HSV-1 and probed with an anti-gB antibody (A) or an anti-GFP antibody (B) 1 day postinfection. Equal volumes of lysates were loaded. The gB antiserum weakly cross-reacts with glycoprotein C, which migrates just below gB (Patricia Spear, personal communication).
Tracking the motion of individual HSV-1 particles in living cells first required an assessment of GFP-gB structural incorporation and fluorescence emissions from viral particles. This was accomplished in two ways. First, HSV-1 wild-type, GFP-env, and RFP-cap/GFP-env extracellular particles were purified and examined by Western blot analysis. A GFP antibody detected a single band at the expected size for the fluorescent fusion protein in both the GFP-env mono- and dual-fluorescence virions, confirming the structural incorporation of the GFP-gB fusion protein (Fig. 3A); however, gB-specific antisera yielded a 3- or 5-fold-decreased reactivity with GFP-gB in HSV-1 GFP-env or RFP-cap/GFP-env particles, respectively, relative to that of the wild type (Fig. 3B). Whether these results represented a reduction in GFP-gB incorporation or a decreased reactivity of the gB antisera as a result of the GFP fusion was not immediately clear. As a second test of GFP-gB incorporation, extracellular viral particles that had accumulated on bare regions of a coverslip following release from nearby infected Vero cells were imaged by fluorescence microscopy, as previously described (5, 35). This in situ assay allows protein incorporation to be assessed at the level of individual viral particles. Two days following infection of Vero cells with HSV-1 RFP-cap/GFP-env, GFP-gB emissions were detected from an average of 58% of extracellular HSV-1 particles (Fig. 3C and D). This frequency is less than the 85% detection frequency previously obtained with PRV expressing a gD-GFP envelope fusion (5), and in conjunction with the Western blot data, it suggests that the GFP fusion to gB likely results in a decrease in gB incorporation into virions (Fig. 3B). Although the 58% frequency indicated that detection of GFP-gB in intracellular virions would be more challenging than the corresponding experiments previously performed with PRV, the frequency was sufficiently high to encourage us that a conclusive result of HSV-1 particle composition during anterograde axon transport could be achieved. This result provided a critical parameter for time-lapse imaging of individual particles during infection: the frequency of GFP-gB detection in HSV-1 extracellular particles is an approximate upper limit to what should be detected in living cells if capsids are enveloped. As a side note, punctate GFP-gB emissions were frequently detected in the absence of a corresponding RFP-tagged capsid signal in the in situ assay and were interpreted as light particles (64).
FIG. 3.

Incorporation of fluorescent fusion protein into extracellular viral particles. (A) Western blot of sucrose gradient-purified extracellular viral particles from the indicated HSV-1 strain probed with an anti-GFP antibody. (B) Western blot of sucrose gradient-purified extracellular viral particles from the indicated HSV-1 strains probed with anti-gB and anti-VP5 antibodies. The latter was included as a loading control. (C) Examples of GFP and RFP emissions from individual extracellular viral particles released from Vero cells 2 to 3 days postinfection with the indicated HSV-1 strain. The fields are each 24 μm by 24 μm; the bar is 2 μm. (D) GFP-gB incorporation frequency (GFP + RFP particles/total RFP particles) determined from images as represented in panel C. Averages from five individual experiments are shown, with error bars representing the standard deviation (n, number of capsid-containing particles imaged).
Accumulation and transport frequency in axons of cultured neurons.
Monofluorescent GFP-gB punctae were visible in chick DRG axons as early as 9 h postinfection (hpi). Similar to the case with PRV gD-GFP, these HSV-1 GFP-gB signals frequently moved bidirectionally in axons independent of capsids and were highly dynamic (5). These GFP-gB-containing structures may represent prelight particles in the axon secretory pathway or may simply be secretory vesicles containing glycoproteins (42). In contrast to GFP-gB, HSV-1 RFP-capsids were infrequently detected in axons, and the majority of these were static. This low frequency of HSV-1 capsid anterograde transport in axons is in contrast to reports of PRV, where anterograde moving capsids are readily detected in axons (5, 12, 21, 34, 35, 57). The relative frequencies of HSV-1 and PRV capsid egress in axons were compared using a previously described dual-fluorescence strain of PRV encoding a red fluorescent capsid and GFP fused to gD (referred to as “PRV RFP-cap/GFP-env”) (5). Low-density chick DRG sensory neuron cultures were infected with an equal number of PFU of either HSV-1 RFP-cap/GFP-env or PRV RFP-cap/GFP-env, and axons were imaged at 9, 12, 15, 18, and 21 hpi. Only axons from neurons that were at the egress stage of infection (based on red fluorescent nuclei) were imaged. PRV capsids were present in axons more frequently than HSV-1 capsids and peaked at the 18-hpi time point. In contrast, HSV-1 maintained a low level of capsid axon localization throughout the experiment, with a small increase at the 21-hpi time point (Fig. 4A and B). A similar trend was noted in infected rat DRG sensory neurons and in neurons infected with the HSV-1 RFP-cap monofluorescent virus (data not shown). A low frequency of HSV-1 axon egress relative to that of PRV was suspected previously and is supported by these findings (54).
FIG. 4.

