

Abstract
7SL RNA promotes the formation of the signal recognition particle that targets secretory and membrane proteins to the endoplasmic reticulum. 7SL RNA is also selectively packaged by many retroviruses, including HIV-1. Here, we demonstrate that 7SL RNA is an integral component of the viral ribonucleoprotein (RNP) complex containing Gag, viral genomic RNA, and 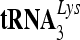 . Only the potent anti-HIV-1 cytidine deaminases can bind to 7SL RNA and target to HIV-1 RNP. A conserved motif in the amino-terminal region of A3G is important for 7SL RNA interaction. The weak anti-HIV-1 A3C did not interact with 7SL RNA and failed to target to viral RNPs, despite efficient virion packaging. However, a chimeric construct of A3C plus the 7SL-binding amino terminus of A3G did target to viral RNPs and showed enhanced anti-HIV-1 activity. 7SL RNA binding is a conserved feature of human anti-HIV-1 cytidine deaminases. Thus, potent anti-HIV-1 cytidine deaminases have evolved to possess a unique RNA-binding ability for precise HIV-1 targeting and viral inhibition.
. Only the potent anti-HIV-1 cytidine deaminases can bind to 7SL RNA and target to HIV-1 RNP. A conserved motif in the amino-terminal region of A3G is important for 7SL RNA interaction. The weak anti-HIV-1 A3C did not interact with 7SL RNA and failed to target to viral RNPs, despite efficient virion packaging. However, a chimeric construct of A3C plus the 7SL-binding amino terminus of A3G did target to viral RNPs and showed enhanced anti-HIV-1 activity. 7SL RNA binding is a conserved feature of human anti-HIV-1 cytidine deaminases. Thus, potent anti-HIV-1 cytidine deaminases have evolved to possess a unique RNA-binding ability for precise HIV-1 targeting and viral inhibition.
APOBEC proteins belong to a family of cytidine deaminases that edit RNA or DNA (13). APOBEC3G (A3G) (50) and the related human APOBEC3 are potent inhibitors of human immunodeficiency virus type 1 (HIV-1) in the absence of the viral protein Vif. A major result of the packaging of A3G into virions is the induction of C-to-U mutations in the minus-strand viral DNA during reverse transcription (19, 30, 37, 38, 54, 62, 66). Human APOBEC3 proteins have been identified as broad antiviral factors against HIV-1, simian immunodeficiency viruses (SIV), murine leukemia virus (MuLV), adeno-associated virus, and hepatitis B virus, as well as endogenous retroelements (9, 14, 17, 18, 36, 46).
In order to successfully replicate, HIV-1 encodes the Vif protein, which induces polyubiquitination and degradation of multiple APOBEC3 molecules (11, 31, 32, 39, 40, 51, 53, 63). Vif molecules of HIV-1 and SIV are substrate receptor proteins that assemble with Cul5, Elongin B, Elongin C, and Rbx to form an E3 ubiquitin ligase (63) through a virus-specific BC box motif (40, 64) and another highly conserved HX2YFXCFX4ΦX2AΦX7-8CX5H motif (35, 41, 59, 60).
The human cytidine deaminases vary significantly in their anti-HIV-1 activities. However, the reasons for this variation in potency are not well understood. Also, the mechanisms governing the packaging of cytidine deaminases into HIV-1 virions are as yet poorly defined. In the absence of the Vif protein, A3G can be packaged into a wide range of retroviruses and mediates potent antiviral functions in the newly infected target cells. Encapsidation of A3G into HIV-1 particles is mediated by the RNA-binding nucleocapsid (NC) domain of the Gag molecules (2, 6, 7, 15, 34, 43, 49, 57, 65). While some studies have reported that viral genomic RNA is required for efficient A3G packaging (28, 55), other studies have found that viral genomic RNA is dispensable for its packaging (2, 6, 7, 15, 34, 43, 49, 65). A role for cellular RNA in the virion packaging of A3G has also been proposed, but there is no consensus regarding what cellular RNA is involved (3, 57).
In the present study, we examined the specific 7SL RNA binding of various human cytidine deaminases and found that only the more potent anti-HIV-1 cytidine deaminases A3G and, to a lesser extent, A3B and A3F could interact with 7SL RNA. We also identified a unique motif (RLYYF/YW) in the amino-terminal region of A3G that mediates this selective interaction with 7SL RNA. 7SL RNA is an integral component of the viral RNP complex that contains HIV-1 Gag proteins, viral genomic RNA, and primer 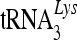 . We have also demonstrated that nonfunctional anti-viral APOBEC proteins lack the RLYYF/YW motif, interact poorly with 7SL RNA, and are either packaged poorly into HIV-1 virions or fail to target to 7SL RNA- and HIV-1 genomic-RNA-containing RNPs. Furthermore, the addition of 7SL RNA-binding capacity to the weakly anti-HIV-1 A3C (in the form of an A3G amino terminus/A3C chimera) was able to convert it to a more potent anti-HIV-1 protein.
. We have also demonstrated that nonfunctional anti-viral APOBEC proteins lack the RLYYF/YW motif, interact poorly with 7SL RNA, and are either packaged poorly into HIV-1 virions or fail to target to 7SL RNA- and HIV-1 genomic-RNA-containing RNPs. Furthermore, the addition of 7SL RNA-binding capacity to the weakly anti-HIV-1 A3C (in the form of an A3G amino terminus/A3C chimera) was able to convert it to a more potent anti-HIV-1 protein.
MATERIALS AND METHODS
Plasmid construction.
The infectious molecular clone of the Vif mutant (pNL4-3ΔVif) construct and the expression vectors for AID, APOBEC1, and APOBEC2 were obtained from the AIDS Research Reagents Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). Expression vectors for A3A-hemagglutinin (HA) and A3C-HA were generously provided by Michael Malim. The expression vector for A3B-HA was generously provided by Ned Landau and that for A3Gn/3A by Klaus Strebel. The A3G-HA and A3F-HA expression vectors have been described previously (63). The A3G mutant constructs derived from the A3G-HA expression vector were constructed by the PCR-based mutagenesis method using the indicated primers: A3GY91A Forward, 5′-TATGAGGTCACCTGGGCAATATCCTGGAGTCCC-3′; A3GY91A Reverse, 5′-GGGACTCCAGGATATTGCCCAGGTGACCTCATA-3′; A3GI92A Forward, 5′-GAGTATGAGGTCACCTGGTACGCATCCTGGAGCCCCTG-3′; A3GI92A Reverse, 5′-CAGGGGCTCCAGGATGCGTACCAGGTGACCTCATACTC-3′; A3GS93A Forward, 5′-GAGTATGAGGTCACCTGGTACATAGCCTGGAGCCCCTGCAC-3; A3GS93A Reverse, 5′-GTGCAGGGGCTCCAGGCTATGTACCAGGTGACCTCATACTC-3; A3GW94A Forward, 5′-GGTCACCTGGTACATATCCGCGAGCCCCTGCACAAAG-3′; A3GW94A Reverse, 5′-CTTTGTGCAGGGGCTCGCGGATATGTACCAGGTGACC-3′; A3GS95A Forward, 5′-CACCTGGTACATATCCTGGGCCCCCTGCACAAAGTGTAC-3′; A3GS95A Reverse, 5′-GTACACTTTGTGCAGGGGGCCCAGGATATGTACCAGGTG-3′; A3GR122A Forward, 