

Abstract
The distribution of the severe acute respiratory syndrome coronavirus (SARS-CoV) receptor, an angiotensin-converting enzyme 2 (ACE2), does not strictly correlate with SARS-CoV cell tropism in lungs; therefore, other cellular factors have been predicted to be required for activation of virus infection. In the present study, we identified transmembrane protease serine 2 (TMPRSS2), whose expression does correlate with SARS-CoV infection in the upper lobe of the lung. In Vero cells expressing TMPRSS2, large syncytia were induced by SARS-CoV infection. Further, the lysosome-tropic reagents failed to inhibit, whereas the heptad repeat peptide efficiently inhibited viral entry into cells, suggesting that TMPRSS2 affects the S protein at the cell surface and induces virus-plasma membrane fusion. On the other hand, production of virus in TMPRSS2-expressing cells did not result in S-protein cleavage or increased infectivity of the resulting virus. Thus, TMPRSS2 affects the entry of virus but not other phases of virus replication. We hypothesized that the spatial orientation of TMPRSS2 vis-a-vis S protein is a key mechanism underling this phenomenon. To test this, the TMPRSS2 and S proteins were expressed in cells labeled with fluorescent probes of different colors, and the cell-cell fusion between these cells was tested. Results indicate that TMPRSS2 needs to be expressed in the opposing (target) cell membrane to activate S protein rather than in the producer cell, as found for influenza A virus and metapneumoviruses. This is the first report of TMPRSS2 being required in the target cell for activation of a viral fusion protein but not for the S protein synthesized in and transported to the surface of cells. Our findings suggest that the TMPRSS2 expressed in lung tissues may be a determinant of viral tropism and pathogenicity at the initial site of SARS-CoV infection.
Angiotensin-converting enzyme 2 (ACE2) has been shown to be the functional receptor for severe acute respiratory syndrome coronavirus (SARS-CoV) (18, 24), the etiological agent of an acute infectious disease that spreads mainly via the respiratory route. Although ACE2 is present in the vascular endothelial cells of all organs, SARS-CoV is highly pathogenic only in the lungs (12). Furthermore, while ACE2 expression in the lung has been shown for both type I and type II pneumocytes (12), cell tropism of SARS-CoV does not strictly correlate with ACE2 expression, suggesting that other factors are required to explain the pathogenesis of this disease (8, 32). One such factor is the critical role played by host cellular proteases in the process of viral entry into cells. For example, a variety of proteases such as trypsin, tryptase Clara, mini-plasmin, human airway trypsin-like protease (HAT), and TMPRSS2 (transmembrane protease, serine 2) are known to cleave the glycoprotein hemagglutinin (HA) of influenza A viruses, a prerequisite for the fusion between viral and host cell membranes and viral cell entry. Cleavage of HA is critical for viral infection, with the tissue distribution of proteases determining cell tropism of virus strains (16). There are two major mechanisms responsible for proteolytic activation of viral glycoproteins. For many viruses, such as the human immunodeficiency virus (HIV) and Nipah virus, cellular proteases (e.g., furin or cathepsin) cleave the glycoprotein during biogenesis, separating receptor binding and fusion subunits and converting the precursor glycoprotein to its fusion-competent state (36). Alternatively, for other viruses such as Ebola and SARS-CoV, cleavage of the viral glycoprotein by endosomal proteases induces conformational changes during viral entry following receptor binding and/or endocytosis (6, 20, 28, 31).
Three proteases—trypsin, cathepsin L, and elastase—have been previously reported to activate the spike (S) protein of SARS-CoV (3, 13, 20, 30, 31). In the absence of proteases at the cell surface, SARS-CoV enters cells by an endosomal pathway, and S protein is activated for fusion by cathepsin L in the endosome (14, 30, 37). Conversely, in the presence of proteases at the cell surface, such as trypsin and elastase, viral S proteins attached to the receptor at the host cell surface are activated, inducing envelope-plasma membrane fusion and direct entry of SARS-CoV into cells (21). Viral replication in the latter case has been shown to be 100 times higher than replication via the endosomal pathway (21), suggesting that the higher infectivity of SARS-CoV in the lungs could be due to an enhancement of direct viral cell entry mediated by proteases. In this study we test the possibility that TMPRSS2 is an activator of SARS-CoV entry into host cells. TMPRSS2 is highly expressed at epithelial cells in human lungs (9, 26) and activates influenza A virus and metapneumovirus in culture cells (4, 5, 7, 29). Here, we present data indicating that the distribution of TMPRSS2 correlates with SARS-CoV infection in the lung and that this protease can efficiently activate SARS-CoV S protein to induce virus-cell membrane fusion at the cell surface.
MATERIALS AND METHODS
Animal experiments.