Frequency of capsid trafficking in axons during egress. (A) Dissociated chick DRG sensory neurons were infected with PRV RFP-cap/GFP-env (circles) or HSV-1 RFP-cap/GFP-env (triangles), and seven neurons were imaged during the indicated times postinfection. For each neuron, a proximal (P) and distal (D) region of the axon were imaged in a 58.5-μm by 78-μm field, and the number of individual capsids present in the axon segment was determined. Bars indicate mean values. (B) Summary of the data presented in panel A. The total number of capsids present in axons at the each time postinfection for PRV RFP-cap/GFP-env (circles; solid lines) or HSV-1 RFP-cap/GFP-env (triangles; dashed lines) at proximal (open symbols) or distal (closed symbols) sites are indicated. (C) Comparison of total anterograde transport events captured as part of the current HSV-1 RFP-cap/GFP-env study or from a previous study of PRV RFP-cap/GFP-env (5). Imaging for both studies was during peak egress (PRV, 10 to 13 h postinfection; HSV-1, 18 to 24 h postinfection) in dissociated chick DRG sensory neurons. The number of moving capsids (PRV = 181 capsids; HSV-1 = 123 capsids) recorded during each independent experiment (for PRV, 9 experiments; for HSV-1, 35 experiments) is indicated. Bars indicate mean values.
Progeny HSV-1 particles are typically imaged in axons of neurons fixed at 18 to 24 hpi, which is considered to be the peak of HSV-1 axon egress (16, 41). To better assess the relative frequencies of HSV-1 and PRV axon transport during peak times of egress, the number of moving PRV capsids per live-cell imaging session was determined based on our previously reported data recorded between 10 and 13 hpi (5) and compared to the number of moving HSV-1 capsids recorded per imaging session between 18 and 24 hpi. The average number of moving capsids per imaging session was 20 for PRV and 3 for HSV-1, with many imaging sessions failing to detect any moving HSV-1 capsids in axons (Fig. 4C). Although this was not a side-by-side comparison but rather was a summary of data acquisition conducted over a year, both sets of experiments were conducted with chick DRG sensory neurons under equivalent conditions. Together, these results confirm that HSV-1 egress in axons is infrequent relative to that of PRV.
HSV-1 capsids transport in axons with the gB envelope marker.
Low-density chick DRG neurons infected with HSV-1 RFP-cap/GFP-env were imaged from 18 to 24 hpi, and actively transporting capsid and gB signals were tracked in axons using continuous 100-ms exposures of RFP and GFP emissions. The analysis focused on viral particles moving through axon regions away from branch points, varicosities, and the initial axon segment, which were proposed as sites of capsid envelopment in a previous study (54). Although HSV-1 transport events were significantly less common than with PRV, infrequent HSV-1 capsids displayed bidirectional transport with net anterograde motion similarly to PRV (57). The majority of HSV-1 capsids preferentially moved either anterograde or retrograde during the course of a recording. However, unlike PRV, a fraction of HSV-1 capsids underwent multiple reversal events during a single recording, making the determination of overall transport direction sometimes difficult. Capsids that either showed no net directionality or underwent three or more reversal events over the course of a single recording were therefore scored as having “unclear” transport directionality. Of 123 HSV-1 capsids tracked in chick DRG axons, 58 were moving anterograde and 28 retrograde, and 37 were classified as unclear (Fig. 5B). The proportion of HSV-1 retrograde-transported capsids during egress (1 in 3 capsids) was greater than that previously reported for PRV (1 in 7 capsids) (5, 57).
FIG. 5.

Capsids are transported with gB during egress in axons. Dissociated DRG sensory neurons from chick (A) or rat (C) or a differentiated mouse CNS cell line (CAD) (D) were infected with HSV-1 RFP-cap/GFP-env, and axons were imaged following replication from 18 to 24 h postinfection. Montages are frames of alternating RFP-capsid (R) and GFP-gB (G) emissions captured with continuous 100-ms exposures. Each montage documents the motion of a single viral particle toward the axon terminal. Frames are 2.5 μm by 20.5 μm (A), 2.5 μm by 21 μm (C), or 2.5 μm by 11 μm (D). (B) The numbers of RFP-capsids that are transported with (gray) or without (black) GFP-gB in chick DRG sensory neurons. (E) Percentages of anterograde moving capsids that are cotransported with GFP-gB in different neuronal cell types (left) and the percentage of GFP-gB incorporation into extracellular viral particles as derived from Fig. 3 (right). n, number of capsids tracked.
Seventy percent of anterograde-transported HSV-1 capsids were associated with the GFP-gB membrane protein in chick DRG axons (40 of 58 capsids emitted detectable GFP fluorescence) (Fig. 5A and B). This is similar to our previous finding that PRV capsids traffic as a complex with the gD membrane protein (5). In contrast to PRV, where only the anterograde population was associated with glycoprotein, HSV-1 capsids were associated with GFP-gB regardless of transport direction (Fig. 5B).
To determine if the results obtained in avian neurons were consistent with HSV-1 egress transport in mammalian neurons, two additional sets of experiments were conducted. First, dissociated rat DRG sensory neurons were infected with HSV-1 RFP-cap/GFP-env and imaged between 18 and 24 hpi. Because the results obtained in avian neurons required a year of experimentation due to the low frequency of HSV-1 anterograde transport events, the sample size of transport events subsequently captured in rat DRG neurons was by necessity smaller. Dual-fluorescence time-lapse imaging revealed 68% of anterograde transported capsids were moving in association with GFP-gB (19 of 28 capsids emitted GFP fluorescence) (Fig. 5C and E). Similar to the capsid dynamics observed in chick DRG, approximately 1 in 3 particles were moving retrograde in rat DRG axons during egress time points; however, of these, only 25% were being transported in association with the GFP-gB envelope marker (3 of 12 capsids emitted GFP fluorescence). Second, a mouse central nervous system (CNS) cell line (CAD cells) (52) was differentiated in culture to project neurites and used to study HSV-1 capsid and envelope trafficking during egress. Unlike primary cells, in which capsid transport events were infrequent (Fig. 4), HSV-1 capsid transport was surprisingly common in a subset of the CAD cell neurites. Of 136 HSV-1 capsids tracked in CAD neurites, 131 capsids were traveling anterograde, and 64% of these were associated with the GFP-gB membrane protein (84 of 131 capsids emitted GFP fluorescence) (Fig. 5D and E).