5′-CCATCTTCGTTGCCGCCCTCTACTACTTCTGGG-3′; A3GR122A Reverse, 5′-CCCAGAAGTAGTAGAGGGCGGCAACGAAGATGG-3′; A3GL123A Forward, 5′-CTTCGTTGCCCGCGCCTACTACTTCTGGGACCC-3′; A3GL123A Reverse, 5′-GGGTCCCAGAAGTAGTAGGCGCGGGCAACGAAG-3′; A3GY124A Forward, 5′-CGTTGCCCGCCTCGCCTACTTCTGGGACCCAGA-3′; A3GY124A Reverse, 5′-TCTGGGTCCCAGAAGTAGGCGAGGCGGGCAACG-3′; A3GY125A Forward, 5′-GTTGCCCGCCTCTACGCCTTCTGGGACCCAGATT-3′; A3GY125A Reverse, 5′-AATCTGGGTCCCAGAAGGCGTAGAGGCGGGCAAC-3′; A3GF126A Forward, 5′-GCCCGCCTCTACTACGCCTGGGACCCAGATTAC-3′; A3GF126A Reverse, 5′-GTAATCTGGGTCCCAGGCGTAGTAGAGGCGGGC-3′; A3GF126Y Forward, 5′-GCCCGCCTCTACTACTACTGGGACCCAGATTAC-3′; A3GF126Y Reverse, 5′-GTAATCTGGGTCCCAGTAGTAGTAGAGGCGGGC-3′; A3GW127A Forward, 5′-CGCCTCTACTACTTCGCTGACCCAGATTACCAG-3′; A3GW127A Reverse, 5′-CTGGTAATCTGGGTCAGCGAAGTAGTAGAGGCG-3′; A3GW127F Forward, 5′-GTTGCCCGCCTCTACTACTTCTTCGACCCAGATTACCAGGA-3′; A3GW127F Reverse, 5′-TCCTGGTAATCTGGGTCGAAGAAGTAGTAGAGGCGGGCAAC-3′; A3GW127L Forward, 5′-GTTGCCCGCCTCTACTACTTCCTGGACCCAGATTACCAGGA-3′; A3GW127L Reverse, 5′-TCCTGGTAATCTGGGTCCAGGAAGTAGTAGAGGCGGGCAAC-3′; A3GW127Y Forward, 5′-GTTGCCCGCCTCTACTACTTCTACGACCCAGATTACCAGGA-3′; A3GW127Y Reverse, 5′-TCCTGGTAATCTGGGTCGTAGAAGTAGTAGAGGCGGGCAAC-3′; A3GP129A Forward, 5′-CTCTACTACTTCTGGGACGCAGATTACCAGGAGGCG-3′; A3GP129A Reverse, 5′-CGCCTCCTGGTAATCTGCGTCCCAGAAGTAGTAGAG-3′; A3GD130A Forward, 5′-CTACTTCTGGGACCCAGCTTACCAGGAGGCGC-3′; A3GD130A Reverse, 5′-GCGCCTCCTGGTAAGCTGGGTCCCAGAAGTAG-3′; A3GY131A Forward, 5′-CTTCTGGGACCCAGATGCCCAGGAGGCGCTTC-3′; A3GY131A Reverse, 5′-GAAGCGCCTCCTGGGCATCTGGGTCCCAGAAG-3′; A3GQ132A Forward, 5′-TTCTGGGACCCAGATTACGCGGAGGCGCTTCGC-3′; A3GQ132A Reverse, 5′-GCGAAGCGCCTCCGCGTAATCTGGGTCCCAGAA-3′; A3GE133A Forward, 5′-GGACCCAGATTACCAGGCGGCGCTTCGCAGC-3′; A3GE133A Reverse, 5′-GCTGCGAAGCGCCGCCTGGTAATCTGGGTCC-3′; A3GR136A Forward, 5′-CCAGGAGGCGCTTGCCAGCCTGTGTCAGAAAAGA-3′; A3GR136A Reverse, 5′-TCTTTTCTGACACAGGCTGGCAAGCGCCTCCTGG-3′; A3GS137A Forward, 5′-GAGGCGCTTCGCGCCCTGTGTCAGAAAAGAGACG-3′; A3GS137A Reverse, 5′-CGTCTCTTTTCTGACACAGGGCGCGAAGCGCCTC-3′; A3GL138A Forward, 5′-GCGCTTCGCAGCGCGTGTCAGAAAAGAGACGGT-3′; and A3GL138A Reverse, 5′-ACCGTCTCTTTTCTGACACGCGCTGCGAAGCGC-3′.
A3G-N was amplified with the following primers: Forward, 5′-GTACGCTAGCGCCATGAAGCCTCACTTCAGAAACACAGT-3′, and Reverse, 5′-GTACAAGCTTTCACGCGTAATCTGGGACGTCGTAAGGGTATCTGAGAATCTCCCCCAGCATG-3′, containing SalI and NotI sites, respectively, and C-terminal HA tags. The PCR product was cloned into VR1012 to generate A3G-N-HA. A3G-C was amplified using a SalI site-anchored Forward primer, 5′-GTACGCTAGCGCCATGCACTCGATGGATCCACCCAC-3′; a NotI site-anchored Reverse primer, 5′-GTACAAGCTTTCACGCGTAATCTGGGACGTCGTAAGGGTAGTTTTCCTGATTCTGGAGAATGGCC-3′; and a C-terminal HA tag. The PCR product was cloned into VR1012 to generate A3G-C-HA. A3Gn/3A-HA was amplified with the following primers: Forward 5′-CTCGAGACCATGAAGCCTCACTTCAGAA-3′, and Reverse, 5′-GAATCCTCACGCGTAATCTGGGACGTCGTAAGGGTAGTTTCCCTGATTCTGGAG-3′, containing XhoI and EcoRI sites, respectively, and C-terminal HA tags, using A3Gn/3A as a template. The PCR product was cloned into pcDNA3.1 to generate A3Gn/3A-HA.
Construction of A3Gn/3C-HA.
The full-length A3G-N and A3C were amplified separately with the following primers: A3G-N Forward, 5′-GTACGCTAGCGCCATGAAGCCTCACTTCAGAAAC-3′; A3G-N Reverse, 5′-TCTGAGAATCTCCCCCAGC-3′; A3C Forward, 5′-GGGAGATTCTCAGAATGAATCCACAGATCAGAAAC-3′; and A3C Reverse, 5′-GTACAAGCTTTCACGCGTAATCTGGGACGTCGTAAGGGTACCTCACTGGAGACTCTCC-3′. The PCR products of A3G-N and A3C were annealed and fused together using an overlapping method. The sequence-verified, amplified DNA was digested with NheI/HindIII and then cloned into pcDNA3.1.
Antibodies and cell culture.
The following antibodies and sera were used for this study: anti-p24 monoclonal antibody (MAb) (AIDS Research and Reference Reagents Program, Division of AIDS, NIAID, National Institutes of Health; catalog number 1513), goat anti-p7 antiserum, HIV-1-positive serum, mouse anti-V5 (Invitrogen; catalog number R96025), mouse anti-HA MAb (Covance [catalog number MMS-101R-1000] and Sigma [catalog number H3663]), and anti-human ribosomal P antigen antibody (Immunovision; catalog number HP0-0100). 293T and MAGI cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) with 10% fetal bovine serum and penicillin/streptomycin (D-10 medium) and passaged upon confluence.
Transfections, virus purification, and virus infectivity assay.
DNA transfection was carried out using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. Virus in cell culture supernatants was cleared of cellular debris by centrifugation at 3,000 rpm for 15 min in a Sorvall RT 6000B centrifuge and filtration through a 0.22-μm-pore-size membrane (Millipore). The virus particles were then concentrated by centrifugation through a 20% sucrose cushion by ultracentrifugation at 100,000 × g for 2 h at 4°C in a Sorvall Ultra80 ultracentrifuge. The viral pellets were resuspended in lysis buffer (phosphate-buffered saline [PBS] containing 1% Triton X-100 and complete protease inhibitor cocktail [Roche]). Viral lysates were analyzed by immunoblotting.
Viral infection was determined by MAGI assay as follows. MAGI-CCR-5 cells were prepared in six-well plates in D-10 medium 1 day before infection; the cells were at 30 to 40% confluence on the day of infection. The cells were infected by removing the medium from each well and adding dilutions of virus in a total volume of 500 μl of complete DMEM with 20 μg of DEAE-dextran per well. After a 2-h incubation at 37°C in a 5% CO2 incubator, 2 ml of complete DMEM was added to each well, and the cells were incubated for 48 h under the same conditions. The supernatants were removed, and 800 μl of fixing solution (1% formaldehyde, 0.2% glutaraldehyde in PBS) was added. After a 5-min incubation, the cells were washed twice with PBS. The staining solution (20 μl of 0.2 M potassium ferrocyanide, 20 μl of 0.2 M potassium ferricyanide, 2 μl of 1 M MgCl2, and 10 μl of 40-mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactosidaseactopyranoside [X-Gal]) was added. The cells were incubated for 2 h at 37°C in a non-CO2 incubator. Staining was stopped by removing the staining solution, and the cells were thoroughly washed twice with PBS. β-Galatosidase activity is under the control of the HIV-1 long terminal repeat (LTR) promoter, which is trans-activated in this system; positive blue dots indicate the presence of integrated virus. The positive blue dots were counted, and viral infectivity was determined after the amount of input virus was normalized in terms of the p24 antigen content. All results represent infections done in triplicate.