Animal experiments with SARS-CoV-infected cynomolgus monkeys were performed as described previously (23). Briefly, 3-year-old male cynomolgus monkeys were intratracheally inoculated with 108 50% tissue culture infective doses (TCID50) of SARS-CoV. On day 7 postinoculation animals were euthanized, and tissue samples of the lungs were collected.
Immunofluorescence staining method.
Detection of TMPRSS2, ACE2, and SARS-CoV antigens was performed on paraffin-embedded sections by a double immunofluorescence staining method modified from a previous protocol (19, 22, 23). Rabbit antibody (Ab) against TMPRSS2 (ab56110; Abcam), goat antibody against recombinant human ACE2 (R&D Systems, MN), and the SKOT9 monoclonal mouse antibody against nucleocapsid protein of SARS-CoV (25) were used as primary antibodies. Briefly, after deparaffinization with xylene, the sections were rehydrated in ethanol and immersed in phosphate-buffered saline (PBS). Antigens were retrieved by hydrolytic autoclaving in the retrieval solution at pH 9.0 (Nichirei). After a cooling step, normal donkey or goat serum was used to block background staining. To detect the TMPRSS2 or ACE2 antigen, the sections were immersed in PBS and then incubated with primary antibodies overnight at 4°C. After three washes in PBS, the sections were incubated with monoclonal antibody for 60 min at 37°C to detect the SARS-CoV protein. After three additional washes in PBS, the sections were incubated with anti-rabbit (goat origin) or anti-goat (donkey origin) Alexa Fluor 546 and anti-mouse (goat or donkey origin) Alexa Fluor 488 (Molecular Probes, Eugene, OR) for 60 min at 37°C to detect the primary antibodies,. The sections were mounted with ProLong antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI; Molecular Probes), and images were captured and analyzed by confocal laser microscopy (Fluoview FV1000-D; Olympus, Tokyo, Japan).
Cells and viruses.
Vero E6 and Vero cells and Vero cells expressing TMPRSS2 (Vero-TMPRSS2) (29) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco/BRL), supplemented with 5% fetal bovine serum (Gibco/BRL). The SARS-CoV Frankfurt 1 strain, kindly provided by J. Ziebuhr (University of Würzburg, Germany), and the recombinant vaccinia virus containing the gene encoding the spike protein of SARS-CoV (Dis/SARS-S), kindly provided by K. Ishii (NIID, Japan), were propagated and assayed by using Vero E6 and 293T cells, respectively.
Virus entry assay.
Vero or Vero-TMPRSS2 cells in 96-well plates were treated with DMEM containing the reagents indicated in the legends of Fig. 3 at 37°C for 30 min and then chilled on ice for 10 min. Approximately 105 PFU (PFU) of virus in DMEM was used to infect 106 cells on ice. After an adsorption period of 30 min, the virus was removed, and infected cells were cultured in DMEM with the reagents at 37°C for 5 h. Viral mRNAs were isolated from cells with the addition of 200 μl of Isogen (Nippon gene). Real-time PCR was performed to estimate the amounts of newly synthesized mRNA9 as described previously (21).
Western blotting.
Expression of S protein in Vero E6 cells was analyzed by Western blotting. Preparation of cell lysates, SDS-PAGE, and electrical transfer of the protein onto a transfer membrane were described elsewhere (21). S protein was detected with anti-S antibody (IMG-557; Imgenex, San Diego) and anti-rabbit IgG-horseradish peroxidase (HRPO). Influenza virus HA was detected with anti-HA Ab, Udorn virus HA (29), and anti-goat IgG-HRPO. Bands were visualized by using enhanced chemiluminescence reagents (ECL Plus; Amersham Pharmacia) on an LAS-1000 instrument (Fuji).
Cell-cell fusion.
Effector cells (Vero or Vero-TMPRSS2) were infected with Dis/SARS-S at a multiplicity of infection (MOI) of 1 for 1 h or transfected with pKS/SARS-S by using the Fugene 6 reagent (Roche) and then labeled with 2 μM CellTracker Green (5-chloromethylfluorescein diacetate [CMFDA] Invitrogen) for 30 min and washed three times with medium. Meanwhile, Vero or Vero-TMPRSS2 target cells were labeled with 2 μM orange chloromethyl tetramethylrhodamine (CMTMR; Invitrogen) for 30 min and washed three times with medium. Effector cells were collected with nonenzymatic cell dissociation solution (Sigma) and then overlaid on target cells. Cells were incubated for 20 h and observed by fluorescence microscopy (BioZero, Keyence) for cell-cell fusion. The extent of syncytium formation was semiquantified by counting the number of nuclei in the S-protein-expressing cells (10 syncytia).
RESULTS
Distribution of TMPRSS2 in the SARS-CoV-infected cynomolgus lung.
