

Abstract
The nonstructural protein NS1 of influenza A virus counteracts the interferon (IFN) system and thereby promotes viral replication. NS1 has acquired different mechanisms to limit induction of IFN. It prevents double-stranded RNA (dsRNA) and RIG-I-mediated activation of interferon regulatory factor 3 (IRF3), and it blocks posttranscriptional processing of cellular mRNAs by binding to the cleavage and polyadenylation specificity factor (CPSF). Using a mouse-adapted A/PR/8/34 virus and reverse genetics to introduce specific mutations in NS1 which eliminate one or both functions, we determined the relative contributions of these two activities of NS1 to viral virulence in mice. We found that a functional RNA-binding motif was required for IFN suppression and virulence. Restoration of CPSF binding in the NS1 protein of wild-type A/PR/8/34 virus, which cannot bind CPSF due to mutations in the central binding motif at positions 103 and 106, resulted in enhanced virulence. Surprisingly, if CPSF binding was abolished by substituting glycine for arginine at position 184 in the classical NS1-CPSF binding motif, the mutant virus replicated much more slowly in mice, although the mutated NS1 protein continued to repress the IFN response very efficiently. Our results show that a functional RNA-binding motif is decisive for NS1 of A/PR/8/34 virus to suppress IFN induction. They further demonstrate that in addition to its contribution to CPSF binding, glycine 184 strongly influences viral virulence by an unknown mechanism which does not involve the IFN system.
The virulence of influenza A virus (FLUAV) is determined by several factors, including the viral polymerase complex, the viral glycoproteins hemagglutinin (HA) and neuraminidase (NA), and the two nonstructural proteins PB1-F2 and NS1 (40, 42). The nonstructural protein NS1 suppresses induction and action of the interferon (IFN) system (14, 22, 29). Accordingly, mutations in NS1 create viruses with strong IFN-inducing capacity that are attenuated in IFN-competent cells and animals (15, 28, 38).
The anti-IFN activity of NS1 depends on multiple functions that affect IFN induction as well as the action of IFNs and IFN-induced genes (20, 26). The NS1 molecule is divided into an N-terminal RNA-binding domain (amino acids 1 to 73) and a C-terminal effector domain (amino acids 74 to 230) (Fig. 1A) (2). It forms homodimers that directly interact with double-stranded RNA (dsRNA), primarily via an invariant arginine at position 38 (R-38) (6, 47, 60). NS1 binding of dsRNA interferes with RIG-I-mediated induction of IFN (35, 43). Additionally, the association of NS1 with TRIM25 prevents RIG-I activation (13). Even though NS1 binding to TRIM25 is independent of its binding to dsRNA, mutations that disrupt dsRNA binding also disrupt TRIM25 binding (13). Two motifs in the effector domain of NS1 are involved in binding of the 30-kDa subunit of the cleavage and polyadenylation specificity factor (CPSF), which is involved in the 3′ processing of cellular mRNAs. Association of NS1 with CPSF thereby provokes a general shutoff of cellular gene expression, including expression of IFNs, by blocking the processing of cellular mRNAs (7, 41). Structural analysis of NS1-CPSF complexes revealed a distinct CPSF-binding pocket in NS1 with a central glycine at position 184 (7). Additional contact points at the surface of the NS1 molecule involving the hydrophobic residues phenylalanine (F-103) and methionine (M-106) stabilize this interaction (7). The residues lining the CPSF-binding pocket in NS1 are highly conserved. However, NS1 sequences of some virus strains, including A/PR/8/34, show variations in the hydrophobic residues at positions 103 and 106, resulting in attenuation or loss of CPSF binding (26, 30). However, the significance of these variations for viral virulence is not understood sufficiently well.
FIG. 1.

A/PR/8/34 viruses with mutations in the NS1 gene. (A) Schematic representation of functional domains of NS1 and the inactivating mutations. The functional amino acids are indicated by bold letters. The RNA-binding domain (positions 1 to 73) was inactivated by mutation R38A. CPSF binding in the effector domain (positions 74 to 230) was abolished by mutations F103S, M106I, and G184R. NS1(R/SI/G) represents wild-type NS1 of A/PR/8/34. (B) Subcellular localization of NP and NS1 in virus-infected cells. A549 cells were infected with the recombinant viruses at an MOI of 0.25. At 8 h p.i. the cells were fixed and stained with a polyclonal rabbit antiserum directed against NP and a monoclonal mouse antibody (1A7) directed against NS1. One representative result out of three independent experiments is shown.
NS1 also affects the IFN-induced antiviral state (24, 26, 52). The effector domain of NS1 suppresses mRNA export into the cytoplasm (12, 44) by interacting with poly(A)-binding protein II (5) and with components of the cellular mRNA export machinery (51), thereby blocking the expression of IFNs and IFN-stimulated genes. In addition, NS1 prevents the activation of RNase L and of the dsRNA-activated protein kinase (PKR) (1, 31, 36, 37). By interacting with the cellular translation initiation factor eIF4GI via its central domain (amino acids 74 to 113), NS1 is able to stimulate translation of viral transcripts (3, 10). NS1 also directly influences viral replication independently of its effects on the IFN system (11, 37), and coprecipitation studies suggested an association of NS1 with the viral polymerase complex (30, 33). Furthermore, NS1 affects viral replication by activation of the cellular phosphatidylinositol 3-kinase/Akt pathway, thereby suppressing apoptosis in virus-infected cells (9, 19, 53).
It is unclear how the multiple functions of NS1 contribute to pathogenesis of FLUAV in the infected host. We hypothesize that a virus possessing both anti-IFN mechanisms in NS1, i.e., inhibition of RIG-I activation and of CPSF functions, would increase its capacity to suppress IFN-induced host defense and therefore gain enhanced virulence. Here, we addressed this assumption by generating recombinant PR8 viruses carrying NS1 with amino acid exchanges in the RNA-binding motif and in the two CPSF interaction sites. Analysis of these viruses revealed a dominant contribution of the RNA-binding site for enhanced virulence, which correlated with efficient control of the IFN system. Most surprisingly, however, a mutation of glycine 184 to arginine in the CPSF-binding pocket of NS1 strongly attenuated the recombinant viruses without affecting the IFN-suppressive activity. This severe attenuation was not seen in viruses expressing an NS1 protein defective in CPSF binding due to exchanges at positions 103 and 106, indicating a considerable contribution of glycine 184 to virulence of A/PR/8/34 virus irrespective of its role in CPSF binding.
(This work was conducted by Sabine Steidle in partial fulfillment of the requirements for an M.D. degree from the Medical Faculty of the University of Freiburg, Freiburg, Germany.)