The initial characterization of GFP-gB incorporation into HSV-1 particles (Fig. 3) indicated that if HSV-1 egressed in axons as enveloped viral particles, approximately 58% of anterograde moving capsids should emit sufficient green fluorescence to detect the presence of GFP-gB. The analysis of HSV-1 anterograde axon transport surpassed this baseline and consistently yielded 65 to 70% GFP-gB association with capsids in each neuron cell type (Fig. 5E). These results support a model of HSV-1 anterograde transport in axons as fully assembled virions.
HSV-1 and PRV capsids are transported in axons using the secretory pathway.
To further examine the composition of viral particles during anterograde axon transport, HSV-1 and PRV encoding RFP-capsid (mRFP1-VP26) were modified to also express GFP-tagged cellular markers (Table 1). This was achieved by recombining a cytomegalovirus immediate-early promoter driving the expression of either the neuropeptide Y (NPY) signal sequence fused to GFP (NPYss-GFP) or vesicle-associated membrane protein 2 (Vamp2) fused to GFP (Vamp2-GFP) into either the gJ (HSV-1) or gG (PRV) locus. The NPY signal sequence is sufficient to direct GFP into the lumen of Golgi-derived vesicles (18). In contrast, the Vamp2-GFP fusion serves as a marker for vesicle membranes in axons (2, 46). All four reporter viruses were used to infect low-density chick DRG neuron cultures, and anterograde transport in axons was imaged from 10 to 13 hpi for PRV and from 18 to 24 hpi for HSV-1.
The frequency and brightness of NPYss-GFP and Vamp2-GFP labeled vesicles varied from cell to cell and differed from findings in studies with PRV versus HSV-1. As with the HSV-1 GFP-gB and PRV gD-GFP membrane markers, NPYss-GFP and Vamp2-GFP were frequently observed trafficking in axons apart from HSV-1 or PRV capsids. However, NPYss-GFP and Vamp2-GFP vesicles were less frequent in HSV-1-infected neurons in general (data not shown). Several time-lapse recordings were discarded due to a complete lack of GFP punctae in axons (more often the case with NPYss-GFP) or because the GFP background levels were so high that coincidental association with capsids was inevitable (more often the case with Vamp2-GFP). From the remaining recordings, 76 capsids were recorded traveling toward axon terminals in neurons infected with PRV RFP-cap/NPYss-GFP. Of these, 84% emitted detectable GFP fluorescence during time-lapse imaging (Fig. 6A and Fig. 6C). For PRV RFP-cap/Vamp2-GFP, 59 capsids were recorded traveling toward axon terminals and 66% yielded detectable emissions from the GFP vesicle marker (Fig. 6C). These findings are consistent with previous reports that PRV capsids acquire membrane before the onset of anterograde transport in axons (5, 21). For the corresponding HSV-1 recombinant viruses, 47% (n = 30) of capsids had detectable NPYss-GFP and 52% (n = 23) had detectable Vamp2-GFP fluorescence while traveling toward axon terminals (Fig. 6B and C). Like capsid cotransport with GFP-gB, HSV-1 capsids showed a similar association with both NPYss-GFP and Vamp2-GFP regardless of transport direction (data not shown). Although the vesicle marker approach yielded variable labeling of HSV-1 and PRV capsids, the presence of NPYss-GFP and Vamp2-GFP with capsids during transport provided support for the GFP-gB findings and indicated that anterograde axon transport of HSV-1 occurs by way of the neuronal secretory pathway.
FIG. 6.
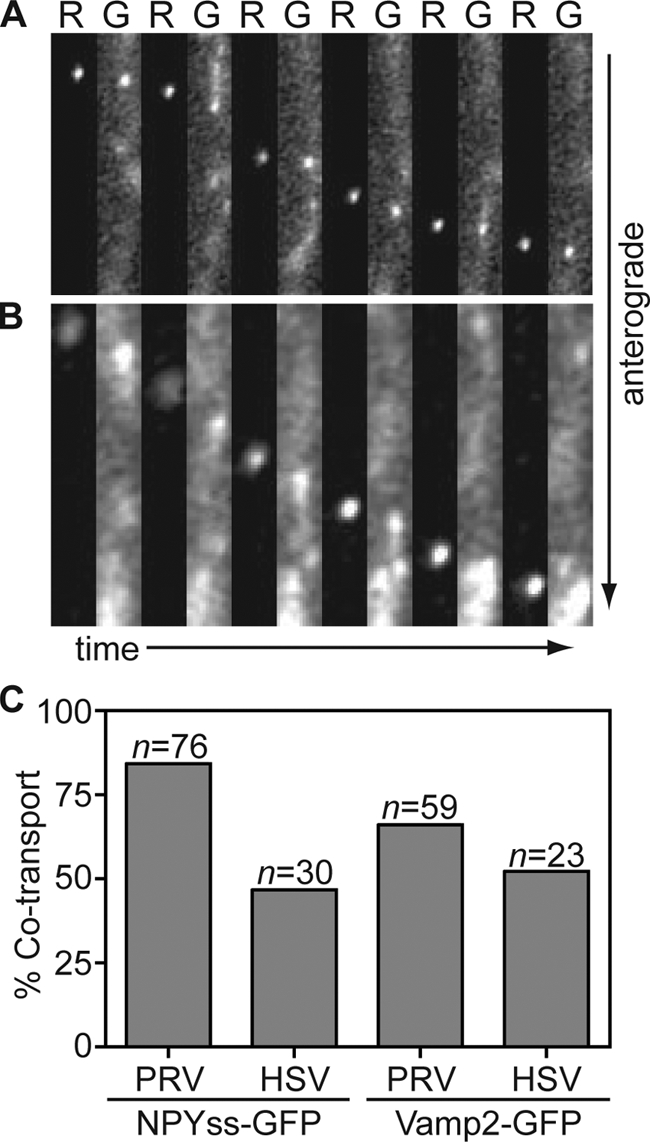
Capsids are transported with labeled cellular vesicles during egress in axons. Dissociated chick DRG sensory neurons infected with PRV RFP-cap/NPYss-GFP (A) or HSV-1 RFP-cap/NPYss-GFP (B) were imaged during peak times postinfection (PRV, 10 to 13 hpi; HSV, 18 to 24 hpi). Frames of alternating RFP (R) and GFP (G) emissions were captured using continuous 100-ms sequential exposures. Example time-lapse series are shown from left to right where a single progeny capsid is seen moving toward the axon terminal in association with the NPYss-GFP vesicle lumen marker. Frames are 2.4 μm by 13.3 μm (A) or 1.9 μm by 12 μm (B). (C) The percentages of RFP-capsids that are cotransported with NPYss-GFP (left) or Vamp2-GFP (right) vesicle markers are reported.