We examined the immunohistochemical distribution of TMPRSS2 and ACE2 in the uninfected lung of cynomolgus monkeys. TMPRSS2 antigens were detected in type I pneumocytes (Fig. 1A, arrows indicating thin shapes in middle panel), whereas only weak staining of ACE2 antigens was detected in enlarged type II pneumocytes (Fig. 1C, arrows indicating round shapes in middle panel) by the immunofluorescence staining method used. An antibody against SARS-CoV was used as a negative control (Fig. 1A and C, upper left panels). In addition, the histopathology of SARS-CoV-infected lungs was examined on day 7 postinoculation as described previously (23). Mild lesions with some regenerated type II epithelial cells were observed in the upper lobe (Fig. 1B and D,). SARS-CoV antigens were detected in the cytoplasm of type I pneumocytes (Fig. 1B and D, arrowheads), which primarily resembled TMPRSS2-expressing cells (Fig. 1B, upper right) more than ACE2-expressing cells (Fig. 1D, upper right). In contrast, severe pulmonary edema and inflammatory reactions were observed in severe lesions (Fig. 1B and D). Marked immunostaining of TMPRSS2 and ACE2 antigens was detected in the cytoplasm of enlarged type II pneumocytes (Fig. 1B and D, lower middle panels). SARS-CoV antigens were detected in the cytoplasm of many unspecified cells (Fig. 1B and D, lower left), but these cells never merged with either TMPRSS2- or ACE2-expressing cells. These results suggest that the presence of SARS-CoV antigens does not correlate with the presence of either ACE2 or TMPRSS2 antigens in severe lesions.
FIG. 1.

Distribution of TMPRSS2 and ACE2 and histopathology of SARS-CoV-infected cynomolgus lung. The distribution of TMPRSS2 (A, arrows indicating round shapes in middle panel) and ACE2 (C, arrows indicating thin shapes in middle panel) in healthy cynomolgus lung sections stained with antibodies against SARS-CoV, TMPRSS2, or DAPI (upper panels) was detected by an immunofluorescence staining method (see Materials and Methods section). Samples stained with secondary antibodies were used as controls (lower panels). The distribution of TMPRSS2 (B) and ACE2 (D) was also examined in mild (upper row) and severe (lower row) lesions of SARS-CoV-infected lungs stained with antibodies against SARS-CoV, TMPRSS2, or DAPI (nuclear staining).
Syncytium formation by SARS-CoV infection of TMPRSS2-expressing cells.
Vero cells have been generally used for infection assays of SARS-CoV (10). SARS-CoV S protein-expressing Vero cells form syncytia if treated with trypsin but not in its absence (Fig. 2B, frames a and c). Here, we examined whether Vero cells stably expressing TMPRSS2 (VERO-TMPRSS2) undergo syncytium formation induced by SARS-CoV infection or S-protein expression. After 36 h of SARS-CoV infection at an MOI of 0.1, large syncytia were observed in Vero-TMPRSS2 cells (Fig. 2A, frame b) but not in Vero cells (Fig. 2A, frame a). To determine if expression of S protein induced syncytium formation, Vero-TMPRSS2 cells were infected with vaccinia virus encoding the SARS-CoV S gene, Dis/SARS-S, which resulted in the formation of large syncytia at 20 h after infection (Fig. 2B, frame b). In addition, plasmid-based expression of S protein also induced syncytia (see Fig. 5B). To further assess if TMPRSS2 can proteolytically activate S protein, Vero-TMPRSS2 cells expressing S protein were treated with various concentrations of leupeptin (Fig. 2C), a known inhibitor of serine and cysteine proteases. The results show that syncytium formation was moderately inhibited at a concentration of 500 μM (Fig. 2C, frame b, and D) and completely inhibited at a concentration of 5 mM (Fig. 2C, frame c, and D), suggesting that leupeptin inhibits the activity of TMPRSS2, suppressing S-protein-induced cell-cell fusion.
FIG. 2.

Cytopathic changes on SARS-CoV S-protein-expressing cells. (A) Vero cells or Vero-TMPRSS2 cells were infected with SARS-CoV at an MOI of 0.1 and incubated at 37°C for 36 h. Cells were stained with crystal violet. (B) Vero cells or Vero-TMPRSS2 cells were infected with Dis/SARS-S at an MOI of 0.1 and incubated at 37°C for 20 h, after which the cells were treated with 10 μg/ml trypsin at 37°C for 30 min and then incubated for another 3 h. (C) Vero-TMPRSS2 cells were infected with Dis/SARS-S at an MOI of 0.1 and incubated at 37°C for 20 h in the absence (a) or presence of 500 μM (b) or 5 mM (c) leupeptin. Cells not infected with Dis/SARS-S were used as a control (d). (D) The size of syncytia in the absence and presence of 5 μM, 50 μM, 500 μM, and 5 mM leupeptin was quantified by counting the number of nuclei in the fused cells. The error bars are standard deviations.