MATERIALS AND METHODS
Generation of recombinant viruses.
A mixed genetic background was used to generate A/PR/8/34 viruses. cDNA plasmids encoding segments 1, 2, 3, 5, and 7 from a low-virulence A/PR/8/34 (lvPR8) and segments 4, 6, and 8 from a high-virulence PR8 variant (hvPR8) were combined as described previously (17). Mutations in NS1 were introduced into the cDNA of segment 8 of hvPR8 by a PCR-based strategy without affecting the open reading frame of the NEP/NS2 gene. The resulting cDNAs were cloned into the ambisense expression vector pDZ, which contains a human RNA polymerase I promoter and an RNA polymerase II-driven chicken β-actin promoter (45). For the rescue of recombinant viruses, a mixture of eight pDZ plasmids (0.5 μg each) was transfected into cocultures of 293T and Madin-Darby canine kidney (MDCK) cells. Recombinant viruses in the supernatant of transfected cells were plaque purified on MDCK cells in the presence of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma). Virus stocks were prepared in 8-day-old embryonated chicken eggs and stored at −80°. The presence of the mutations was confirmed by reverse transcription-PCR (RT-PCR) and sequencing of segments 8 of the individual virus stocks.
Virus growth was determined on MDCK and human A549 cells by infecting the cells with the recombinant viruses at multiplicities of infection (MOIs) of 0.001 and 0.01, respectively, in the presence of 0.5 μg/ml of trypsin in the medium. Virus titers in the supernatants were determined by infecting MDCK cells with 10-fold serial dilutions. Fluorescent cell foci detected after staining with a rabbit antiserum specific for NP were counted. Virus titers are expressed as focus-forming units (FFU). All infection experiments were confirmed with a second, independently generated recombinant virus variant.
Determination of IFN induction in cell culture.
Mouse embryo fibroblasts (MEF) prepared from IFN-βdelβ-luc/delβ-luc reporter mice (32) were infected with the PR8 viruses at an MOI of 0.5 for 20 h. Firefly luciferase activity was determined in the cell lysates using the luciferase assay system (Promega).
To determine type I IFN production in infected cells, human A549 cells were infected at an MOI of 0.5 for 20 h. Supernatants were dialyzed against 0.1 M glycine (pH 2.0) for 10 h and subsequently against phosphate-buffered saline (PBS) (pH 7.5) for 24 h. 293T cells transfected with a reporter plasmid encoding firefly luciferase under the control of the Mx1 promoter (25) were incubated with the acid-treated supernatants for 18 h, and luciferase activity in the cell lysates was then determined.
Coimmunoprecipitation and Western blot analysis. (i) Binding of NS1 to poly(I·C).
293T cells were infected with the viruses at an MOI of 0.5 for 16 h before the cells were lysed in a mixture of 50 mM Tris (pH 8.0), 250 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.5% NP-40, 1 mM dithiothreitol (DTT), and protease inhibitors. The cleared lysates were incubated with poly(I·C) (Sigma) coupled to CNBr-activated Sepharose beads (Roche) for 2 h at 4°C. As a control, the lysates were incubated with mock-treated Sepharose beads. After five washes with binding buffer, precipitated NS1 proteins were eluted and detected by Western blotting.
(ii) NS1/TRIM25 coimmunoprecipitation.
293T cells were transfected with an expression construct encoding V5-tagged TRIM25 for 30 h as described previously (13) before the cells were infected with the various viruses at an MOI of 2.0 for 18 h. The cells were then lysed in a mixture of 50 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, and protease inhibitors, and the cleared lysates were incubated with an antibody directed against the V5 tag (Abcam, Cambridge, MA) for 4 h at 4°C and then with protein A-Sepharose (Roche) for an additional 2 h. After three washes with binding buffer, the precipitated proteins were eluted and analyzed by Western blotting using the V5-specific antibody and an NS1-specific antiserum.
(iii) NS1/CPSF coprecipitation.
293T cells were transfected with an expression construct encoding the full-length human CPSF C-terminally fused to a Flag tag or encoding the F2F3 domain of CPSF N-terminally fused to glutathione S-transferase (GST) under the control of the chicken β-actin promoter (CAGGS) (26). At 24 h posttransfection, the cells were infected with the various viruses at an MOI of 0.5 for 20 h. The cells were then lysed in a mixture of 50 mM Tris (pH 8.0), 200 mM NaCl, 0.2 mM EDTA, 10% glycerol, 0.5% NP-40, 0.5% Triton X-100, 1 mM DTT, protease inhibitors, and 100 U/ml of Benzonase (Novagen, Merck Chemicals, Nottingham, United Kingdom) as described previously (26). The cleared lysates were incubated with 1 μg of a monoclonal anti-Flag antibody (Sigma) and 25 μl of protein A-Sepharose beads or with glutathione agarose beads (Sigma) for 2 h at 4°C. After three washes with binding buffer, precipitated NS1 proteins were eluted and detected by Western blotting using a monoclonal antibody directed against Flag or GST (Sigma-Aldrich, St. Louis, MO) and an NS1-specific antiserum.
For Western blot analysis, the precipitated proteins were eluted from the beads by boiling in SDS sample buffer. The eluted proteins were separated by 12% denaturing gel electrophoresis and detected by Western blotting and by using polyclonal antisera directed against viral NP and NS1 (54) and a monoclonal mouse antibody for β-actin (Sigma). Horseradish peroxidase-conjugated secondary antibodies, the SuperSignal bioluminescence substrate (Thermo Scientific, Rockford, IL), and ChemiDoc equipment (Bio-Rad, Munich, Germany) were used to detect the primary antibodies.
Immunofluorescence analysis.
A549 cells were seeded onto glass coverslips and infected with the various viruses at an MOI of 0.25 for 8 h. The cells were then fixed with 3% paraformaldehyde, permeabilized with 0.5% Triton X-100, and stained using a polyclonal rabbit antiserum directed against NP and a mouse monoclonal NS1-specific antibody (1A7). Fluorophore-conjugated donkey anti-rabbit (Alexa-555; Invitrogen) and anti-mouse (Alexa-488) antibodies were used as secondary antibodies. Pictures were recorded using epifluorescence with a Zeiss Axioplan-2 microscope.
Mice.