Immunoblot analysis.
Cells were collected 48 h after transfection. Cell and viral lysates were prepared as previously described (63). The samples were lysed in 1× loading buffer (0.08 M Tris, pH 6.8, with 2.0% SDS, 10% glycerol, 0.1 M dithiothreitol, and 0.2% bromophenol blue) and boiled for 5 min, and proteins were separated by SDS-PAGE. The membranes were probed with various primary antibodies against the proteins of interest. The secondary antibodies were alkaline phosphatase-conjugated anti-human, anti-goat, or anti-mouse (Jackson Immunoresearch, Inc.) antibodies, and staining was carried out with 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and nitroblue tetrazolium (NBT) solutions prepared from chemicals obtained from Sigma.
Immunoprecipitation.
To identify A3G-binding RNA, A3G and mutants were transfected into 293T cells. At 48 h after transfection, the cells were harvested and washed twice with cold PBS and then lysed with lysis buffer (PBS containing 1% Triton X-100, complete protease inhibitor cocktail [Roche], and RNase inhibitor [New England BioLabs]) at 4°C for 30 min. Cell lysates were clarified by centrifugation at 10,000 × g for 30 min at 4°C. Anti-HA agarose beads (Roche; catalog number 190-119) were mixed with the precleared cell lysates and incubated at 4°C for 3 h on an end-over-end rocker. The reaction mixtures were then washed six times with cold lysis buffer. Part of each bead pellet was resuspended in 1× loading buffer for immunoblotting; another part was resuspended in 1 ml Trizol (Invitrogen), and RNA was extracted according to the manufacturer's instructions.
To identify APOBEC3-binding RNAs, an A3A-HA, A3B-HA, A3C-HA, A3F-HA, or A3G-HA expression vector or AID-V5, APOBEC2-V5, or A3G-V5 was transfected into 293T cells. At 48 h after transfection, cells were harvested and washed twice with cold PBS and then lysed with lysis buffer (PBS containing 1% Triton X-100, complete protease inhibitor cocktail, and RNase inhibitor) at 4°C for 30 min. Cell lysates were clarified by centrifugation, mixed with anti-HA agarose beads, incubated, and analyzed by immunoblotting or used for RNA extraction as described above.
Homology modeling of the N-terminal domain of A3G.
Homology modeling of the N-terminal domain of A3G was carried out using the program Modeler. The availability of two experimentally determined structures of homologous cytidine deaminases, the crystal structure of APOBEC2, and the nuclear magnetic resonance (NMR) structure of the C-terminal domain (CTD) of A3G improved the quality of the modeling. Structure-based sequence alignment of the two models was carried out in Modeler.
Density and step gradient analyses.
For sucrose density gradient centrifugation, purified viruses were lysed in STE buffer (10 mM Tris-HCl [pH 7.4] containing 100 mM NaCl, 1 mM EDTA, 1% Triton X-100, and RNase inhibitor [New England BioLabs]) at 37°C for 30 min and then loaded onto a linear 20% to 70% (wt/vol) sucrose gradient and centrifuged at 100,000 × g for 16 h using a Beckman SW-41 rotor. Twelve fractions were collected from the top of each gradient, and viral proteins were analyzed by immunoblotting. Samples (100 μl) from each fraction were added to 1 ml Trizol, and RNA was extracted according to the manufacturer's instructions; the samples were also analyzed for viral genomic RNA, 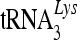 , and 7SL RNA.
, and 7SL RNA.
For step sucrose gradient analysis, purified virus was dissolved in PBS buffer, exposed briefly to 0.1% Triton X-100, and loaded onto a step sucrose gradient as previously described (57). Three fractions, one containing soluble proteins (F1), a buffer fraction (F2), and the virus-core containing fraction (F3), were harvested. The individual gradient fractions were then subjected to immunoblot analysis.
Quantitative real-time PCR (qRT-PCR).
RNA samples were derived from fractions of viral gradient or immunoprecipitated samples and treated with DNase by incubation in 10 μl of diethyl pyrocarbonate (DEPC)-treated water with 1× RQ1 RNase-Free DNase Buffer, l μl RQ1 RNase-Free DNase (Promega), and 4 U RNase inhibitor (New England BioLabs) for 30 min at 37°C. The DNase was inactivated by the addition of 1 μl of RQ1 DNase Stop Solution and incubated at 65°C for 10 min. RNA was reverse transcribed using random primers and the Multiscribe reverse transcriptase from the High Capacity cDNA Archive Kit (Applied Biosystems) according to the manufacturer's instructions. The cDNA was either undiluted or serially diluted in DEPC-treated water before being input into the real-time reaction mixture to ensure that amplification was within the linear range of detection.
The ABI 7000 sequence detection system (Applied Biosystems) was used for real-time PCR amplifications. All primers were synthesized by Invitrogen, and fluorescence-tagged probes were synthesized by Applied Biosystems. Agarose gel analysis was used to verify that each primer pair produced single amplicons, and the identities of the PCR products were verified by cloning and sequencing. qRT-PCR was performed using either TaqMan fluorescent probes or the SYBR green method.
For the TaqMan method, each 20-μl reaction mixture contained 1 μl of each forward and reverse specific primer (10 μM), 1 μl of fluorescent TaqMan probes (5 μM), 10 μl of 2× Universal TaqMan PCR Master Mix, 4 μl of RNase-free water, and 3 μl of template cDNA. The reactions were carried out under the following conditions: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. The target sequences were amplified using the following primer pairs and probes: 7SL RNA, forward, 5′-ATCGGGTGTCCGCACTAAG-3′, reverse, 5′-CACCCCTCCTTAGGCAACCT-3′, and probe, 5′-6-carboxyfluorescein (FAM)-CATCAATATGGTGACCTCC-6-carboxytetramethylrhodamine (TAMRA)-3′; HIV RNA, forward, 5′-TGTGTGCCCGTCTGTTGTGT-3′, reverse, 5′-GAGTCCTGCGTCGAGAGAGC-3′, and probe 5′- FAM-CAGTGGCGCCCGAACAGGGA-TAMRA-3′.
For the SYBR green method, each 20 μl of reaction mixture contained 1 μl of each forward and reverse specific primer (10 μM), 10 μl of 2× SYBR green PCR Master Mix, 5 μl of RNase-free water, and 3 μl of template cDNA. The reactions were performed under the following conditions: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min, followed by a dissociation protocol. Single peaks in the melting curve analysis indicated specific amplicons. The target sequences amplified by the SYBR green method used the following primer pairs: 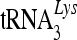 , forward, 5′-GCCCGGATAGCTCAGTCG-3′, and reverse, 5′-TGGCGCCCGAACAGG-3′; GAPDH (glyceraldehyde-3-phosphate dehydrogenase), forward, 5′-GCAAATTCCATGGCACCGT-3′, and reverse, 5′-TCGCCCCACTTGATTTTGG-3′ .
, forward, 5′-GCCCGGATAGCTCAGTCG-3′, and reverse, 5′-TGGCGCCCGAACAGG-3′; GAPDH (glyceraldehyde-3-phosphate dehydrogenase), forward, 5′-GCAAATTCCATGGCACCGT-3′, and reverse, 5′-TCGCCCCACTTGATTTTGG-3′ .
The copy numbers of the target cDNA in the qRT-PCR assay were determined by using a standard curve of 10-fold serial dilutions of nonlinearized plasmid DNA containing the target sequence (ranging from 5 or 10 copies to 5 × 106 or 10 × 106 copies). Absolute RNA copy numbers were calculated by using standard dilution curves of plasmids containing the target sequence. If the template cDNA was diluted before being added to the reaction mixture, the copy number of the target transcript was adjusted by the dilution factor.
RESULTS
Characterization of 7SL RNA-binding residues in A3G.
A3G selectively interacts with 7SL RNA, which has been suggested to play a role in the packaging of the deaminase into HIV-1 virions (57). In the present study, the N-terminal region of A3G was modeled on the basis of the crystal structure (48) of APOBEC2 (Fig. 1A). Similar structure for the N-terminal region of A3G was achieved using the recently determined structure of the A3G C-terminal region (8, 21) as a model (Fig. 1B).
FIG. 1.