Immunofluorescence detection of capsids is restricted in intact virions.
The live-cell imaging data reported here support findings in TEM studies in which HSV capsids enveloped and resident in the neuronal secretory pathway within axons were observed (13, 26, 31, 33, 37, 46a). However, these findings contrast previous immunofluorescence analysis of HSV-1 capsid egress in neurites of a cultured neuroblastoma cell line (60-62). There are several technical differences between the published immunofluorescence studies and the live-cell imaging described in this report that may contribute to the apparent divergent results between these studies (see Discussion). One possible source of the discrepancy comes from studies demonstrating that antibody detection of individual capsids in cells is specifically attenuated for cytoplasmic capsids but not nuclear capsids (45). Because nuclear capsids of HSV-1 are not enveloped, while cytoplasmic capsids typically are, this work raises the question of whether immunofluorescence detection of HSV-1 particles in axons would selectively enrich for nonenveloped capsids while simultaneously overlooking enveloped capsids in the secretory pathway.
To address this question, the previously described immunofluorescence protocol used for detection of HSV-1 particles in neurites was applied verbatim to axons of primary neuronal cells, paying careful attention to use the same antibodies and conditions as previously reported (62). To first characterize antibody detection of individual HSV-1 particles in infected cells, Vero cells and chick DRG neurons were infected with either HSV-1 wild type or RFP-cap for 24 h and then fixed, permeabilized, and stained with a combination of two mouse monoclonal antibodies directed against the VP5 major capsid protein. With both wild-type-infected (data not shown) and RFP-cap-infected cells, the VP5 antibody mix labeled capsids in the nuclei of Vero cells but reacted poorly with cytoplasmic RFP-capsids (Fig. 7A), recapitulating previous findings obtained with HSV-1-infected Vero cells (45). In axons from chick DRG, 41.3% (n = 75) of RFP-capsids were detected with the VP5 antibody mix (Fig. 7B). Because these results provided preliminary support for the idea that capsid detection with antibodies could be restricted by the presence of surrounding tegument and envelope, antibody detection of capsids in isolated virus particles was next examined. Three types of HSV-1 RFP-cap particles were included in this study: (i) viral particles that were released onto coverslips in situ from infected cells (as described above), (ii) purified extracellular heavy particles (which includes mature virions), and (iii) capsids isolated from the nuclei of infected Vero cells. Particles were absorbed to glass coverslips and processed using the same immunofluorescence protocol that was applied to Vero cells and sensory neurons. In addition to the VP5 antibody mix, an antibody directed against the gC envelope protein was included in the study. As expected, nucleocapsids (which have not acquired an envelope) were not labeled by the gC antibody; unexpectedly, only 32% of RFP-capsid extracellular heavy particles that were purified using a dextran gradient displayed gC reactivity. Both of these viral particle preparations yielded 100% labeling with the anti-VP5 antibody mix. In contrast, 92% of in situ-released RFP-capsids containing viral particles were labeled for gC, but only 46% of these had detectable anti-VP5 reactivity (Fig. 7C). Two conclusions can be made from these results. First, purification of enveloped viral particles can alter virion integrity and allow more effective antibody access to the capsid, as observed with the HSV-1 H-particle preparation. Second, and more relevant to examining viral particle composition in axons, capsids within enveloped virions that have been fixed and permeabilized are not consistently accessible to VP5 antibodies, as shown by the in situ extracellular HSV-1 sample. These observations suggest that while unenveloped capsids should be efficiently identified in axons by antibody detection, enveloped virions would often be overlooked. Since enveloped virions in cells are further enclosed in a vesicle membrane that is tightly juxtaposed to the viral envelope (9, 13), antibody detection may be further hindered.
FIG. 7.