Infection studies were performed with either wild-type C57BL/6 or congenic B6.A2G-Mx1 mice carrying intact Mx1 alleles (55), B6.DKO-Mx1 mice lacking functional type I and III IFN receptors (38), PKR0/0 knockout mice with the mixed 129/SvxC57BL/6J background (63), or IFN-βdelβ-luc/delβ-luc knock-in reporter mice expressing firefly luciferase under the control of the IFN-β promoter (32). Six- to 8-week-old animals were used for the experiments, which were performed in accordance with the guidelines of the local Animal Care Committee. For infection, animals were anesthetized by intraperitoneal injection of a mixture of ketamine (100 μg per gram of body weight) and xylazine (5 μg per gram of body weight) and infected intranasally with the indicated doses of virus in 50 μl of PBS containing 0.3% bovine serum albumin. Animals were killed if severe symptoms developed or if the weight loss approached 30%. Fifty percent lethal dose (LD50) values were calculated as described previously (46).
Determination of virus titers and luciferase activity in lungs.
Lung homogenates were prepared in 0.8 ml PBS using a FastPrep-24 (Molecular Probes) homogenizer, and tissue debris was removed by low-speed centrifugation. Virus titers in supernatants were determined on MDCK cells by serial 10-fold dilution and staining with specific antibodies as described above. For determination of the luciferase activity, the supernatants were diluted in an equal volume of 2× passive lysis buffer and luciferase activity was determined using the luciferase assay system (Promega).
Minireplicon assay.
293T cells were transfected using Nanofectin (PAA Laboratories, Pasching, Austria) with pCAGGS expression plasmids for PB2, PB1, and PA (125 ng each) and NP (300 ng) of lvPR8 and with pPOLI-FFLuc-RT (50 ng) encoding firefly luciferase in the negative-sense orientation under the control of the noncoding regions of FLUAV segment 8 (48). In this construct, expression of the viral minigenome is under the control of the human RNA polymerase I promoter and terminator, respectively. The cells were cotransfected with pCAGGS expression constructs for NS1 with the splice acceptor mutation (57). In addition, an expression plasmid (40 ng) encoding Renilla luciferase under the control of the simian virus 40 (SV40) promoter (pRL-SV40) was cotransfected. At 24 h after transfection, the cells were harvested and luciferase activities were determined using the dual luciferase assay system (Promega). Firefly luciferase activity was normalized to the Renilla luciferase activity.
RESULTS
dsRNA and CPSF binding of PR8-NS1.
To evaluate the contribution of NS1 to virulence, we used a mouse-adapted A/PR/8/34 virus and Mx1-positive mice. This animal model system benefits from the exclusive induction of Mx1 by IFNs (38) and from the strong antiviral activity of Mx1 against influenza A viruses (58), which mimics the antiviral response of influenza virus infection in natural, Mx-competent hosts, including humans. In this system, even slight changes in virus-induced IFN levels are expected to result in altered expression of Mx1, thereby strongly affecting virus replication and pathogenesis. For the current study we used a recombinant PR8 virus of intermediate virulence in Mx1-positive mice (17).
To generate this parental virus by reverse genetics, we used plasmids for segments 4 (HA), 6 (NA), and 8 (NS1/NEP) of a highly virulent variant of PR8 and for segments 1 (PB2), 2 (PB1), 3 (PA), 5 (NP), and 7 (M1/M2) of regular PR8 (17). We then introduced various mutations into the NS1 protein of this virus. First, we inactivated the RNA-binding motif in NS1 by changing arginine at position 38 to alanine. This amino acid exchange has previously been shown to reduce dsRNA binding (8, 60) and interaction with TRIM25, a cofactor of RIG-I-mediated signaling (13). Second, to restore CPSF binding of the NS1 protein of PR8, the central interaction motif was modified by changing serine at position 103 to phenylalanine and isoleucine at position 106 to methionine. According to our previous studies, these changes should restore functional association between NS1 and CPSF (26). Third, we examined the role of the C-terminal CPSF-binding pocket in NS1 around glycine 184 by converting this residue to arginine. This mutation should result in loss of CPSF binding (7). None of the mutations introduced into segment 8 resulted in changes of the NEP/NS2 amino acid sequence. Figure 1A summarizes the different NS1 sequences of the eight recombinant viruses used in this study, which carry all possible combinations in the three functional NS1 motifs. NS1(R/SI/G) corresponds to wild-type PR8-NS1. All recombinant viruses grew to comparable titers in 8-day-old embryonated eggs.
The subcellular localization of the NS1 mutants was first analyzed in virus-infected cells. For this, human alveolar epithelial A549 cells were infected with the different viruses for 8 h and then analyzed for NS1 localization. At this time point, all NS1 variants showed predominantly nuclear localization (Fig. 1B). In three independent experiments, no significant differences in the subcellular distributions of the various NS1 mutants were detected.
Infection of Madin-Darby canine kidney (MDCK) and human A549 cells resulted in replication to high titers with comparable kinetics (Fig. 2A and B). At later time points the titers of the viruses in the cell culture supernatants differed by at most 10-fold, but these variations in virus growth did not correlate with one of the specific mutations introduced in the NS1 gene.
FIG. 2.
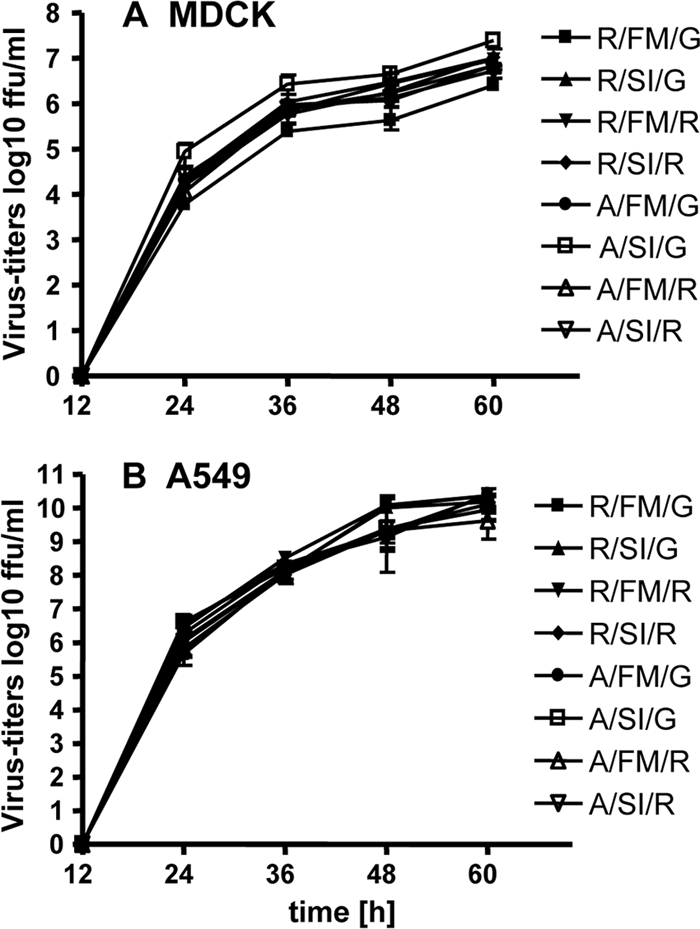
Growth of the recombinant viruses in cell culture. MDCK (A) and A549 (B) cells in six well plates were infected in triplicates with the viruses at MOIs of 0.001 and 0.01, respectively. At the indicated time points, supernatants were harvested and stored at −70°C. Virus titers were determined on MDCK cells. Error bars indicate standard deviations.