Characterization of 7SL RNA-binding residues in A3G. (A) The N-terminal region of A3G, modeled on the crystal structure of APOBEC2. Zinc-coordinating residues H65, C97, and C100 are shown in red and E67 in light blue. The loop region, RLYYFW, is shown in dark blue; helix 4 in orange; and amino acids Y91 to S95 (SWS motif) of A3G in green. (B) Very similar models resulted from using the A3G CTD structure as the template, especially for the homologous region, where the motif RLYYFW is located, which imparts confidence in the homology modeling. (C) A3G single-amino-acid substitution mutants created in the loop region. (D) Effects of single-amino-acid substitutions within the loop region of A3G on the interaction with 7SL RNA. 293T cells were transfected with the A3G-HA or A3G mutant expression vector, and cell lysates from the transfected cells were immunoprecipitated with anti-HA antibody-conjugated agarose and analyzed by immunoblotting using an anti-HA antibody. RNAs were extracted from the coprecipitated samples and analyzed by qRT-PCR using primers specific for 7SL RNA. The level of binding of 7SL RNA to wild-type A3G was set to 100. Nonspecific binding of 7SL RNA from 293T cells to the immunoprecipitation system was also assessed. The error bars indicate standard deviations. (E) The various A3G127 mutants constructed. (F) Interaction of various A3G127 mutants with 7SL RNA, analyzed as described in the legend to panel D.
The cytidine deaminases that target free cytidine or cytosine are present as either dimers or tetramers. The dimer interfaces of these enzymes are formed by the helices that contain the zinc coordination residues, and the compact structure of these cytidine deaminases prevents the zinc coordination residues from participating in interactions with large substrates, such as RNA or DNA. The subunits of another APOBEC, APOBEC2, interact with distinct interfaces formed by β-sheet 2 (48). Consequently, helices 2 and 3 in APOBEC2, which contain the zinc coordination residues, protrude from the surface of the dimer rather than being buried at the subunit interface. This structural feature may explain why AID/APOBEC proteins can bind long polynucleotide substrates. Also, unlike the structure of the free nucleotide cytidine deaminases, the crystal structure of APOBEC2 has revealed the presence of a unique loop sequence and an additional α-helix 4 after β-sheet 4 that are in close proximity to the zinc coordination site (48). Similarly, the crystal structure of a tRNA-adenosine deaminase (TadA) complexed with an RNA substrate has revealed a loop sequence following β-strand 4 that binds oligoribonucleotides directly (33). A similar loop seems to be conserved among AID/APOBEC cytidine deaminases and has been proposed to mediate their interaction with RNA/DNA (12).
The loop region of A3G is highly exposed on the surface (Fig. 1). To characterize the residues that are critical for 7SL RNA binding, A3G mutants containing individual amino acid substitutions in the loop region and the downstream α-helix region were first generated and characterized (Fig. 1C and 2C). The interaction of these A3G mutants with 7SL RNA was compared to that of the parental A3G by using coimmunoprecipitation analysis as previously described (57). Mutation of amino acids in the loop region affected the binding of 7SL RNA to various degrees after adjustment to the immunoprecipitated protein levels (Fig. 1D). Mutation of R122 and Y124 in A3G reduced its 7SL RNA binding more profoundly than did mutation of L123 and Y125 (Fig. 1D). F126 was also found to contribute to 7SL RNA binding (Fig. 1D). All these A3G mutants maintained substantial abilities to bind cellular mRNA and GAPDH (Fig. 1D). Consistent with our previous observations (57), W127 was also important for 7SL RNA binding by A3G (Fig. 1F). The results for various amino acid substitutions at the hydrophobic residue W127 (Fig. 1E) indicated that substitution of F or Y for W was better tolerated than a change to A or L (Fig. 1F), suggesting that an aromatic residue at this position is favored for 7SL RNA binding. The RLYYFW motif has been reported to be important for A3G virion packaging and anti-HIV-1 function (5, 24).
FIG. 2.

Helix 4 and the SWS motif are not required for 7SL RNA interaction. (A) Illustration of the A3G mutants constructed. (B) Interaction of wild-type and mutant A3G-HA with 7SL, analyzed as described in the legend to Fig. 1D. The error bars indicate standard deviations. (C) A3G single-amino-acid substitution mutants created in the SWS motif. (D) Effects of single-amino-acid substitution mutants in A3G SWS motif on the interaction with 7SL, analyzed as described in the legend to Fig. 1D.
In contrast to the results for the loop region, amino acid substitutions in the α-helix 4 region (P129 to L138) of A3G (Fig. 2A) had little effect on the A3G-7SL RNA interaction (Fig. 2B). AID/APOBEC cytidine deaminases also contain an SWS motif, which is predicted to be located in close proximity to the zinc coordination site (Fig. 1A). To determine whether the SWS motif might also be involved in 7SL RNA binding, we generated A3G mutants containing individual amino acid substitutions in the SWS motif (Fig. 2C). Most of the Ala substitutions in the SWS motif (I92 to S95) did not significantly affect the interaction with 7SL RNA (Fig. 2D). However, A3GY91A showed a significantly reduced ability to interact with 7SL RNA compared to the parental A3G (Fig. 2D). Unlike A3GW127A, which maintained the ability to interact with mRNAs, A3GY91A was also unable to bind mRNAs (data not shown). It is not clear whether Y91 of A3G directly participates in RNA binding or is important for protein folding. Unlike W127, which is exposed on the surface, Y91 is predicted to be buried within the A3G molecule (67). Thus, of the three unique features of AID/APOBEC cytidine deaminases (the SWS motif, the loop sequence after β-sheet 4, and α-helix 4), the loop sequence appears to be the most important for 7SL RNA binding. This loop region (RLYYFW) is expected to be exposed on the surface of A3G (Fig. 1). This finding is reminiscent of the reported interaction of TadA with target RNA, in which the loop sequences following β-strand 4 make direct contact with oligoribonucleotides (33). Whether the loop region of A3G directly binds RNA remains to be determined (5, 23).
7SL RNA binding maps to the amino-terminal cytidine deamination domain of A3G.
The RLYYFW motif, which is important for the interaction of A3G with 7SL RNA, is present only in the amino-terminal cytidine deamination domain (N-CDD) of A3G, suggesting that the carboxyl-terminal CDD (C-CDD) is not important for RNA binding. This observation is also consistent with the idea that the N-CDD of A3G mediates RNA binding (25, 43, 57) and virion packaging (7, 24, 34, 57, 67) and the C-CDD of A3G mediates DNA deamination (29, 43, 44). To test this hypothesis, we generated A3G N-CDD and C-CDD expression vectors (Fig. 3A) and then compared the interaction of the N-terminal fragment, the C-terminal fragment, and the full-length A3G with 7SL RNA. Amino acids 1 to 194 of A3G maintained ∼50% or more of the 7SL RNA-binding activity seen for the full-length A3G (Fig. 3B). In contrast, amino acids 195 to 384 of A3G showed a reduction of at least 100-fold in their 7SL RNA-binding activity compared to the full-length A3G (Fig. 3B). Collectively, these data indicate that the N-CDD of A3G mediates the binding of A3G to 7SL RNA.
FIG. 3.

7SL RNA-binding maps to the amino-terminal cytidine deamination domain of A3G. (A) The truncated A3G constructs used in this study. (B) Interaction of A3G-HA, A3G-N-HA, and A3G-C-HA with 7SL RNA, analyzed as described in the legend to Fig. 1D. The error bars indicate standard deviations.
The 7SL RNA-binding and virion-packaging efficiencies of the various human AID/APOBEC cytidine deaminases are widely divergent.
Since a wide range of AID/APOBEC proteins are known to bind RNAs (26, 42), we aligned and compared the amino acid sequences of the loop structures from various human AID/APOBEC proteins (Fig. 4A). The loop sequences (RLYYYW) of A3B and A3F were very similar to that of A3G (RLYYFW) (Fig. 4A). Like A3G and A3F, A3B also interacted with 7SL RNA (Fig. 4B). The 7SL binding efficiencies of A3B and A3F were approximately 4- to 10-fold lower than that of A3G. The residue before Trp is a Tyr in A3B and A3F and a Phe in A3G (Fig. 4A). However, the F126Y mutant of A3G did not show a significantly different level of interaction with 7SL RNA than the parental A3G (Fig. 2D). These results suggest that additional amino acid differences between A3G and A3B/A3F, located elsewhere in the molecule, may contribute to the differences in their 7SL RNA-binding efficiencies. A Trp residue that is conserved in these three 7SL RNA-binding cytidine deaminases (A3B, A3F, and A3G) is missing from the other human cytidine deaminases (A3A, A3C, AID, APOBEC1, and APOBEC2) (Fig. 4A). The loop sequences of these five other cytidine deaminases are also highly divergent from the RLYYF/YW sequence (Fig. 4A). Although A3C, APOBEC1, and AID are known to interact with RNAs, these proteins interacted more poorly with 7SL RNA than did A3G (Fig. 4B). We did not detect any interaction between A3A or APOBEC2 and 7SL RNA (Fig. 4B).