Anti-VP5 antibodies are partially excluded from labeling intact virions but not unenveloped capsids. (A and B) Vero cell (A) or dissociated chick DRG sensory neuron (B) fixed at 24 h postinfection with HSV-1 RFP-cap (red) and stained with a combination of two antibodies directed against VP5 (green). VP5 antibodies labeled capsids present in the nucleus but displayed poor staining of RFP-capsids present in the cytoplasm (A) or axon (B). Frames are 30 μm by 30 μm (A) or 31.5 μm by 10.5 μm (B). (C) Imaging of individual viral nucleocapsids isolated from HSV-1 RFP-cap-infected nuclei (left panel), extracellular virions purified from HSV-1 RFP-cap-infected Vero cell supernatants (middle panel), or viral particles released from Vero cells onto coverslips at 2 to 3 days postinfection with HSV-1 RFP-cap (right panel) are shown. Nucleocapsids and purified virions were seeded on coverslips for 2 h before fixation and were stained with either a combination of two VP5-specific antibodies or an antibody directed against gC. Examples of mRFP1-VP26 (red-capsid) and anti-VP5 or anti-gC (green) emissions from the same 24-μm by 24-μm field are shown. Graphs below each panel represent the fraction of mRFP1 particles that emit green antibody fluorescence (n, number of RFP-capsids).
DISCUSSION
One defining feature of neuroinvasive herpesviruses is the use of intracellular axon transport to travel within their hosts. While several viruses are known to use the microtubule cytoskeleton to move within cells (reviewed in reference 55), viruses such as HSV-1 and PRV are remarkable for two reasons: intracellular viral particles sustain microtubule transport over long distances, and the net direction of transport reverses between initial infection (retrograde transport to the neural soma) and late infection (anterograde transport to the distal axon). Global cellular changes resulting from infection do not account for the switch from retrograde to anterograde trafficking, but instead, differences in the composition of the actively moving viral structure appear to dictate the transport direction (58). Therefore, studies of herpesvirus intracellular transport and its regulation require an understanding of the composition of the actively trafficking viral particles at each stage of infection.
During initial infection, HSV and PRV both engage the cellular retrograde transport machinery following fusion-mediated entry into axons (4, 31, 58, 67), which for both viruses results in loss of the viral envelope (5, 23, 24, 36, 37, 40, 44, 47, 59) and a subset of tegument proteins (4, 23, 25, 35, 40). However, the VP1/2 tegument protein, which binds directly to the capsid surface (11, 49), and the UL37 tegument protein, which binds VP1/2 (29, 69), remain associated with HSV-1 and PRV capsids during the trip to the neural soma (4, 35). Retrograde transport events in neurons immediately following infection with either HSV-1 (strain F) or PRV (strain Becker) are equally abundant and kinetically indistinguishable (4). The dynein motor complex is required for viral retrograde transport (17); whether VP1/2, UL37, a component of the capsid, or a combination thereof recruits the dynein motor complex is an active area of investigation.
For PRV, the switch to anterograde trafficking during late infection is accompanied by a compositional shift in the transported viral particle. Electron microscopy imaging studies demonstrate that an abundance of viral particles are present in axons during late infection and that these particles are predominantly membrane bound and resident within the lumen of vesicles (10, 15, 21, 22, 38, 39). Initial support for the model that PRV virions transport to distal axons in vesicles came from observations that impairing viral membrane glycoprotein delivery to axons late during infection, either by treatment with brefeldin A or by infection of neurons with mutant PRV that does not express the Us9 gene product, coincidentally blocked capsid localization to axons (10, 15, 38). Ultimately, direct evidence that the membrane-bound viral particles are engaged in anterograde axon transport was obtained by live-cell fluorescence microscopy studies of cultured neurons (5, 21).
Finding HSV-1 particles in axons during the egress phase of infection, particularly in distal regions, has proved challenging (13, 37, 50, 54) and was suggested to be a difference between HSV-1 and PRV infection (14, 16). The small amount of HSV-1 axon transport relative to that of PRV is formally documented in a side-by-side experiment as part of this study (Fig. 4A and B). Nevertheless, several different laboratories using electron microscopy to resolve the structure of HSV particles in axons during late infection have either reported the presence of enveloped capsids (13, 33, 37), unenveloped capsids (32, 50), or both (26, 31, 46a, 54). As with PRV, HSV-1 particles of differing composition reside within axons during the egress stage of infection; however, the HSV-1 reports are inconsistent regarding whether enveloped virions or naked capsids are the more prevalent species. Furthermore, the lack of live-cell imaging studies has precluded formal conclusions of which type of particle, enveloped or naked, moves anterograde in axons.
Using time-lapse fluorescence microscopy, we provide evidence that enveloped HSV-1 virions are transported in axons by the neuronal secretory pathway. This conclusion is supported by three lines of evidence. First, using the HSV-1 RFP-cap/GFP-env reporter virus, approximately 65 to 70% of anterograde-transported HSV-1 capsids were associated with a GFP-gB signal in either chick or rat DRG sensory neurons or in a mouse CNS cell line (CAD) that was differentiated in culture to project neurites. While it is possible that the remaining 30 to 35% of capsids were transported toward the distal axon without an envelope, imaging results of static extracellular virions indicated that the absence of colocalization was a limitation of the GFP-gB fusion as an envelope marker. Therefore, rather than HSV-1 having two distinct mechanisms of anterograde transport, it seems more likely that only enveloped virions in host-derived vesicles actively move anterograde toward the distal axon. Second, imaging of an HSV-1 recombinant expressing a secreted GFP (NPYss-GFP) demonstrated that the actively transported HSV-1 particles were often associated with this luminal marker of the neuronal secretory pathway. Third, the axon-targeted synaptic vesicle protein Vamp2 was frequently cotransported with HSV-1 capsids during anterograde transport. The latter finding is notable, since it not only provides an additional line of support for a secretory model of HSV-1 axon transport but also provides the first indication of a class of vesicle (presynaptic) that HSV-1 can hijack to be transported to the distal axon. Although the frequency of detection of capsids associated with vesicle markers was lower than with GFP-gB, this was not necessarily surprising since expression of the NPYss-GFP luminal marker and that of the Vamp2-GFP membrane marker were more variable during infection. Also noteworthy was that recombinants of PRV that expressed the vesicle markers yielded colocalization with capsids during transport more frequently than the HSV-1 counterparts. While this may indicate a difference between the two viruses, such a conclusion is premature, since the different infection kinetics of the two viruses necessitated examining transport at different times postinfection. This impacted the amount of GFP signal associated with virions during transport and the background GFP apart from virions, both of which impacted our ability to effectively monitor the composition of particles during transport. Nevertheless, these points do not detract from the larger finding: HSV-1 capsids were cotransported with the GFP-gB viral envelope marker, NPYss-GFP luminal marker, and Vamp2-GFP vesicle membrane marker during anterograde axon transport by time-lapse imaging. Collectively, these results support transport of HSV-1 by the secretory pathway in neurons.