Viral entry into TMPRSS2-expressing cells.
The above observations suggest that TMPRSS2 activates S protein at the cell surface, enabling viral entry into the cell. To assess this possibility, we first examined the effects of EST (Calbiochem), which is known to inhibit endosomal cathepsins and thereby block infection of Vero cells by SARS-CoV (35). Viral entry into cells was estimated by the newly synthesized viral mRNA9, which was quantified by real-time PCR as described previously (21). As shown in Fig. 3, treatment of Vero cells with EST at a concentration of 50 μM caused a 1,000-fold decrease of SARS-CoV cell entry via the endosomal pathway. In contrast, EST failed to suppress viral entry into Vero-TMPRSS2 cells. Similarly, treatment with bafilomycin A (Sigma) at a concentration of 500 μM reduced infectivity by 100-fold in Vero cells although only mild suppression (10%) of viral infection was observed in Vero-TMPRSS2 cells. Furthermore, we examined the effect of heptad repeat 2 (HR2) peptide, kindly provided by R. S. Hodges (33), on SARS-CoV infectivity in Vero-TMPRSS2 cells. HR2 peptide is known to prevent trypsin-activated cell entry of SARS-CoV via the cell surface; presumably an S protein activated by cell surface receptor exposes the HR1 region, and the HR2 peptide then interferes with the six-helix bundle formation (34). Treatment with HR2 peptide at a concentration of 5 μM efficiently suppressed viral entry into Vero-TMPRSS2 cells but not into Vero cells. These results suggest that TMPRSS2 activates S protein and facilitates viral entry via the cell surface.
FIG. 3.
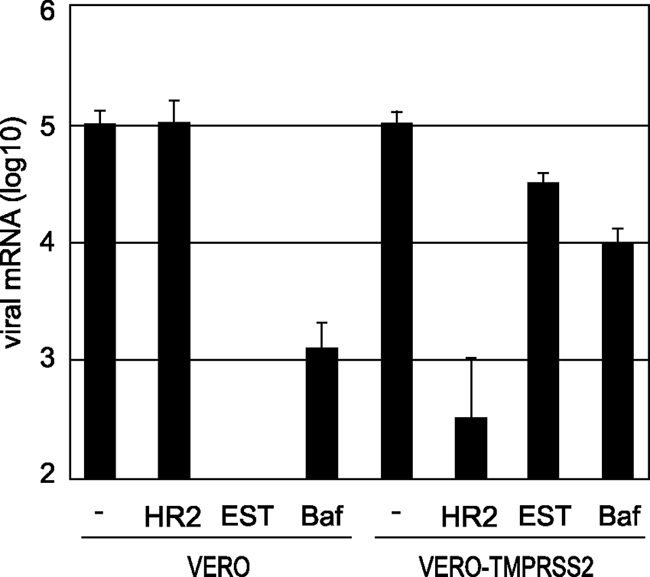
Effect of TMPRSS2 on virus entry into cells. Vero or Vero-TMPRSS2 cells were infected with SARS-CoV at an MOI of 0.1 in the presence of 5 μM HR2 peptide, 50 μM EST, or 500 μM bafilomycin A1 and then cultured for 5 h. The amount of viral mRNA9 was measured by real-time PCR. Cells not treated with reagents were used as controls. The error bars are standard deviations of at least six independent measurements.
TMPRSS2 does not cleave SARS-CoV S during virus production.
It has been previously shown that TMPRSS2 expressed in cells promoted cleavage of glycoprotein of influenza A virus and metapneumovirus, increasing infectivity (5, 29). Here, we examined whether the S protein of SARS-CoV is cleaved and activated in Vero-TMPRSS2 cells. After 36 h of infection of Vero and Vero-TMPRSS2 cells with SARS-CoV, cells and medium were collected, and Western blotting was performed to detect S protein. As shown in Fig. 4A (right), the HA of influenza virus was markedly cleaved in Vero-TMPRSS2 cells at 18 h after transfection. However, S proteins synthesized in either Vero or Vero-TMPRSS2 cells were not cleaved when examined in cell lysates or in virions released into the medium (Fig. 4A, left). Furthermore, the infectious titers of SARS-CoV produced in Vero and Vero-TMPRSS2 (on Vero E6 cells) were not markedly different, and in both cases infection was inhibited by bafilomycin A1 but not HR2 peptide (Fig. 4B). These results suggest that SARS-CoV produced in Vero-TMPRSS2 cells has no fusion-competent, cleaved S protein entering into cells via the endosomal pathway.
FIG. 4.