IFN-inducing capacity of viruses with altered NS1 genes.
We and others recently described the importance of the RNA/TRIM25-binding region and the CPSF interaction motif of NS1 for suppression of type I IFN (13, 26, 29). To determine which NS1 mutants might have lost this activity, we infected MEF that express firefly luciferase under the control of the IFN-β promoter (32). As a control, we used a PR8 mutant virus that lacks NS1 altogether (delNS1) (28). This analysis revealed that all PR8 viruses carrying NS1 proteins with a functional RNA-binding motif (R-38) suppressed the activity of the IFN-β promoter rather efficiently. Infection with these viruses resulted in 10- to 30-fold activation of the reporter gene compared to the results for the mock-treated control (Fig. 3A). Under these conditions, the delNS1 virus induced the reporter gene about 450-fold (Fig. 3A). Inactivation of the CPSF-binding motif in NS1 did not significantly alter the IFN-suppressing properties of mutants carrying R-38. In contrast, viruses encoding NS1 proteins with a defective RNA/TRIM25-binding domain (A-38 mutants) showed strong reporter gene activation, with induction values ranging from 200- to 400-fold (Fig. 3A). Of note is that the A-38 mutants failed to suppress reporter gene expression irrespective of whether a functional CPSF interaction motif was present or not. Although there were some variations, Western blot analysis of cell lysates confirmed comparable degrees of infection of the indicator cells by the different viruses and comparable production of the various NS1 proteins (Fig. 3A).
FIG. 3.

IFN-inducing capacity of recombinant A/PR/8/34 with mutations in NS1. (A) Activation of the IFN-β promoter in cell culture. MEF expressing firefly luciferase (FF-luc) under the control of the IFN-β promoter were infected at an MOI of 0.5 for 20 h. Luciferase activity of the cell lysates was determined. (B) Induction of type I IFN. A549 cells were infected at an MOI of 0.5 for 20 h. The cell culture supernatants were dialyzed against low-pH buffer to inactivate the viruses as described previously (27). 293T cells expressing firefly luciferase under the control of the Mx1 promoter (25) were then incubated with the treated supernatants for 18 h. Luciferase activity in the lysates of the indicator cells was determined. Data from three independent experiments are shown, and error bars indicate standard deviations. Viral proteins and β-actin in the whole-cell lysates of infected MEF or A549 cells were analyzed by Western blotting using specific antibodies directed against NS1, NP, and β-actin.
Using human A549 cells, we evaluated the capacities of the various viruses to induce secretion of type I IFN. For this, the culture supernatants of the infected cells were first treated with low-pH buffer to inactivate virus and then tested for acid-stable IFN activity using a bioassay which is based on 293T cells carrying the firefly luciferase gene under the control of the mouse Mx1 gene promoter (25). All virus variants encoding NS1 proteins with a defective RNA/TRIM25-binding domain (A-38 mutants) induced large amounts of IFN, comparable to the case for the delNS1 virus (Fig. 3B). In contrast, all viruses encoding NS1 proteins with a functional RNA-binding domain (R-38) efficiently suppressed IFN synthesis in infected cells (Fig. 3B).
Properties of NS1 proteins with mutations in RNA- and CPSF-binding motifs.
The N-terminal RNA/TRIM25-binding domain of NS1 was first tested for binding to poly(I·C) and coprecipitation with TRIM25. Lysates of infected 293T cells were incubated with poly (I·C) covalently attached to Sepharose beads as described previously (4). All NS1 proteins with a functional RNA-binding motif (R-38) were precipitated with poly(I·C)-Sepharose, whereas all NS1 mutants with a defective RNA-binding motif (A-38) did not (Fig. 4A).
FIG. 4.

NS1 interaction with cellular factors. (A) NS1 binding to dsRNA. 293T cells were infected with the recombinant viruses at an MOI of 0.5. At 16 h p.i. cell lysates were incubated with poly(I·C) immobilized on Sepharose beads. Poly(I·C)-bound proteins were isolated and analyzed by Western blotting (IB) using antibodies directed against NS1. Whole-cell extracts (WCL) were analyzed for NS1 and NP expression using antibodies directed against NP and NS1. (B) NS1 association with TRIM25. 293T cells were transfected with an expression construct encoding V5-tagged TRIM25 and infected 30 h later with the recombinant viruses at an MOI of 2. TRIM25 was immunoprecipitated at 18 h p.i. with an antibody directed against the V5 tag, and associated NS1 was analyzed by Western blotting as described above. (C) NS1 interaction with CPSF. Cells were transfected with an expression construct encoding GST fusion protein with the F2F3 domains of CPSF. At 24 h posttransfection, the cells were infected with the recombinant viruses at an MOI of 0.5. At 20 h p.i. cell lysates were subjected to CPSF precipitation using glutathione-agarose. The precipitated proteins (GST-PD) and proteins in the whole-cell lysates (WCL) were analyzed by Western blotting using antibodies directed against NS1, NP, and the GST-tag. All results show representative data from three independent experiments.
NS1 inhibits RIG-I activation by binding to TRIM25, and this interaction is abolished by the R38A mutation (13). To test our NS1 mutants for TRIM25 binding, 293T cells were transfected with an expression construct encoding V5-tagged TRIM25 before the cells were infected with the recombinant viruses. Coimmunoprecipitation with a V5-specific antibody revealed that the R-38 but not the A-38 mutants bound to TRIM25 (Fig. 4B).
We further measured the interaction of the different NS1 proteins with CPSF. For this, we transfected an expression construct encoding the GST-tagged second and third zinc finger domains (F2F3) of CPSF, which were shown to comprise the NS1 interaction site (7). GST pulldown precipitated only NS1 proteins that possess functional middle (F-103/M-103) and C-terminal (G-184) interaction motifs, whereas NS1 proteins possessing either the S-103/I-106 or R-184 mutations did not interact with CPSF (Fig. 4C). In addition, we evaluated the binding of NS1 mutants R/FM/G and R/SI/G to full-length Flag-tagged CPSF using the same approach. Consistent with the results shown in Fig. 4C, only the R/FM/G variant and not the R/SI/G variant was coimmunoprecipitated (see Fig. S1 in the supplemental material), indicating the requirement of both the central and the classical, C-terminal interaction motifs for association of NS1 with CPSF in infected cells.