The source of the controversy surrounding this topic is undoubtedly multifaceted (27, 32, 41, 43, 48, 50, 60-62). The primary complication of HSV-1 studies is the low frequency of anterograde transport events, which has the effect of proportionally increasing the number of static and retrograde moving viral particles in axons of cultured cells. The latter particles may arise from nonproductive fusion of the virion being transported with the surrounding vesicle membrane, reinfection of the axon, or second rounds of infection of adjacent neurons. Although the axon egress frequency may mechanistically be a small difference between how HSV-1 and PRV function, it could manifest as a large perceivable distinction that makes the viruses appear to function by discrete mechanisms. Consistent with this interpretation, a recent comparative TEM analysis of three HSV-1 strains found in all cases that 75% of virions in axons during late infection are enveloped in vesicles (46a).
Analysis of HSV-1 composition by immunofluorescence assay during anterograde trafficking has also led to the conclusion that capsids are transported separately from viral glycoproteins in neurites of human neuroblastoma SK-N-SH cells (60-62). However, these immunofluorescence results should be considered in context with analogous studies of PRV-infected neurons, which have also produced images in which capsids appear to lack viral glycoproteins in axons (19, 57). The latter result is hard to reconcile given the ease of imaging enveloped PRV particles in axons by TEM and time-lapse microscopy. We therefore evaluated the effectiveness of immunofluorescence to assess the composition of individual intracellular viral particles. Antibodies directed toward the HSV-1 capsid, which previously were reported to show little overlap with envelope signals in neurites (60-62), labeled nonenveloped (nuclear) capsids consistently but were restricted in the ability to label enveloped capsids. These findings support those of a previous study that suggest capsid epitopes may be blocked in enveloped virions (45). Given the heterogeneity of HSV-1 particle composition in axons, these findings imply that the use of capsid-specific antibodies selectively enriches for the detection of unenveloped capsids. The prior immunofluorescence studies also used an HSV-1 GFP-capsid virus in conjunction with antibodies against viral membrane glycoproteins to show a lack of membrane markers with capsids in axons. The latter discrepancy may be explained if antibody accessibility to glycoproteins in axonal virions are restricted by the surrounding vesicle membrane and associated proteins, similar to the restriction of capsid antibodies by the viral envelope and tegument. Testing this hypothesis would require the technical achievement of purifying intracellular virion-containing vesicles, which was not included in this study since it is beyond our current means.
This study addresses a longstanding controversy regarding the envelopment state of neurotropic herpesviruses during egress in axons, and these results are in agreement with observations made with HSV-1-infected neurons more than 25 years ago (37). The use of time-lapse fluorescence microscopy in living isolated neurons demonstrated that while PRV is not a precise model of all aspects of HSV-1 anterograde trafficking in axons, particularly with regard to the frequency of transport events, the two viruses share a fundamentally conserved process for transport to the distal axon via the neuronal secretory pathway.
ADDENDUM IN PROOF
While this manuscript was in press, a related study appeared online as an in press manuscript (J. Huang, H. M. Lazear, H. M. Friedman, Virology, 30 October 2010, doi: 10.1016/j.virol.2010.10.009). Applying electron microscopy to primary sympathetic neurons grown in culture chambers, the authors observed that HSV-1, HSV-2, and PRV particles are enveloped and resident in the lumen of axon vesicles.
Acknowledgments
We thank Jenifer Klabis and Kevin Bohannon for help with the construction of dual-fluorescence HSV-1 strains. We are also grateful to several individuals for sharing reagents that made this study possible: Julie Luisi and Gary Banker for the peVamp2-GFP and peNPYss-no Met-GFP plasmids, Yasushi Kawaguchi for the pYEbac102 infectious clone, Nikolaus Osterrieder for the pEP-EGFP-in and pEP-mRFP1-in plasmids, Patricia Spear for antibodies against HSV-1 gB and gC, Dona Chikaraishi and Kristen Verhey for the CAD cell line, and Lynn Enquist for the pALM104 plasmid. We also thank Lynn Enquist for suggestions to improve the manuscript and for his encouragement.
This work was supported by a grant from the Cold Sore Research Foundation and NIH grants R01AI056346 (to G.A.S.) and NIH R01MH066179 (to Gary Banker). S.E.A. was supported in part by the Cellular and Molecular Basis of Disease training program in the National Institutes of Health (T32 GM08061).
Footnotes
Published ahead of print on 1 September 2010.