TMPRSS2 does not affect virus production. (A) SDS-PAGE and Western blot analysis were performed to detect S protein in cell lysates (cell) and culture medium (med) of Vero or Vero-TMPRSS2 (Vero-TM2) cells at 20 h after SARS-CoV infection (left panel). Cells treated with 10 μg/ml trypsin (tryp) at 37°C for 5 min were used as a cleaved-S control. Influenza virus (Flu)-HA produced by plasmid transfection was also used as a cleavage control (right panel). (B) Infectivity of SARS-CoV produced in Vero or Vero-TMPRSS2 cells. Vero-E6 cells were infected with SARS-CoV produced in either Vero or Vero-TMPRSS2 cells in the presence of 5 μM HR2 peptide or 500 nM bafilomycin A1 and then cultured for 5 h. The amount of viral mRNA9 was measured by real-time PCR. Cells not treated with reagents were used as controls. The error bars are standard deviations of at least six independent measurements.
Productive activation of S by TMPRSS2 requires TMPRSS2 expression in target cells.
The previous results suggest that TMPRSS2 affects viral S protein attached to receptors at the cell surface but not newly synthesized S proteins either during transport to the plasma membrane or incorporation into the virion. We hypothesized that the spatial orientation of TMPRSS2 against S protein might be critical for S protein activation. To demonstrate this spatial dependence between TMPRSS2 and S protein, a cell-cell fusion assay was conducted. Our design is diagramed in Fig. 5A. Vero and Vero-TMPRSS2 cells were either Dis/SARS-S infected (Fig. 5B, frames a, b, c, and d) or pKS/SARS-S transfected (Fig. 5B, frames e, f, g, and h) to express S protein and then stained with green as described in the methodology. The green-labeled Vero and Vero-TMPRSS2 cells were collected with nonenzymatic cell dissociation solution and overlaid onto the orange-labeled target cells, Vero and Vero-TMPRSS2. As shown in Fig. 5B, the formation of syncytia was not induced in cells not expressing TMPRSS2 (Fig. 5B, frames a and e). Also, syncytia were not formed between green cells expressing both TMPRSS2 and S protein and the orange Vero cells without TMPRSS2 (Fig. 5B, frames b and f). However, large syncytia were formed when TMPRSS2 was expressed either in the orange target cells (Fig. 5B, frames c and g) or both in the orange target and green producer cells (Fig. 5B, frames d and h). The extent of syncytium formation was semiquantified by counting the number of nuclei in the fused cells infected with Dis/SARS-S (Fig. 5C). It was previously reported that ACE2 is downregulated in S-expressing cells (11, 17), indicating that green producer cells are refractory to self-fusion due to a lack of receptors at the cell surface. These results suggest that the spatial orientation of TMPRSS2 must be opposite that of S protein (namely, the two proteins must reside in opposite membranes) to induce membrane fusion. This observation is in agreement with the idea that TMPRSS2 activates only viral S protein already attached to receptors at the cell surface but not during virus maturation.
FIG. 5.

TMPRSS2 dependence on spatial orientation for the activation of SARS-CoV S protein. (A) Schematic diagrams of S-expressing effector cells (green) and acceptor cells (orange) shown in panel B. (B) To detect cell-cell fusion of S-expressing cells, Dis/SARS-S-infected or pKS/SARS-S-transfected Vero or Vero-TMPRSS2 cells (effector cells) were collected by nonenzymatic cell dissociation solution and then overlaid onto the orange target Vero or Vero-TMPRSS2 cells, respectively. After 20 h of incubation, cells were fixed with 4% formaldehyde and observed by fluorescence microscopy. White arrows indicate fused cells. (C) The sizes of syncytia indicated in the upper row of panel B were quantified by counting the number of nuclei in the fused cells. The error bars are standard deviations.
DISCUSSION
In the present study we showed the distribution of ACE2 in mild and severe inflammatory lesions of cynomolgus lungs infected by SARS-CoV. We found that type II pneumocytes, which are frequently observed in regenerated tissues during inflammation and which express high levels of ACE2, were refractory to SARS-CoV infection, whereas type I pneumocytes, which do not express detectable levels of ACE2, were readily infected by SARS-CoV. This inconsistency may be explained by the downregulation of ACE2 in SARS-CoV-infected cells, as previously reported (11, 17). Here, we showed that the localization of TMPRSS2-expressing cells in normal lung tissues, rather than ACE2-expressing cells, is closely tied to SARS-CoV infection in mild lesions, indicating that TMPRSS2 may determine viral tropism at an early stage of SARS-CoV infection. However, TMPRSS2 expression occurred in cells adjacent to virus-infected cells, not in infected cells, suggesting that TMPRSS2 may also be downregulated in infected cells, as observed for ACE2. We have previously reported that SARS-CoV infectivity was enhanced in culture cells by the addition of exogenous elastase, which enabled virus entry via the cell surface (21). Elastase is a major protease produced by neutrophils during inflammation and may be relevant to the high level of SARS-CoV replication in lungs that results in severe pneumonia in the mouse model (1). We have also reported that in the late stage of SARS-CoV infection in the cynomolgus lung, which remarkably occurs with severe inflammation in the lower lobe, SARS-CoV is distributed in both type I and type II pneumocytes (23), indicating that elastase may trigger viral entry via the surface of these cells and determine SARS-CoV pathogenicity in this late stage of infection.