Effect of RNA and CPSF binding on IFN-induction in vivo.
To test IFN induction in vivo, we infected reporter mice that express firefly luciferase under the control of the IFN-β promoter with 5 × 104 FFU of the various viruses for 24 h. It was shown previously that luciferase expression in lungs of infected mice correlates with induction of IFN-β (32). About an 800-fold-increased luciferase activity was detected after infection with the delNS1 virus, whereas viruses carrying a functional RNA-binding motif were all inefficient IFN inducers. We noted only 20- to 100-fold-increased luciferase activity compared to that in mock-treated animals (Fig. 5A).
FIG. 5.
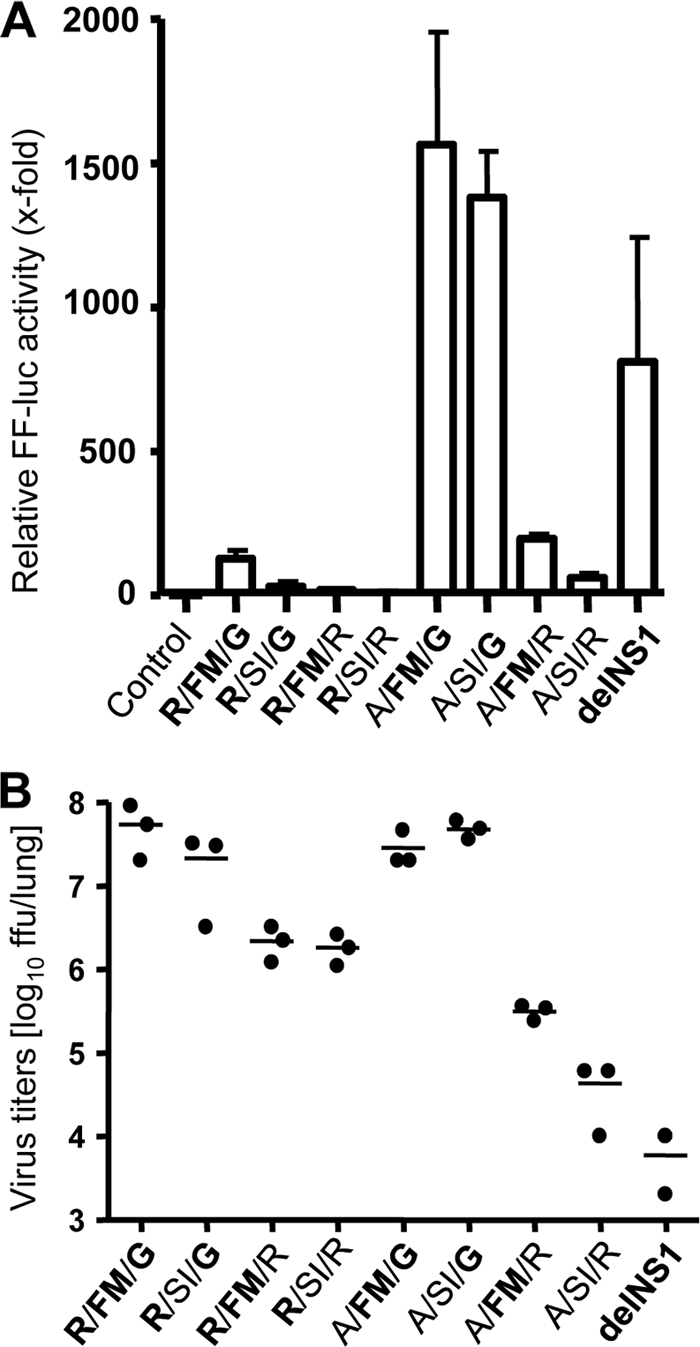
IFN-inducing capacity of recombinant A/PR/8/34 mutants in vivo. (A) Groups of reporter mice (n = 3) expressing firefly luciferase under the control of the IFN-β promoter were infected intranasally with 5 × 104 FFU of the mutant viruses or their counterparts lacking NS1 (delNS1). At 24 h p.i. luciferase activity (FF-luc) in lung homogenates was determined. Error bars indicate standard deviations. (B) In addition, virus titers in lung homogenates of the reporter mice were determined.
The A/FM/G and A/SI/G virus variants, which feature a defective RNA-binding motif, activated the IFN-β promoter efficiently, with induction values of about 1,400-fold (Fig. 5A), as expected from the cell culture experiments. Unexpectedly, however, infection of reporter mice with the A/FM/R and A/SI/R variants did not result in significant IFN-β promoter activation (Fig. 5A), although these viruses lack a functional RNA-binding motif. Titrations showed that during the 24-h infection period, the A/FM/G and A/SI/G viruses grew to at least 100-fold-higher titers in lungs of reporter mice than the A/FM/R and A/SI/R variants (Fig. 5B), which may explain the impaired IFN-inducing capacity of the latter viruses. Interestingly, the R/FM/R and R/SI/R variants, which possess a functional RNA-binding motif but have a mutated C-terminal CPSF-binding motif, also grew less well than R/FM/G and R/SI/G, in which the C-terminal CPSF-binding motif is intact (Fig. 5B), suggesting that G-184 of NS1 determinates viral fitness in mouse lungs.
Importance of RNA and CPSF binding for virulence in mice.
To determine if the observed strong in vivo attenuation of viruses carrying the G184R mutation in NS1 is dependent on the IFN-induced antiviral effector protein PKR, we infected PKR0/0 mice (63). The PKR gene, which is basically expressed in the absence of IFN, was previously shown to play a role in the antiviral host defense against FLUAV (1, 49, 61). Due to a shortage of these animals, we compared only the four viruses having a functional central CPSF-binding site. Mice were infected intranasally with 1,000 FFU of the various viruses, and lung titers were determined at 24 h postinfection (p.i.). Viruses R/FM/G and A/FM/G, carrying a functional C-terminal CPSF interaction motif, grew to about 1 × 107 FFU in lungs of PKR0/0 mice (Fig. 6A). However, viruses R/FM/R and A/FM/R, carrying the G184R mutation in NS1, remained inefficient and grew to 10- to 100-fold-lower titers in the lungs of PKR-deficient mice (Fig. 6A), confirming a reduction of viral fitness of the NS1(G184R)-encoding viruses that is not due to the action of PKR.
FIG. 6.