FIG. 4.

Divergent loop sequences and 7SL RNA-binding abilities of human AID/APOBEC cytidine deaminases. (A) Alignment of the amino acid sequences of the loop structure and helix 4 from various human AID/APOBEC proteins. (B) Interaction of various human AID/APOBEC proteins with 7SL RNA, analyzed as described in the legend to Fig. 1D. The error bars indicate standard deviations. (C) Virion packaging of A3A was less efficient than that of A3G. HIV-1 Vif virions were generated by transfecting 293T cells in T25 flasks with NL4-3ΔVif (4 μg) plus 4-fold serial dilutions of A3G-HA or A3A-HA. Cell and viral lysates were harvested from each sample and analyzed by immunoblotting at 48 h posttransfection. A3G-HA and A3A-HA proteins were detected with the anti-HA antibody and viral Gag proteins with an anti-p24 antibody. (D) Virion packaging of APOBEC2 was less efficient than that of A3G. HIV-1 Vif virions were generated by transfecting 293T cells with NL4-3ΔVif (4 μg) plus 4-fold serial dilutions of A3G-V5 or APOPEC2-V5. Cell and viral lysates were harvested from each sample and analyzed by immunoblotting at 48 h posttransfection. A3G-V5 and APOPCE2-V5 proteins were detected with anti-V5 antibody and viral Gag proteins with anti-p24 antibody. (E and F) Virion packaging of A3C (E), AID, or APOBEC1 (F) was analyzed as described for panel C.
We then compared the virion packaging of A3A and APOBEC2 to that of A3G. Decreasing expression levels of A3G and A3A were achieved by using decreasing amount of expression vectors during the transfection of 293T cells. Clearly, virion packaging of A3A was less efficient than that of A3G (Fig. 4C). When A3G was expressed in transfected 293T cells at a level similar to that of endogenous A3G in H9 CD4+ T cells, efficient packaging of A3G could still be detected (Fig. 4C, lane 6). At the same time, almost no A3A packaging could be detected (Fig. 4C, lane 3). We estimated that the A3A-packaging efficiency was at least 10- to 20-fold lower than that of A3G at lower intracellular levels. Similarly, the virion packaging of APOBEC2 was also 10- to 20-fold less efficient than that of A3G (Fig. 4D). Thus, 7SL RNA-binding-defective APOBEC2 and A3A are not packaged efficiently into HIV-1 virions. Our data also indicate that when A3A is overexpressed (Fig. 4C, lane 1), it can be detected in HIV-1 virions, possibly as a result of weak interactions with some viral components or diffusion into the assembling particles. HIV-1 virion-associated A3A molecules are largely excluded from viral cores (1, 16), suggesting a more random nature of this A3A packaging. Unlike A3A and APOBEC2, A3C (Fig. 4E) and AID and APOBEC1 (Fig. 4F) could be packaged efficiently into HIV-1 virions.
Fusion of the N-CDD of A3G with A3A (Fig. 5A) allowed the fusion protein A3Gn/3A to interact with 7SL RNA (Fig. 5B) and to be packaged more efficiently than was A3A alone (Fig. 5C). The efficient packaging of A3Gn/3A into HIV-1 virions is consistent with a previous report (1, 16).
FIG. 5.

(A) Illustration of the A3A-A3G fusion construct. The N-terminal region of A3G (amino acids 1 to 194) was fused with A3A (amino acids 13 to 199). (B) Interaction of A3G-HA or A3Gn/3A-HA with 7SL RNA, analyzed as described in the legend to Fig. 1D. The error bars indicate standard deviations. (C) Fusion of the N terminus of A3G to A3A enhances A3A packaging. HIV-1ΔVif virions were generated by transfecting 293T cells in T25 flasks with NL4-3ΔVif (4 μg) plus 4-fold serial dilutions of A3A or A3Gn/3A. Cell lysates and viral lysates were harvested from each sample and analyzed by immunoblotting at 48 h posttransfection. A3A-HA and A3Gn/3A-HA proteins were detected with the anti-HA antibody, and viral Gag proteins were detected with the anti-p24 antibody.
Association with 7SL RNA mediates the targeting of antiviral cytidine deaminases to viral genomic-RNA-containing RNP complexes.
Although A3C does not interact with 7SL RNA, it is also packaged efficiently into HIV-1 virions, suggesting that it uses a packaging mechanism different from that of A3G and A3F (29, 58, 61). While A3C has been detected in HIV-1 virions, it has only weak antiviral activity compared to A3G (29, 61). This lack of antiviral activity of A3A could be attributed to its inefficient packaging (Fig. 4C) and lack of association with viral core structures. However, we found that virion-packaged A3C was largely associated with HIV-1 core structures (Fig. 6A). A3C is a weak antiviral factor and induced much less cytidine deamination in HIV-1 DNA than did A3G (61). A3C in HIV-1 virions has intrinsic deaminase activity (61) and is as efficient as A3G in inducing mutations in Escherichia coli DNA (20). Since the more potent antiviral cytidine deaminase A3G interacted with 7SL RNA and A3C did not, we speculated that the binding of cytidine deaminases to 7SL RNA may target them to the viral genomic-RNA-containing RNP complexes within the viral core structures. An association with viral genomic-RNA-containing complexes during uncoating and/or partial core disassembly would in turn allow more efficient cytidine deamination of newly synthesized viral single-stranded DNA (ssDNA) during reverse transcription.
FIG. 6.

Association of 7SL RNA with viral genomic-RNA-containing RNP complexes. (A) Association of A3C molecules with viral cores. NL4-3 Vif was cotransfected with the A3C expression vector. At 48 h posttransfection, virus supernatants were collected and analyzed on stepwise sucrose gradients. Three fractions were harvested and analyzed by immunoblotting with an HIV-positive human serum to detect CAp24 and Pr55Gag, anti-Map17 and anti-NCp7, or with anti-HA to detect A3C-HA. (B) A3C is not associated with the viral RNP complexes containing viral genomic RNA and primer 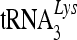 . 293T cells were transfected with NL4-3ΔVif and the A3C-HA expression vector. Purified virions were treated with nonionic detergent (1% Triton X-100) to disrupt the viral cores. Viral lysates were separated on a linear 20 to 70% (wt/vol) sucrose gradient; 12 fractions were collected from the top of each gradient, and A3C-HA and CAp24 were analyzed by immunoblotting. RNAs were extracted from each fraction and analyzed for the presence of viral genomic RNA and
. 293T cells were transfected with NL4-3ΔVif and the A3C-HA expression vector. Purified virions were treated with nonionic detergent (1% Triton X-100) to disrupt the viral cores. Viral lysates were separated on a linear 20 to 70% (wt/vol) sucrose gradient; 12 fractions were collected from the top of each gradient, and A3C-HA and CAp24 were analyzed by immunoblotting. RNAs were extracted from each fraction and analyzed for the presence of viral genomic RNA and 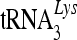 . (C) 7SL RNA molecules are associated with viral genomic RNA and other RNP components inside the viral core structure. Fractions 9 to 12 containing peak viral genomic RNAs were analyzed by qRT-PCR to detect 7SL RNA and by immunoblotting to detect NCp7 and Pr55Gag. The error bars indicate standard deviations.
. (C) 7SL RNA molecules are associated with viral genomic RNA and other RNP components inside the viral core structure. Fractions 9 to 12 containing peak viral genomic RNAs were analyzed by qRT-PCR to detect 7SL RNA and by immunoblotting to detect NCp7 and Pr55Gag. The error bars indicate standard deviations.