Activation of viral glycoprotein by TMPRSS2 has been previously reported for influenza A virus and metapneumovirus (4, 5, 7, 29). The most distinctive difference between these viruses and SARS-CoV is the stage during virus replication in which viral glycoproteins are cleaved by proteases. In influenza A virus and metapneumovirus, the protease makes a simple cut in the glycoprotein during maturation, in a manner similar to that made by furin. In contrast, SARS-CoV S protein is cleaved by the protease following receptor-induced conformational changes. The protease cleavage site in S protein, located nearer the C-terminal region than predicted for a cleavage site, is thought to be exposed only after receptor binding (2, 35). In support of this model, we recently reported that the S protein of mouse hepatitis virus type 2 (MHV-2), which is highly similar to the S protein of SARS-CoV (27), requires two-step conformational changes mediated by sequential receptor binding and proteolysis to be activated for fusion (20). Such a mechanism allows for tight temporal control over fusion by protecting the activating cleavage site from premature proteolysis yet allowing efficient cleavage upon binding to the receptor on target cells.
Previous studies have clearly demonstrated that the SARS-CoV S protein requires proteolytic cleavage for S-mediated cell-cell or virus-cell fusion (3, 13, 21, 30, 31). Recently, the cleavage of S protein by the airway transmembrane protease, TMPRSS11a, was also reported (15). Treatment with a purified soluble form of TMPRSS11a following receptor binding to the pseudotyped SARS-CoV S protein strongly enhanced viral infection. Moreover, proteolytic cleavage by TMPRSS11a was observed at the same position as cleavage by trypsin in the purified soluble form of S protein (15). While the reported concentration of the previously mentioned proteases required to induce membrane fusion is extremely high (3, 15, 21 ), the amount of TMPRSS2 expressed in Vero-TMPRSS2 cells is thought to be low (29). Still, our results showed the formation of massive syncytia when S protein was expressed (Fig. 2), suggesting that TMPRSS2 may be more efficient than previously characterized proteases. The inhibition of cell-cell fusion in TMPRSS2-expressing cells in the presence of a protease inhibitor (Fig. 2) strongly suggests that TMPRSS2 proteolytically affects S protein. Because we could not detect a considerable amount of cleaved S protein in cells expressing both S and TMPRSS2, even when massive syncytia were observed in these cells, we hypothesized that proteolytic activation of S protein by TMPRSS2 occurs only during cell entry, following receptor binding. The results shown in Fig. 5 support this hypothesis and clearly show that the spatial orientation of TMPRSS2 in relation to S protein is a key mechanism underling this phenomenon: TMPRSS2 must be expressed in the opposing cell membrane to activate S protein and induce cell-cell fusion. We speculate that the encounter of receptor-bound S protein with TMPRSS2 at the right time and in the correct spatial orientation at the cell surface results in efficient cleavage of S protein and subsequent membrane fusion. Therefore, only a small amount of S protein needs to be cleaved to enable viral or cell-cell membrane fusion. These cleavage products would be difficult to detect by Western blot analysis.
Additionally, we have previously reported that HR2 peptide inhibits SARS-CoV entry into Vero E6 cells in the presence, but not the absence, of trypsin. Likewise, inhibition of SARS-CoV cell entry does not occur when the virus is allowed to use the normal endosomal, cathepsin L-dependent, entry pathway (34). Here, we show (Fig. 3) that HR2 peptide efficiently inhibits viral entry into TMPRSS2-expressing cells. This result provides further evidence that TMPRSS2 is efficient for viral entry and is localized at the cell surface, exposing the HR2 peptide binding site before endocytosis can occur. Thus, even if HR2 peptide does not efficiently work in commonly used tissue cell cultures, it may be a suitable candidate antiviral for the inhibition of SARS-CoV infection of lung cells that express membrane-associated proteases.
Acknowledgments
We thank S. E. Delos and J. M. White (University of Virginia) for valuable comments on this work. We also thank R. S. Hodges (University of Colorado, Denver, CO) and K. Ishii (National Institute of Infectious Diseases, Japan) for providing us with HR2 peptide and Dis/SARS-S, respectively.