Growth of recombinant A/PR/8/34 mutants in vivo. Lung titers in PKR knockout mice (A) and IFNAR/IL28R double-knockout mice (B) are shown. Mice were infected intranasally with 1,000 FFU of the recombinant viruses. Virus titers in lung homogenates were determined at either 24 or 96 h postinfection as indicated.
We next determined virus growth kinetics in Mx-IFNAR10/0 IL28Rα0/0 mice, which lack functional receptors for both type I and type III IFN (38). At 24 h postinfection, virus R/SI/G, which binds dsRNA but not CPSF, and A/FM/G and A/SI/G, which do not bind dsRNA, grew slightly less well than R/FM/G and reached titers of about 5 × 106 to 1 × 107 FFU (Fig. 6B and data not shown). The R/FM/G virus reached lung titers of about 5 × 107 FFU as early as 24 h postinfection in these mice (Fig. 6B). However, all viruses in which the C-terminal CPSF-binding motif was destroyed by the G184R mutation grew substantially slower and to only low titers (Fig. 6B and data not shown). However, virus R/FM/R titers, which were below 106 FFU at 24 h postinfection, reached values of 107 to 108 FFU by 96 h (Fig. 6B). A similar phenomenon was observed with the virus pair A/FM/G and A/FM/R. Both mutant viruses reached high lung titers at 96 h postinfection but with strikingly reduced kinetics (Fig. 6B).
To further evaluate the fitness of the various viruses, we next determined the lethal dose in Mx1-negative C57BL/6 and congenic B6.A2G-Mx1 mice carrying functional copies of the IFN-induced Mx1 gene. Mx1 is a strong antiviral host factor against influenza virus (17, 23, 56, 58). Mice were infected intranasally with 10-fold dilutions of the different virus variants and monitored for onset of disease and death. Table 1 specifies the mouse 50% lethal doses (LD50) of the different viruses. In C57BL/6 animals, the R/FM/G virus with functional RNA-binding as well as CPSF-binding motifs showed a low LD50 value (Table 1). Inactivation of the central CPSF-binding domain in R/SI/G did not change the LD50, whereas inactivation of the C-terminal CPSF-binding site in R/FM/R and R/SI/R or the RNA/TRIM25-binding site in A/FM/G and A/SI/G decreased virulence by about 10-fold. Inactivation of both the RNA-binding and C-terminal CPSF-binding domains in the A/FM/R and A/SI/R viruses decreased virulence by about 100-fold (Table 1).
TABLE 1.
Virulence of PR8 viruses with mutations in NS1 in mice
| Virus strain | RNA binding | CPSF binding | LD50 (FFU)a in: |
|
|---|---|---|---|---|
| C57BL/6 mice | B6-Mx1+/+ mice | |||
| R/FM/G | + | + | 5 | 5 × 102 |
| R/SI/G | + | − | 5 | 3 × 103 |
| R/FM/R | + | − | 50 | >106 |
| R/SI/R | + | − | 50 | >106 |
| A/FM/G | − | + | 20 | 5 × 105 |
| A/SI/G | − | − | 50 | 5 × 105 |
| A/FM/R | − | − | 500 | >106 |
| A/SI/R | − | − | 500 | >106 |
LD50 values were determined by infecting groups (n = 4) of mice with various doses of the indicated viruses. Animals were killed if they were severely ill or if weight loss approached 30%.
The attenuating effect of the various NS1 mutations was much more pronounced in Mx1-positive animals (Table 1). The parental virus R/FM/G showed an LD50 value of 500 FFU. Inactivation of the central CPSF-binding motif in R/SI/G reduced the virulence by about 6-fold, resulting in an LD50 of 3,000 FFU. Inactivation of the RNA binding in A/FM/G and A/SI/G resulted in an additional 100-fold increase of the LD50 value to 5 × 105 FFU, which correlated well with the strong IFN-inducing capacities of these two virus variants (Fig. 5A). Inactivation of the C-terminal CPSF-binding site in R/FM/R, R/SI/R, A/FM/R, and A/SI/R reduced viral virulence dramatically, resulting in LD50 values of more than 106 FFU in Mx1-positive mice (Table 1).
To evaluate whether the G184R mutation in NS1 variants might reduce viral polymerase activity, we analyzed the NS1 variants in a minireplicon system. 293T cells were transfected with expression constructs coding for the three viral polymerase subunits and NP and with a virus-like minigenome encoding firefly luciferase (48). In this system, reporter gene expression is a measure for the activity of the reconstituted polymerase complex. Coexpression of NS1 increased polymerase activity slightly compared to that with the mock control (see Fig. S2 in the supplemental material), presumably due to a general enhancing effect of NS1 on translation via the RNA-binding domain (50). The different NS1 variants did not differ significantly in this assay (see Fig. S2 in the supplemental material). Thus, G184 does not seem to influence viral polymerase activity.
DISCUSSION
Influenza A viruses have established two main strategies to limit IFN expression in infected cells. The viral NS1 protein sequesters dsRNA via its N-terminal RNA-binding domain around arginine at position 38 (8, 47) and inhibits TRIM25-mediated modification of RIG-I (13). Furthermore, the effector domain of NS1 binds to CPSF and thereby prevents processing of cellular pre-mRNAs, including transcripts of IFN genes (29). We hypothesized that a virus possessing both anti-IFN mechanisms in NS1 would increase its capacity to suppress IFN-induced host defense and therefore gain enhanced virulence in mice that possess a complete antiviral defense system, including the influenza virus-specific Mx1 gene (56). We could confirm our assumption by analyzing recombinant PR8 viruses carrying NS1 genes manipulated in the RNA/TRIM25-binding site as well as in the motifs responsible for CPSF binding. We demonstrated that a functional RNA-binding motif is decisive for viral virulence and suppression of the IFN system. If NS1 was further able to bind CPSF, viral replication and pathogenesis in the infected animals were increased. Unexpectedly, however, we observed strong attenuation of viruses carrying NS1 proteins with a glycine-to-arginine substitution at position 184 that cannot be explained by loss of CPSF binding. This finding suggests an additional, prominent function of the C-terminal NS1 region around glycine 184 for viral replication.