REFERENCES
- 1.Abramoff, M. D., P. J. Magelhaes, and S. J. Ram. 2004. Image processing with ImageJ. Biophotonics Int. 11:36-42. [Google Scholar]
- 2.Ahmari, S. E., J. Buchanan, and S. J. Smith. 2000. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat. Neurosci. 3:445-451. [DOI] [PubMed] [Google Scholar]
- 3.Ali, M. A., M. Butcher, and H. P. Ghosh. 1987. Expression and nuclear envelope localization of biologically active fusion glycoprotein gB of herpes simplex virus in mammalian cells using cloned DNA. Proc. Natl. Acad. Sci. U. S. A. 84:5675-5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antinone, S. E., and G. A. Smith. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 84:1504-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antinone, S. E., and G. A. Smith. 2006. Two modes of herpesvirus trafficking in neurons: membrane acquisition directs motion. J. Virol. 80:11235-11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai, W. H., B. Gu, and S. Person. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99:7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Card, J. P., L. Rinaman, R. B. Lynn, B. H. Lee, R. P. Meade, R. R. Miselis, and L. W. Enquist. 1993. Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of viral replication, transport, and pathogenesis. J. Neurosci. 13:2515-2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Card, J. P., L. Rinaman, J. S. Schwaber, R. R. Miselis, M. E. Whealy, A. K. Robbins, and L. W. Enquist. 1990. Neurotropic properties of pseudorabies virus: uptake and transneuronal passage in the rat central nervous system. J. Neurosci. 10:1974-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ch'ng, T. H., and L. W. Enquist. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 79:10875-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coller, K. E., J. I. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81:11790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coller, K. E., and G. A. Smith. 2008. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 9:1458-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook, M. L., and J. G. Stevens. 1973. Pathogenesis of herpetic neuritis and ganglionitis in mice: evidence for intra-axonal transport of infection. Infect. Immun. 7:272-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curanovic, D., and L. W. Enquist. 2009. Directional transneuronal spread of α-herpesvirus infection. Future Virol. 4:591-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.del Rio, T., T. H. Ch'ng, E. A. Flood, S. P. Gross, and L. W. Enquist. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 79:3903-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diefenbach, R. J., M. Miranda-Saksena, M. W. Douglas, and A. L. Cunningham. 2008. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 18:35-51. [DOI] [PubMed] [Google Scholar]
- 17.Dohner, K., A. Wolfstein, U. Prank, C. Echeverri, D. Dujardin, R. Vallee, and B. Sodeik. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 13:2795-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Meskini, R., L. Jin, R. Marx, A. Bruzzaniti, J. Lee, R. Emeson, and R. Mains. 2001. A signal sequence is sufficient for green fluorescent protein to be routed to regulated secretory granules. Endocrinology 142:864-873. [DOI] [PubMed] [Google Scholar]
- 19.Enquist, L. W., M. J. Tomishima, S. Gross, and G. A. Smith. 2002. Directional spread of an alpha-herpesvirus in the nervous system. Vet. Microbiol. 86:5-16. [DOI] [PubMed] [Google Scholar]
- 20.Farnsworth, A., T. W. Wisner, M. Webb, R. Roller, G. Cohen, R. Eisenberg, and D. C. Johnson. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. U. S. A. 104:10187-10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feierbach, B., M. Bisher, J. Goodhouse, and L. W. Enquist. 2007. In vitro analysis of transneuronal spread of an alphaherpesvirus infection in peripheral nervous system neurons. J. Virol. 81:6846-6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Field, H. J., and T. J. Hill. 1974. The pathogenesis of pseudorabies in mice following peripheral inoculation. J. Gen. Virol. 23:145-157. [DOI] [PubMed] [Google Scholar]
- 23.Fuller, A. O., R. E. Santos, and P. G. Spear. 1989. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 63:3435-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuller, A. O., and P. G. Spear. 1987. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc. Natl. Acad. Sci. U. S. A. 84:5454-5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill, T. J., H. J. Field, and A. P. Roome. 1972. Intra-axonal location of herpes simplex virus particles. J. Gen. Virol. 15:233-235. [DOI] [PubMed] [Google Scholar]
- 27.Holland, D. J., M. Miranda-Saksena, R. A. Boadle, P. Armati, and A. L. Cunningham. 1999. Anterograde transport of herpes simplex virus proteins in axons of peripheral human fetal neurons: an immunoelectron microscopy study. J. Virol. 73:8503-8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato, A., J. Arii, I. Shiratori, H. Akashi, H. Arase, and Y. Kawaguchi. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klupp, B. G., W. Fuchs, H. Granzow, R. Nixdorf, and T. C. Mettenleiter. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knapp, A. C., and L. W. Enquist. 1997. Pseudorabies virus recombinants expressing functional virulence determinants gE and gI from bovine herpesvirus 1.1. J. Virol. 71:2731-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristensson, K., B. Ghetti, and H. M. Wisniewski. 1974. Study on the propagation of herpes simplex virus (type 2) into the brain after intraocular injection. Brain Res. 69:189-201. [DOI] [PubMed] [Google Scholar]
- 32.LaVail, J. H., A. N. Tauscher, J. W. Hicks, O. Harrabi, G. T. Melroe, and D. M. Knipe. 2005. Genetic and molecular in vivo analysis of herpes simplex virus assembly in murine visual system neurons. J. Virol. 79:11142-11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LaVail, J. H., K. S. Topp, P. A. Giblin, and J. A. Garner. 1997. Factors that contribute to the transneuronal spread of herpes simplex virus. J. Neurosci. Res. 49:485-496. [PubMed] [Google Scholar]
- 34.Liu, W. W., J. Goodhouse, N. L. Jeon, and L. W. Enquist. 2008. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS One 3:e2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luxton, G. W., S. Haverlock, K. E. Coller, S. E. Antinone, A. Pincetic, and G. A. Smith. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lycke, E., B. Hamark, M. Johansson, A. Krotochwil, J. Lycke, and B. Svennerholm. 1988. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch. Virol. 101:87-104. [DOI] [PubMed] [Google Scholar]