This work was supported by an MEXT grant and by grants from the Uehara Memorial Foundation and the Ichiro Kanehara Foundation.
Footnotes
Published ahead of print on 6 October 2010.
REFERENCES
- 1.Ami, Y., N. Nagata, K. Shirato, R. Watanabe, N. Iwata, K. Nakagaki, S. Fukushi, M. Saijo, S. Morikawa, and F. Taguchi. 2008. Co-infection of respiratory bacterium with severe acute respiratory syndrome coronavirus induces an exacerbated pneumonia in mice. Microbiol. Immunol. 52:118-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belouzard, S., V. C. Chu, and G. R. Whittaker. 2009. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. U. S. A. 106:5871-5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosch, B. J., W. Bartelink, and P. J. M. Rottier. 2008. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 82:8887-8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Böttcher, E., T. Matrosovich, M. Beyerle, H. Klenk, W. Garten, and M. Matrosovich. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80:9896-9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaipan, C., D. Kobasa, S. Bertram, I. Glowacka, I. Steffen, T. S. Tsegaye, M. Takeda, T. H. Bugge, S. Kim, Y. Park, A. Marzi, and S. Pölmann. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83:3200-3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandran, K., N. J. Sullivan, U. Felbor, S. P. Whelan, and J. M. Cunningham. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi, S., S. Bertram, I. Glowacka, Y. W. Park, and S. Pölmann. 2009. Type II transmembrane serine proteases in cancer and viral infections. Trends Mol. Med. 15:303-312. [DOI] [PubMed] [Google Scholar]
- 8.Ding, Y., H. Wang, H. Shen, Z. Li, J. Geng, H. Han, J. Cai, X. Li, W. Kang, D. Weng, Y. Lu, D. Wu, L. He, and K. Yao. 2003. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J. Pathol. 200:282-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donaldson, S. H., A. Hirsh, D. C. Li, G. Holloway, J. Chao, R. C. Boucher, and S. E. Gabriel. 2002. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 277:8338-8345. [DOI] [PubMed] [Google Scholar]
- 10.Drosten, C., S. Günther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L. Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguière, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Müller, V. Rickerts, M. Stürmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz, and H. W. Doerr. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348:1967-1976. [DOI] [PubMed] [Google Scholar]
- 11.Glowacka, I., S. Bertram, P. Herzog, S. Pfefferle, I. Steffen, M. O. Muench, G. Simmons, H. Hofmann, T. Kuri, F. Weber, J. Eichler, C. Drosten, and S. Pölmann. 2010. Differential downregulation of ACE2 by the spike proteins of SARS-coronavirus and human coronavirus NL63. J. Virol. 84:1198-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamming, I., W. Timens, M. L. C. Bulthuis, A. T. Lely, G. J. Navis, and H. van Goor. 2004. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203:631-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang, I., B. J. Bosch, F. Li, W. Li, K. H. Lee, S. Ghiran, N. Vasilieva, T. S. Dermody, S. C. Harrison, P. R. Dormitzer, M. Farzan, P. J. M. Rottier, and H. Choe. 2006. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 281:3198-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue, Y., N. Tanaka, Y. Tanaka, S. Inoue, K. Morita, Z. Min, T. Hattori, and K. Sugamura. 2007. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 81:8722-8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kam, Y., Y. Okumura, H. Kido, L. F. P. Ng, R. Bruzzone, and R. Altmeyer. 2009. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PloS One 4:e7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kido, H., Y. Okumura, E. Takahashi, H. Pan, S. Wang, J. Chida, T. Q. Le, and M. Yano. 2008. Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J. Mol. Gen. Med. 3:167-175. [PMC free article] [PubMed] [Google Scholar]
- 17.Kuba, K., Y. Imai, S. Rao, H. Gao, F. Guo, B. Guan, Y. Huan, P. Yang, Y. Zhang, W. Deng, L. Bao, B. Zhang, G. Liu, Z. Wang, M. Chappell, Y. Liu, D. Zheng, A. Leibbrandt, T. Wada, A. Slutsky, D. Liu, C. Qin, C. Jiang, and J. Penninger. 2005. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11:875-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, W., M. Moore, N. Vasilieva, J. Sui, S. K. Wong, M. Berne, M. Somasundaran, J. Sullivan, K. Luzuriaga, T. Greenough, H. Choe, and M. Farzan. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liem, N. T., N. Nakajima, L. P. Phat, Y. Sato, H. N. Thach, P. V. Hung, L. T. San, H. Katano, T. Kumasaka, T. Oka, S. Kawachi, T. Matsushita, T. Sata, K. Kudo, and K. Suzuki. 2008. H5N1-infected cells in lung with diffuse alveolar damage in exudative phase from a fatal case in Vietnam. Jpn. J. Infect. Dis. 61:157-160. [PubMed] [Google Scholar]