The importance of the N-terminal RNA/TRIM25-binding site of NS1 for suppression of IFN induction has been elaborated extensively (8, 13, 57, 62). Structural examination of the N-terminal part of NS1 in complex with dsRNA demonstrated the critical function of Arg-38 for binding of dsRNA (6, 60). The crystal structure of full-length NS1 revealed the formation of long NS1 filaments. Three NS1 filaments assemble into a tubular structure with basic residues critical for dsRNA binding, including Arg-38, directed to the center of this tube (2). It is thought that by sequestration of the dsRNA in this tunnel, NS1 prevents exposure of dsRNA to cytoplasmic RNA sensors such as RIG-I. In addition, it has recently been demonstrated that Arg-38 is critical for binding of NS1 to TRIM25, an ubiquitin ligase that is involved in RIG-I activation. Mutant NS1(R38A) did not interact with TRIM25 and was therefore not able to suppress IFN induction in virus-infected cells (13). NS1 additionally associates with RIG-I, possibly via dsRNA (35, 43), indicating the formation of a huge complex of at least dsRNA, RIG-I, and TRIM25 whose activity is controlled by NS1 in virus-infected cells. Our data now indicate that a functional RNA/TRIM25 interaction site in NS1 is most critical for suppression of IFN induction and virulence of a highly pathogenic mouse-adapted PR8 virus. These results are in agreement with previous publications that demonstrated a strong attenuation of influenza viruses with mutated NS1(R38A) in vivo due to loss of RNA binding and enhanced IFN induction (8). Arg-38 is part of the first nuclear localization sequence (NLS1) of NS1, encoded by amino acids 34 to 41 (16, 34). Accordingly, it was reported that NS1(R38A) of A/WSN/33 showed enhanced cytoplasmic accumulation in transfected and infected cells (34, 36). However, examination of PR8-NS1(R38A) in cells transfected with the respective cDNA expression constructs (57) or in infected cells (Fig. 1B) demonstrated nuclear accumulation of NS1(R38A) that was indistinguishable from that of wild-type PR8-NS1. This suggests that C-terminal basic amino acids between positions 203 and 230, most likely a stretch of arginine and lysines at positions 217 to 220, might serve as a second NLS, NLS2 (16), that is sufficient for nuclear accumulation of PR8-NS1(R38A). The RNA-binding domain of NS1 is also part of a dimerization motif which is required for efficient dsRNA binding (39, 60). However, intensive mutational analysis and structural examination of the RNA-binding domain revealed that the arginine at position 38 is not involved in intermolecular dimer formation of the N-terminal part of NS1 (6, 60), indicating that the amino acid exchange at position 38 specifically abolishes dsRNA and TRIM25 binding but does not affect other functions of the NS1 protein. In addition to the N terminus, the C-terminal effector domain contributes to NS1 oligomerization (2, 39). This C-terminal contribution to NS1 dimerization seems to have a critical role in suppression of the IFN system at least in vivo. Likewise, recombinant influenza viruses with C-terminal truncations in the NS1 proteins were attenuated in infected mice (27, 28, 61). However, viruses encoding the NS1 RNA-binding domain fused to a heterologous dimerization domain regained virulence in infected animals (61), indicating the importance of dimerization via the C-terminal effector domain for a functional RNA-binding site.
Another prominent function of the effector domain of NS1 is the inhibition of 3′-end processing of cellular pre-mRNAs by binding and inactivation of the 30-kDa subunit of CPSF (41). Two sites in NS1 are involved in this interaction: (i) a C-terminal binding pocket formed mostly by hydrophobic amino acids and involving Gly-184 and (ii) two amino acids outside this pocket, Phe-103 and Met-106, that stabilize the NS1-CPSF complex (7). Comparisons of 1,520 NS1 sequences of mammalian and avian influenza A virus isolates revealed an invariable glycine residue at position 184 within the CPSF-binding pocket. However, the hydrophobic residues at positions 103 and 106 show some, at least infrequent, variations. The NS1 sequences of A/HK/483/97, isolated from a fatal human infection during the H5N1 outbreak in Hong Kong in 1997, and some other avian viruses have a leucine instead of phenylalanine at position 103 and an isoleucine instead of methionine at position 106, which weakens the association between NS1 and CPSF (59). The NS1 of A/PR/8/34 has a very rare Ser-103 combined with Ile-106 that completely abolishes CPSF binding (26). Interestingly, the NS1 of A/HK/483/97 regained interaction with CPSF and suppression of the IFN system in the presence of its cognate viral polymerase complex, indicating that the polymerase complex is involved in NS1-CPSF interaction (30, 59). In contrast, PR8-NS1 containing Ser-103 combined with Ile-106 did not regain CPSF binding in the presence of the viral polymerase complex (Fig. 4C). Nevertheless, this PR8 virus, R/SI/G, showed efficient control of the IFN system in infected cell cultures and animals. However, reconstitution of CPSF binding by changing Ser-103 to phenylalanine and Ile-106 to methionine in PR8-NS1 further increased virulence, indicating that NS1-CPSF interaction has some virulence-enhancing function in addition to the RNA-binding capacity of NS1 in the context of the PR8 virus. The impact of CPSF binding on virulence in mice appears to be strain specific, as this function did not enhance virulence in the context of the new pandemic H1N1 virus (21).
The most intriguing result was the remarkable attenuation of PR8 viruses carrying NS1(G184R). Structural analysis of A/Udorn/72-NS1(G184R) showed that this amino acid substitution has only little or no effect on the overall structure of the NS1 effector domain (7). Inactivation of this C-terminal CPSF interaction site by similar means in A/Udorn/72 (H1N1) resulted in enhanced expression of IFN and attenuation of the recombinant virus (7, 41). However, PR8 viruses with NS1(G184R) did not show pronounced attenuation of virus growth in MDCK and A549 cells (Fig. 2A and B). In addition, in vivo attenuation of NS1(G184R)-expressing viruses was not due to a reduced ability to control the IFN system. Viruses with a functional RNA-binding motif and a defective C-terminal CPSF-binding site, R/FM/R and R/SI/R, suppressed IFN induction as efficiently as the virus that showed NS1-CPSF interaction, R/FM/G, in cell culture and in infected animals (Fig. 3 and 5A). In vivo, viruses with NS1(G184R) were attenuated even in mice lacking PKR expression and in double-knockout mice that lack functional type I and type III receptors and do not express IFN-stimulated genes upon virus infection (Fig. 6A and B). These results indicate that the observed attenuation of the viruses encoding NS1(G184R) is not due to an overactivation of the host IFN response or to an enhanced sensitivity of these viruses to the antiviral effect of PKR. Therefore, we assume that the region around Gly-184 in NS1 serves additional functions in the virus life cycle beside CPSF binding, resulting in this strong attenuation especially under the growth-restricting conditions in Mx1-positive mice (Table 1). A recent structural analysis of the C-terminal effector domain of an avian H7N7 NS1 protein revealed a contribution of the long helix formed by amino acids 171 to 188 in NS1 dimer formation (18). However, particularly Trp-187 but not Gly-184 contributes to this dimeric interface. In addition, efficient dsRNA binding and control of IFN induction by viruses carrying Arg-38 combined with Arg-184 argue against a defect of NS1(G184R) in dimer formation. Since the activity of the viral polymerase complex might be influenced by NS1 (30, 33), we tested the possibility that NS1(G184R) could affect viral polymerase activity. However, coexpression of NS1(G184R) in a minireplicon system for PR8 polymerase did not show any effect on polymerase activity compared to wild-type NS1 (see Fig. S2 in the supplemental material). In summary, our data reveal that viruses encoding NS1(G184R) are comparable to viruses carrying wild-type NS1 with respect to replication in cell culture and controlling IFN induction. However, all viruses carrying NS1(G184R) showed an unexpected strong attenuation in vivo.