To analyze the association of A3C with viral RNP complexes inside the cores, we disrupted the viral cores by means of a brief treatment with nonionic detergent (Fig. 6B). Viral RNP complexes from released mature HIV-1 virions were then resolved by sucrose density gradient centrifugation. HIV-1 genomic RNA- and primer 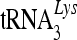 -containing viral RNP complexes were detected in sucrose gradient fractions 9 to 12 (Fig. 6B). CAp24 and A3C were largely separated from the viral RNP complexes (Fig. 6B). We found that 7SL RNA molecules were also associated with the viral RNPs (Fig. 6C). The copy number for 7SL RNA in the viral RNP complexes was about two to three times that of the viral genomic RNA (Fig. 6C). Assuming two copies of viral genomic RNA per viral particle, we estimated that 4 to 6 copies of the 7SL RNA molecules are associated with each viral RNP containing two copies of viral genomic RNA. The viral RNP complexes also contain uncleaved Pr55Gag and NCp7 (Fig. 6C).
-containing viral RNP complexes were detected in sucrose gradient fractions 9 to 12 (Fig. 6B). CAp24 and A3C were largely separated from the viral RNP complexes (Fig. 6B). We found that 7SL RNA molecules were also associated with the viral RNPs (Fig. 6C). The copy number for 7SL RNA in the viral RNP complexes was about two to three times that of the viral genomic RNA (Fig. 6C). Assuming two copies of viral genomic RNA per viral particle, we estimated that 4 to 6 copies of the 7SL RNA molecules are associated with each viral RNP containing two copies of viral genomic RNA. The viral RNP complexes also contain uncleaved Pr55Gag and NCp7 (Fig. 6C).
Addition of a 7SL RNA-binding domain enhances A3C antiviral activity.
In contrast to A3C, the potent antiviral cytidine deaminases A3G (Fig. 7A) and A3F (data not shown) were found to be mainly associated with the viral RNP complexes. These two potent antiviral cytidine deaminases were able to target HIV-1 viral RNPs by binding to 7SL RNA. Since A3C is associated with HIV-1 cores but still lacks potent antiviral activity, we hypothesized that the addition of 7SL RNA-binding ability to A3C might enhance its antiviral activity. Since the N-terminal domain of A3G is able to bind 7SL RNA (Fig. 3B), we generated an expression vector contained amino acids 1 to 194 of A3G fused to A3C (A3Gn/3C) (Fig. 7B). This chimeric protein was expressed in virus-producing cells and was packaged into HIV-1 virions (Fig. 7C). Compared to A3C, A3Gn/3C showed enhanced antiviral activity against HIV-1 Vif (Fig. 7D). Also, unlike A3C, A3Gn/3C was largely associated with viral RNP complexes (Fig. 7E). Thus, addition of a 7SL RNA-binding domain converted A3C to a more potent anti-HIV-1 cytidine deaminase by allowing it to be targeted to viral RNPs.
FIG. 7.

Addition of a 7SL RNA-binding domain from A3G enhances the antiviral activity of A3C. (A) A3G is associated with the viral RNP. 293T cells were transfected with NL4-3ΔVif and the A3G-HA expression vector. Viral lysates were separated, and RNA was extracted as in Fig. 5C. (B) Construction of an A3Gn/3C chimera. A3G-N and A3C were amplified, and the PCR products of A3G-N and A3C were annealed and fused using the overlapping method. (C) The A3Gn/3C chimera associates with the viral RNP as analyzed in Fig. 5C. (D) Chimeric A3Gn/3C is packaged into HIV-1 virions. The error bars indicate standard deviations. (E). A3Gn/3C exhibits increased antiviral activity against HIV-1 compared to parental A3C. Virus infectivity in the absence of A3C or A3Gn/3C was set to 100%. A3C molecules are largely associated with the viral cores but not viral RNPs. A3C cannot be targeted efficiently to the viral genomic-RNA- and primer 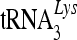 -containing complexes and only inefficiently induces the deamination of newly synthesized viral DNA. (3) The potent anti-HIV-1 cytidine deaminases A3G and A3F interact with 7SL RNA and are packaged efficiently into HIV-1 virions. Their association with 7SL RNA could promote their antiviral cytidine deaminase activity by strong association with viral reverse transcription complexes for efficient DNA substrate recognition and deamination.
-containing complexes and only inefficiently induces the deamination of newly synthesized viral DNA. (3) The potent anti-HIV-1 cytidine deaminases A3G and A3F interact with 7SL RNA and are packaged efficiently into HIV-1 virions. Their association with 7SL RNA could promote their antiviral cytidine deaminase activity by strong association with viral reverse transcription complexes for efficient DNA substrate recognition and deamination.
DISCUSSION
Human AID/APOBEC cytidine deaminases have an intrinsic ability to interact with RNAs (26, 42, 43, 45, 57). However, we have now demonstrated that only the more potent anti-HIV-1 cytidine deaminases, such as A3G and, to a lesser extent, A3B and A3F, are able to interact with 7SL RNA (Fig. 4B). Compared to A3G, the other known RNA-binding cytidine deaminases, A3C, AID, and APOBEC1, interacted poorly with 7SL RNA. The non-anti-HIV-1 cytidine deaminases A3A and APOBEC2 did not interact with 7SL RNA (Fig. 4B) and were packaged into HIV-1 virions at a much lower efficiency than was A3G (Fig. 4C and D). 7SL RNA is preferentially packaged into HIV-1 virions over many other cellular RNAs (22, 27, 47, 56), and it is also one of the more abundant cellular RNAs in HIV-1 virions (47, 57). Although A3G could bind various cellular RNAs, 7SL RNA is preferentially recognized by A3G. Thus, we have proposed the binding of A3G to 7SL RNA as a plausible mechanism for its efficient packaging into virions (56, 57). Consistent with this argument, we have observed that overexpression of the 7SL RNA-binding protein SRP19 inhibits 7SL RNA and A3G packaging into HIV-1 virions (57). SRP19 inhibits A3G packaging more efficiently when A3G expression is controlled at a level similar to that observed in CD4+ T cells (57). Since A3G could bind RNAs in addition to 7SL RNA (4, 10, 43, 57), high levels of A3G expression may result in 7SL RNA-independent virion packaging of A3G (3). However, overexpression of A3G could also lead to increased A3G packaging outside the viral cores (52).
An association with 7SL RNA may not be the sole mechanism for packaging cytidine deaminases into HIV-1 virions. APOBEC1, AID, and A3C did not interact efficiently with 7SL RNA but were still packaged into HIV-1 virions (Fig. 4). Unlike A3G, A3C is packaged into HIV-1 particles through an NC-independent, 7SL RNA-independent mechanism (58). Virion-packaged A3C showed only weak antiviral activity and induced a low level of cytidine deamination in HIV-1 DNA. We found that A3C molecules are associated with viral cores. Thus, an association with viral core structures is apparently not sufficient to mediate robust antiviral activity by cytidine deaminases. On the other hand, an association with viral RNP complexes inside viral cores predicts the potency of the anti-HIV-1 activity displayed by these proteins. We observed that although A3C was associated with viral cores, it was largely separated from the viral RNP complexes containing viral genomic RNA and primer 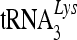 (Fig. 6C). In contrast, the potent antiviral cytidine deaminases A3G (Fig. 7A) and A3F (data not shown) were tightly associated with viral RNP complexes inside viral cores (Fig. 7A). We found that the 7SL RNA molecules were also associated with viral genomic RNA and other RNP components inside the viral core structures (Fig. 6D). Addition of the amino-terminal region of A3G, which contains a 7SL RNA-binding domain to A3C, allowed the fusion protein to associate with viral RNPs and enhanced its anti-HIV-1 function (Fig. 7). An interesting future question is whether A3G recognizes a complex structure containing viral genomic RNA, 7SL RNA, and NC proteins.
(Fig. 6C). In contrast, the potent antiviral cytidine deaminases A3G (Fig. 7A) and A3F (data not shown) were tightly associated with viral RNP complexes inside viral cores (Fig. 7A). We found that the 7SL RNA molecules were also associated with viral genomic RNA and other RNP components inside the viral core structures (Fig. 6D). Addition of the amino-terminal region of A3G, which contains a 7SL RNA-binding domain to A3C, allowed the fusion protein to associate with viral RNPs and enhanced its anti-HIV-1 function (Fig. 7). An interesting future question is whether A3G recognizes a complex structure containing viral genomic RNA, 7SL RNA, and NC proteins.