- 37.Lycke, E., K. Kristensson, B. Svennerholm, A. Vahlne, and R. Ziegler. 1984. Uptake and transport of herpes simplex virus in neurites of rat dorsal root ganglia cells in culture. J. Gen. Virol. 65:55-64. [DOI] [PubMed] [Google Scholar]
- 38.Lyman, M. G., B. Feierbach, D. Curanovic, M. Bisher, and L. W. Enquist. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 81:11363-11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maresch, C., H. Granzow, A. Negatsch, B. G. Klupp, W. Fuchs, J. P. Teifke, and T. C. Mettenleiter. 2010. Ultrastructural analysis of virion formation and anterograde intraaxonal transport of the alphaherpesvirus pseudorabies virus in primary neurons. J. Virol. 84:5528-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maurer, U. E., B. Sodeik, and K. Grunewald. 2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A. 105:10559-10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miranda-Saksena, M., P. Armati, R. A. Boadle, D. J. Holland, and A. L. Cunningham. 2000. Anterograde transport of herpes simplex virus type 1 in cultured, dissociated human and rat dorsal root ganglion neurons. J. Virol. 74:1827-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miranda-Saksena, M., R. A. Boadle, A. Aggarwal, B. Tijono, F. J. Rixon, R. J. Diefenbach, and A. L. Cunningham. 2009. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 83:3187-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miranda-Saksena, M., R. A. Boadle, P. Armati, and A. L. Cunningham. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J. Virol. 76:9934-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan, C., H. M. Rose, and B. Mednis. 1968. Electron microscopy of herpes simplex virus. I. Entry. J. Virol. 2:507-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagel, C. H., K. Dohner, M. Fathollahy, T. Strive, E. M. Borst, M. Messerle, and B. Sodeik. 2008. Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J. Virol. 82:3109-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakata, T., S. Terada, and N. Hirokawa. 1998. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J. Cell Biol. 140:659-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46a.Negatsch, A., H. Granzow, C. Maresch, B. G. Klupp, W. Fuchs, J. P. Teifke, and T. C. Mettenleiter. 2010. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 84:13031-13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicola, A. V., J. Hou, E. O. Major, and S. E. Straus. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohara, P. T., A. N. Tauscher, and J. H. LaVail. 2001. Two paths for dissemination of herpes simplex virus from infected trigeminal ganglion to the murine cornea. Brain Res. 899:260-263. [DOI] [PubMed] [Google Scholar]
- 49.Pasdeloup, D., D. Blondel, A. L. Isidro, and F. J. Rixon. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610-6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Penfold, M. E., P. Armati, and A. L. Cunningham. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. U. S. A. 91:6529-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Potel, C., K. Kaelin, I. Gautier, P. Lebon, J. Coppey, and F. Rozenberg. 2002. Incorporation of green fluorescent protein into the essential envelope glycoprotein B of herpes simplex virus type 1. J. Virol. Methods 105:13-23. [DOI] [PubMed] [Google Scholar]
- 52.Qi, Y., J. K. Wang, M. McMillian, and D. M. Chikaraishi. 1997. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 17:1217-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raviprakash, K., L. Rasile, K. Ghosh, and H. P. Ghosh. 1990. Shortened cytoplasmic domain affects intracellular transport but not nuclear localization of a viral glycoprotein. J. Biol. Chem. 265:1777-1782. [PubMed] [Google Scholar]
- 54.Saksena, M. M., H. Wakisaka, B. Tijono, R. A. Boadle, F. Rixon, H. Takahashi, and A. L. Cunningham. 2006. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 80:3592-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith, G. A., and L. W. Enquist. 2002. Break ins and break outs: viral interactions with the cytoskeleton of mammalian cells. Annu. Rev. Cell Dev. Biol. 18:135-161. [DOI] [PubMed] [Google Scholar]
- 56.Smith, G. A., and L. W. Enquist. 1999. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 73:6405-6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith, G. A., S. P. Gross, and L. W. Enquist. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. U. S. A. 98:3466-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith, G. A., L. Pomeranz, S. P. Gross, and L. W. Enquist. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. U. S. A. 101:16034-16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith, J. D., and E. de Harven. 1974. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. II. An ultrastructural study of viral penetration. J. Virol. 14:945-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Snyder, A., B. Bruun, H. M. Browne, and D. C. Johnson. 2007. A herpes simplex virus gD-YFP fusion glycoprotein is transported separately from viral capsids in neuronal axons. J. Virol. 81:8337-8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Snyder, A., K. Polcicova, and D. C. Johnson. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 82:10613-10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Snyder, A., T. W. Wisner, and D. C. Johnson. 2006. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J. Virol. 80:11165-11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugimoto, K., M. Uema, H. Sagara, M. Tanaka, T. Sata, Y. Hashimoto, and Y. Kawaguchi. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198-5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szilagyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72:661-668. [DOI] [PubMed] [Google Scholar]
- 65.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tischer, B. K., J. von Einem, B. Kaufer, and N. Osterrieder. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191-197. [DOI] [PubMed] [Google Scholar]
- 67.Topp, K. S., L. B. Meade, and J. H. LaVail. 1994. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: relevance for the retrograde transport of herpes simplex virus. J. Neurosci. 14:318-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wisner, T. W., and D. C. Johnson. 2004. Redistribution of cellular and herpes simplex virus proteins from the trans-Golgi network to cell junctions without enveloped capsids. J. Virol. 78:11519-11535. [DOI] [PMC free article] [PubMed] [Google Scholar]