- 20.Matsuyama, S., and F. Taguchi. 2009. Two-step conformational changes in a coronavirus envelope glycoprotein mediated by receptor binding and proteolysis. J. Virol. 83:11133-11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuyama, S., M. Ujike, S. Morikawa, M. Tashiro, and F. Taguchi. 2005. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. U. S. A. 102:12543-12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagata, N., N. Iwata, H. Hasegawa, S. Fukushi, A. Harashima, Y. Sato, M. Saijo, F. Taguchi, S. Morikawa, and T. Sata. 2008. Mouse-passaged severe acute respiratory syndrome-associated coronavirus leads to lethal pulmonary edema and diffuse alveolar damage in adult but not young mice. Am. J. Pathol. 172:1625-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagata, N., N. Iwata, H. Hasegawa, Y. Sato, S. Morikawa, M. Saijo, S. Itamura, T. Saito, Y. Ami, T. Odagiri, M. Tashiro, and T. Sata. 2007. Pathology and virus dispersion in cynomolgus monkeys experimentally infected with severe acute respiratory syndrome coronavirus via different inoculation routes. Int. J. Exp. Pathol. 88:403-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nie, Y., P. Wang, X. Shi, G. Wang, J. Chen, A. Zheng, W. Wang, Z. Wang, X. Qu, M. Luo, L. Tan, X. Song, X. Yin, J. Chen, M. Ding, and H. Deng. 2004. Highly infectious SARS-CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem. Biophys. Res. Commun. 321:994-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohnishi, K., M. Sakaguchi, T. Kaji, K. Akagawa, T. Taniyama, M. Kasai, Y. Tsunetsugu-Yokota, M. Oshima, K. Yamamoto, N. Takasuka, S. Hashimoto, M. Ato, H. Fujii, Y. Takahashi, S. Morikawa, K. Ishii, T. Sata, H. Takagi, S. Itamura, T. Odagiri, T. Miyamura, I. Kurane, M. Tashiro, T. Kurata, H. Yoshikura, and T. Takemori. 2005. Immunological detection of severe acute respiratory syndrome coronavirus by monoclonal antibodies. Jpn. J. Infect. Dis. 58:88-94. [PubMed] [Google Scholar]
- 26.Paoloni-Giacobino, A., H. Chen, M. C. Peitsch, C. Rossier, and S. E. Antonarakis. 1997. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics 44:309-320. [DOI] [PubMed] [Google Scholar]
- 27.Qiu, Z., S. T. Hingley, G. Simmons, C. Yu, J. Das Sarma, P. Bates, and S. R. Weiss. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 80:5768-5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schornberg, K., S. Matsuyama, K. Kabsch, S. Delos, A. Bouton, and J. White. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 80:4174-4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirogane, Y., M. Takeda, M. Iwasaki, N. Ishiguro, H. Takeuchi, Y. Nakatsu, M. Tahara, H. Kikuta, and Y. Yanagi. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J. Virol. 82:8942-8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmons, G., D. N. Gosalia, A. J. Rennekamp, J. D. Reeves, S. L. Diamond, and P. Bates. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 102:11876-11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmons, G., J. D. Reeves, A. J. Rennekamp, S. M. Amberg, A. J. Piefer, and P. Bates. 2004. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U. S. A. 101:4240-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.To, K. F., and A. W. I. Lo. 2004. Exploring the pathogenesis of severe acute respiratory syndrome (SARS): the tissue distribution of the coronavirus (SARS-CoV) and its putative receptor, angiotensin-converting enzyme 2 (ACE2). J. Pathol. 203:740-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tripet, B., D. J. Kao, S. A. Jeffers, K. V. Holmes, and R. S. Hodges. 2006. Template-based coiled-coil antigens elicit neutralizing antibodies to the SARS-coronavirus. J. Struct. Biol. 155:176-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ujike, M., H. Nishikawa, A. Otaka, N. Yamamoto, N. Yamamoto, M. Matsuoka, E. Kodama, N. Fujii, and F. Taguchi. 2008. Heptad repeat-derived peptides block protease-mediated direct entry from the cell surface of severe acute respiratory syndrome coronavirus but not entry via the endosomal pathway. J. Virol. 82:588-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe, R., S. Matsuyama, K. Shirato, M. Maejima, S. Fukushi, S. Morikawa, and F. Taguchi. 2008. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J. Virol. 82:11985-11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White, J., S. Delos, M. Brecher, and K. Schornberg. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang, Z., Y. Huang, L. Ganesh, K. Leung, W. Kong, O. Schwartz, K. Subbarao, and G. J. Nabel. 2004. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78:5642-5650. [DOI] [PMC free article] [PubMed] [Google Scholar]