The association of cytidine deaminases with viral genomic RNA and primer 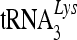 complexes may provide access to the newly synthesized viral DNA substrate during reverse transcription, resulting in more efficient deamination of the HIV-1 DNA. We have now demonstrated for the first time that 7SL RNA, as well as primer
complexes may provide access to the newly synthesized viral DNA substrate during reverse transcription, resulting in more efficient deamination of the HIV-1 DNA. We have now demonstrated for the first time that 7SL RNA, as well as primer 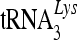 , is strongly associated with viral genomic RNA inside viral cores. We have also detected NCp7 and almost all the virion-associated uncleaved Gag precursor molecules in these viral RNP complexes. The role of 7SL RNA, which is present at two to three times the level of viral genomic RNA in these complexes, remains to be defined. However, it is reasonable to postulate a role for this RNA in mediating the precise targeting of cytidine deaminases to the viral reverse transcription complex for efficient viral DNA deamination.
, is strongly associated with viral genomic RNA inside viral cores. We have also detected NCp7 and almost all the virion-associated uncleaved Gag precursor molecules in these viral RNP complexes. The role of 7SL RNA, which is present at two to three times the level of viral genomic RNA in these complexes, remains to be defined. However, it is reasonable to postulate a role for this RNA in mediating the precise targeting of cytidine deaminases to the viral reverse transcription complex for efficient viral DNA deamination.
It has been proposed that reverse transcription and the degradation of A3G-associated RNA during virus infection are required for maximal activation of the deaminase and its antiviral activity (52). The association of 7SL RNA-binding A3G with viral reverse transcription complexes may also increase the likelihood that A3G-associated RNA will be targeted for degradation by viral RNase H and subsequent enzyme activation. It is interesting that reverse transcription of 7SL RNA has been observed in HIV-1 virions and virus-infected cells (56). Thus, human cytidine deaminases can be separated into three groups. (i) A3A and APOBEC2 do not interact with 7SL RNA and are packaged poorly into HIV-1 virions; the few A3A molecules that are packaged into HIV-1 virions are mainly found outside the core structures. (ii) A3C, AID, and APOBEC1 are RNA-binding proteins; however, they do not interact with 7SL RNA and are packaged into HIV-1 virions through 7SL RNA-independent mechanisms. These three deaminases show poor anti-HIV-1 activity despite the fact that A3C molecules are largely associated with the viral cores. A3C cannot be targeted efficiently to the viral genomic-RNA- and primer 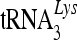 -containing complexes and only inefficiently induce the deamination of newly synthesized viral DNA. We have further demonstrated the importance of the interaction with 7SL RNA by demonstrating that A3C targeting to viral RNPs and anti-HIV-1 function can be enhanced by fusing it to a 7SL RNA-binding domain from A3G (Fig. 7). (iii) The potent anti-HIV-1 cytidine deaminases A3G and A3F interact with 7SL RNA and are packaged efficiently into HIV-1 virions. Their association with 7SL RNA could promote their antiviral cytidine deaminase activities by (a) mediating efficient virion packaging and (b) delivering them to viral reverse transcription complexes for efficient DNA substrate recognition and viral DNA deamination.
-containing complexes and only inefficiently induce the deamination of newly synthesized viral DNA. We have further demonstrated the importance of the interaction with 7SL RNA by demonstrating that A3C targeting to viral RNPs and anti-HIV-1 function can be enhanced by fusing it to a 7SL RNA-binding domain from A3G (Fig. 7). (iii) The potent anti-HIV-1 cytidine deaminases A3G and A3F interact with 7SL RNA and are packaged efficiently into HIV-1 virions. Their association with 7SL RNA could promote their antiviral cytidine deaminase activities by (a) mediating efficient virion packaging and (b) delivering them to viral reverse transcription complexes for efficient DNA substrate recognition and viral DNA deamination.
7SL RNA-binding maps to the amino-terminal CDD of A3G. Our data indicate that amino acids 1 to 194 of A3G maintained more than 50% of the 7SL RNA-binding ability of the full-length A3G, but residues 195 to 384 showed no appreciable interaction with 7SL RNA. It remains to be determined whether other regions in addition to the RLYYFW motif also contribute to its binding to 7SL RNA. The AID/APOBEC proteins contain a unique SWS motif, which is also located close to the zinc coordination domain. However, we have now observed that this SWS motif is largely dispensable for 7SL RNA binding (Fig. 2). Although A3G binding to 7SL RNA was disrupted in the Y91A mutant (Fig. 2), it is not clear whether Y91 directly participates in 7SL RNA binding, since it has been argued that this residue is somewhat buried in the molecule. The AID/APOBEC-unique α-helix 4, which is important for the A3G-Vif interaction, was also dispensable for A3G-7SL RNA binding. In A3G, the N-CDD of A3G contains more positively charged amino acids than does the C-CDD. The potential contribution of these positively charged amino acids in the N-CDD of A3G to 7SL RNA binding still needs to be evaluated. An identification of the structural mechanisms that govern A3G and target RNA recognition will increase our understanding of the functions of these (and perhaps other) cytidine deaminases and may contribute to the development of more potent anti-HIV inhibitors.
Acknowledgments
We thank Michael Malim, Ned Landau, Ruben Harris, Klaus Strebel, and Michael Emerman for critical reagents; the AIDS Research and Reference Reagents Program, Division of AIDS, NIAID, NIH, for the APOBEC1, APOBEC2, and AID expression vectors; and Deborah McClellan for editorial assistance. We also thank Elana Ehrlich for technical assistance and thoughtful discussions.
This work was supported by grants AI062644 and AI071769 from the NIH to X.-F.Yu.
Footnotes
Published ahead of print on 6 October 2010.
REFERENCES
- 1.Aguiar, R. S., N. Lovsin, A. Tanuri, and B. M. Peterlin. 2008. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 283:2518-2525. [DOI] [PubMed] [Google Scholar]
- 2.Alce, T. M., and W. Popik. 2004. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 279:34083-34086. [DOI] [PubMed] [Google Scholar]
- 3.Bach, D., S. Peddi, B. Mangeat, A. Lakkaraju, K. Strub, and D. Trono. 2008. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogerd, H. P., and B. R. Cullen. 2008. Single-stranded RNA facilitates nucleocapsid: APOBEC3G complex formation. RNA 14:1228-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulliard, Y., P. Turelli, U. F. Rohrig, V. Zoete, B. Mangeat, O. Michielin, and D. Trono. 2009. Functional analysis and structural modeling of human APOBEC3G reveal the role of evolutionarily conserved elements in the inhibition of human immunodeficiency virus type 1 infection and Alu transposition. J. Virol. 83:12611-12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnett, A., and P. Spearman. 2007. APOBEC3G multimers are recruited to the plasma membrane for packaging into human immunodeficiency virus type 1 virus-like particles in an RNA-dependent process requiring the NC basic linker. J. Virol. 81:5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cen, S., F. Guo, M. Niu, J. Saadatmand, J. Deflassieux, and L. Kleiman. 2004. The interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem. 279:33177-33184. [DOI] [PubMed] [Google Scholar]
- 8.Chen, K. M., E. Harjes, P. J. Gross, A. Fahmy, Y. Lu, K. Shindo, R. S. Harris, and H. Matsuo. 2008. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452:116-119. [DOI] [PubMed] [Google Scholar]
- 9.Chiu, Y. L., and W. C. Greene. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317-353. [DOI] [PubMed] [Google Scholar]
- 10.Chiu, Y. L., H. E. Witkowska, S. C. Hall, M. Santiago, V. B. Soros, C. Esnault, T. Heidmann, and W. C. Greene. 2006. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U. S. A. 103:15588-15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conticello, S. G., R. S. Harris, and M. S. Neuberger. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009-2013. [DOI] [PubMed] [Google Scholar]
- 12.Conticello, S. G., M. A. Langlois, and M. S. Neuberger. 2007. Insights into DNA deaminases. Nat. Struct. Mol. Biol. 14:7-9. [DOI] [PubMed] [Google Scholar]
- 13.Conticello, S. G., M. A. Langlois, Z. Yang, and M. S. Neuberger. 2007. DNA deamination in immunity: AID in the context of its APOBEC relatives. Adv. Immunol. 94:37-73. [DOI] [PubMed] [Google Scholar]
- 14.Cullen, B. R. 2006. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J. Virol. 80:1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Douaisi, M., S. Dussart, M. Courcoul, G. Bessou, R. Vigne, and E. Decroly. 2004. HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem. Biophys. Res. Commun. 321:566-573. [DOI] [PubMed] [Google Scholar]
- 16.Goila-Gaur, R., M. A. Khan, E. Miyagi, S. Kao, and K. Strebel. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goila-Gaur, R., and K. Strebel. 2008. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haché, G., L. M. Mansky, and R. S. Harris. 2006. Human APOBEC3 proteins, retrovirus restriction, and HIV drug resistance. AIDS Rev. 8:148-157. [PubMed] [Google Scholar]
- 19.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803-809. [DOI] [PubMed] [Google Scholar]
- 20.Harris, R. S., S. K. Petersen-Mahrt, and M. S. Neuberger. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10:1247-1253. [DOI] [PubMed] [Google Scholar]
- 21.Holden, L. G., C. Prochnow, Y. P. Chang, R. Bransteitter, L. Chelico, U. Sen, R. C. Stevens, M. F. Goodman, and X. S. Chen. 2008. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 456:121-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houzet, L., J. C. Paillart, F. Smagulova, S. Maurel, Z. Morichaud, R. Marquet, and M. Mougel. 2007. HIV controls the selective packaging of genomic, spliced viral and cellular RNAs into virions through different mechanisms. Nucleic Acids Res. 35:2695-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huthoff, H., F. Autore, S. Gallois-Montbrun, F. Fraternali, and M. H. Malim. 2009. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 5:e1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huthoff, H., and M. H. Malim. 2007. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and virion encapsidation. J. Virol. 81:3807-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwatani, Y., H. Takeuchi, K. Strebel, and J. G. Levin. 2006. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80:5992-6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarmuz, A., A. Chester, J. Bayliss, J. Gisbourne, I. Dunham, J. Scott, and N. Navaratnam. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79:285-296. [DOI] [PubMed] [Google Scholar]
- 27.Keene, S. E., S. R. King, and A. Telesnitsky. 2010. 7SL RNA is retained in HIV-1 minimal virus-like particles as an S-domain fragment. J. Virol. 84:9070-9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan, M. A., S. Kao, E. Miyagi, H. Takeuchi, R. Goila-Gaur, S. Opi, C. L. Gipson, T. G. Parslow, H. Ly, and K. Strebel. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 79:5870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langlois, M. A., R. C. Beale, S. G. Conticello, and M. S. Neuberger. 2005. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 33:1913-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lecossier, D., F. Bouchonnet, F. Clavel, and A. J. Hance. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112. [DOI] [PubMed] [Google Scholar]
- 31.Liu, B., P. T. Sarkis, K. Luo, Y. Yu, and X. F. Yu. 2005. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 79:9579-9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, B., X. Yu, K. Luo, Y. Yu, and X. F. Yu. 2004. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J. Virol. 78:2072-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Losey, H. C., A. J. Ruthenburg, and G. L. Verdine. 2006. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 13:153-159. [DOI] [PubMed] [Google Scholar]
- 34.Luo, K., B. Liu, Z. Xiao, Y. Yu, X. Yu, R. Gorelick, and X. F. Yu. 2004. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 78:11841-11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo, K., Z. Xiao, E. Ehrlich, Y. Yu, B. Liu, S. Zheng, and X. F. Yu. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444-11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malim, M. H., and M. Emerman. 2008. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe 3:388-398. [DOI] [PubMed] [Google Scholar]
- 37.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99-103. [DOI] [PubMed] [Google Scholar]
- 38.Mariani, R., D. Chen, B. Schrofelbauer, F. Navarro, R. Konig, B. Bollman, C. Munk, H. Nymark-McMahon, and N. R. Landau. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21-31. [DOI] [PubMed] [Google Scholar]
- 39.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398-1403. [DOI] [PubMed] [Google Scholar]
- 40.Mehle, A., J. Goncalves, M. Santa-Marta, M. McPike, and D. Gabuzda. 2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18:2861-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehle, A., E. R. Thomas, K. S. Rajendran, and D. Gabuzda. 2006. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol. Chem. 281:17259-17265. [DOI] [PubMed] [Google Scholar]
- 42.Navaratnam, N., S. Bhattacharya, T. Fujino, D. Patel, A. L. Jarmuz, and J. Scott. 1995. Evolutionary origins of apoB mRNA editing: catalysis by a cytidine deaminase that has acquired a novel RNA-binding motif at its active site. Cell 81:187-195. [DOI] [PubMed] [Google Scholar]
- 43.Navarro, F., B. Bollman, H. Chen, R. Konig, Q. Yu, K. Chiles, and N. R. Landau. 2005. Complementary function of the two catalytic domains of APOBEC3G. Virology 333:374-386. [DOI] [PubMed] [Google Scholar]
- 44.Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa, M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166-170. [DOI] [PubMed] [Google Scholar]
- 45.Niewiadomska, A. M., C. Tian, L. Tan, T. Wang, P. T. Sarkis, and X. F. Yu. 2007. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 81:9577-9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niewiadomska, A. M., and X. F. Yu. 2009. Host restriction of HIV-1 by APOBEC3 and viral evasion through Vif. Curr. Top. Microbiol. Immunol. 339:1-25. [DOI] [PubMed] [Google Scholar]
- 47.Onafuwa-Nuga, A. A., A. Telesnitsky, and S. R. King. 2006. 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA 12:542-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prochnow, C., R. Bransteitter, M. G. Klein, M. F. Goodman, and X. S. Chen. 2007. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature 445:447-451. [DOI] [PubMed] [Google Scholar]
- 49.Schäfer, A., H. P. Bogerd, and B. R. Cullen. 2004. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328:163-168. [DOI] [PubMed] [Google Scholar]
- 50.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646-650. [DOI] [PubMed] [Google Scholar]
- 51.Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404-1407. [DOI] [PubMed] [Google Scholar]
- 52.Soros, V. B., W. Yonemoto, and W. C. Greene. 2007. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12:591-601. [DOI] [PubMed] [Google Scholar]
- 54.Suspène, R., P. Sommer, M. Henry, S. Ferris, D. Guetard, S. Pochet, A. Chester, N. Navaratnam, S. Wain-Hobson, and J. P. Vartanian. 2004. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 32:2421-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Svarovskaia, E. S., H. Xu, J. L. Mbisa, R. Barr, R. J. Gorelick, A. Ono, E. O. Freed, W. S. Hu, and V. K. Pathak. 2004. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 279:35822-35828. [DOI] [PubMed] [Google Scholar]
- 56.Tian, C., T. Wang, W. Zhang, and X. F. Yu. 2007. Virion packaging determinants and reverse transcription of SRP RNA in HIV-1 particles. Nucleic Acids Res. 35:7288-7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang, T., C. Tian, W. Zhang, K. Luo, P. T. Sarkis, L. Yu, B. Liu, Y. Yu, and X. F. Yu. 2007. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 81:13112-13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang, T., W. Zhang, C. Tian, B. Liu, Y. Yu, L. Ding, P. Spearman, and X. F. Yu. 2008. Distinct viral determinants for the packaging of human cytidine deaminases APOBEC3G and APOBEC3C. Virology 377:71-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao, Z., E. Ehrlich, Y. Yu, K. Luo, T. Wang, C. Tian, and X. F. Yu. 2006. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 349:290-299. [DOI] [PubMed] [Google Scholar]
- 60.Xiao, Z., Y. Xiong, W. Zhang, L. Tan, E. Ehrlich, D. Guo, and X. F. Yu. 2007. Characterization of a novel cullin5 binding domain in HIV-1 Vif. J. Mol. Biol. 373:541-550. [DOI] [PubMed] [Google Scholar]
- 61.Yu, Q., D. Chen, R. Konig, R. Mariani, D. Unutmaz, and N. R. Landau. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 279:53379-53386. [DOI] [PubMed] [Google Scholar]
- 62.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11:435-442. [DOI] [PubMed] [Google Scholar]
- 63.Yu, X., Y. Yu, B. Liu, K. Luo, W. Kong, P. Mao, and X. F. Yu. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056-1060. [DOI] [PubMed] [Google Scholar]
- 64.Yu, Y., Z. Xiao, E. S. Ehrlich, X. Yu, and X. F. Yu. 2004. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 18:2867-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zennou, V., D. Perez-Caballero, H. Gottlinger, and P. D. Bieniasz. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 78:12058-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang, H., B. Yang, R. J. Pomerantz, C. Zhang, S. C. Arunachalam, and L. Gao. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang, K. L., B. Mangeat, M. Ortiz, V. Zoete, D. Trono, A. Telenti, and O. Michielin. 2007. Model structure of human APOBEC3G. PLoS One 2:e378. [DOI] [PMC free article] [PubMed] [Google Scholar]