We do not know what has driven the selection of PR8-NS1 in favor of replacing Phe-103 and Met-106 toward residues that disrupt CPSF binding. Clearly, our data demonstrate the importance of the RNA/TRIM25-binding site for the function of PR8-NS1 as a virulence factor. They further show that changes in the CPSF interaction sites have no effect on the control of the IFN system. Reconstitution of a functional central CPSF interaction site clearly enhanced virulence in our animal system. However, our study reveals the importance of the highly conserved C-terminal region of PR8-NS1 that is not necessarily involved in the control of the IFN system but seems to serve extra, yet-unknown functions in addition to CPSF binding. Presently, we are searching for interacting host factors that depend on Gly-184 for association with NS1. Identification of such NS1 partners might help to explain the strong attenuation of viruses carrying NS1(G184R).
Supplementary Material
Acknowledgments
We thank Richard Cadagan for excellent technical assistance, Michaela Gack for providing the TRIM25 expression plasmid, and Peter Palese for the pDZ rescue vector.
Parts of this work were supported by a grant from the Deutsche Forschungsgemeinschaft (Ko 1579/5-1) to G.K. and by funds from the Wissenschaftliche Gesellschaft in Freiburg to G.K., as well as by CRIP, an NIAID-funded Center for Research in Influenza Pathogenesis (contract number HHSN266200700010C) and by NIAID grants RO1AI46954 and U19AI83025 to A.G.-S.
Footnotes
Published ahead of print on 6 October 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Bergmann, M., A. Garcia-Sastre, E. Carnero, H. Pehamberger, K. Wolff, P. Palese, and T. Muster. 2000. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 74:6203-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornholdt, Z. A., and B. V. Prasad. 2008. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature 456:985-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgui, I., T. Aragon, J. Ortin, and A. Nieto. 2003. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J. Gen. Virol. 84:3263-3274. [DOI] [PubMed] [Google Scholar]
- 4.Cardenas, W. B., Y. M. Loo, M. Gale, Jr., A. L. Hartman, C. R. Kimberlin, L. Martinez-Sobrido, E. O. Saphire, and C. F. Basler. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 80:5168-5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, Z., Y. Li, and R. M. Krug. 1999. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 18:2273-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, A., S. M. Wong, and Y. A. Yuan. 2009. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 19:187-195. [DOI] [PubMed] [Google Scholar]
- 7.Das, K., L. C. Ma, R. Xiao, B. Radvansky, J. Aramini, L. Zhao, J. Marklund, R. L. Kuo, K. Y. Twu, E. Arnold, R. M. Krug, and G. T. Montelione. 2008. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 105:13093-13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donelan, N. R., C. F. Basler, and A. Garcia-Sastre. 2003. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 77:13257-13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrhardt, C., T. Wolff, S. Pleschka, O. Planz, W. Beermann, J. G. Bode, M. Schmolke, and S. Ludwig. 2007. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 81:3058-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enami, K., T. A. Sato, S. Nakada, and M. Enami. 1994. Influenza virus NS1 protein stimulates translation of the M1 protein. J. Virol. 68:1432-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falcon, A. M., R. M. Marion, T. Zurcher, P. Gomez, A. Portela, A. Nieto, and J. Ortin. 2004. Defective RNA replication and late gene expression in temperature-sensitive influenza viruses expressing deleted forms of the NS1 protein. J. Virol. 78:3880-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fortes, P., A. Beloso, and J. Ortin. 1994. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J. 13:704-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gack, M. U., R. A. Albrecht, T. Urano, K. S. Inn, I. C. Huang, E. Carnero, M. Farzan, S. Inoue, J. U. Jung, and A. Garcia-Sastre. 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Sastre, A. 2001. Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology 279:375-384. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Sastre, A., A. Egorov, D. Matassov, S. Brandt, D. E. Levy, J. E. Durbin, P. Palese, and T. Muster. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324-330. [DOI] [PubMed] [Google Scholar]
- 16.Greenspan, D., P. Palese, and M. Krystal. 1988. Two nuclear location signals in the influenza virus NS1 nonstructural protein. J. Virol. 62:3020-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimm, D., P. Staeheli, M. Hufbauer, I. Koerner, L. Martinez-Sobrido, A. Solorzano, A. Garcia-Sastre, O. Haller, and G. Kochs. 2007. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc. Natl. Acad. Sci. U. S. A. 104:6806-6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hale, B. G., W. S. Barclay, R. E. Randall, and R. J. Russell. 2008. Structure of an avian influenza A virus NS1 protein effector domain. Virology 378:1-5. [DOI] [PubMed] [Google Scholar]
- 19.Hale, B. G., D. Jackson, Y. H. Chen, R. A. Lamb, and R. E. Randall. 2006. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. U. S. A. 103:14194-14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hale, B. G., R. E. Randall, J. Ortin, and D. Jackson. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359-2376. [DOI] [PubMed] [Google Scholar]
- 21.Hale, B. G., J. Steel, R. A. Medina, B. Manicassamy, J. Ye, D. Hickman, R. Hai, M. Schmolke, A. C. Lowen, D. R. Perez, and A. Garcia-Sastre. 2010. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J. Virol. 84:6909-6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haller, O., G. Kochs, and F. Weber. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344:119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haller, O., S. Stertz, and G. Kochs. 2007. The Mx GTPase family of interferon-induced antiviral proteins. Microbes Infect. 9:1636-1643. [DOI] [PubMed] [Google Scholar]
- 24.Hayman, A., S. Comely, A. Lackenby, S. Murphy, J. McCauley, S. Goodbourn, and W. Barclay. 2006. Variation in the ability of human influenza A viruses to induce and inhibit the IFN-beta pathway. Virology 347:52-64. [DOI] [PubMed] [Google Scholar]
- 25.Jorns, C., D. Holzinger, R. Thimme, H. C. Spangenberg, M. Weidmann, J. Rasenack, H. E. Blum, O. Haller, and G. Kochs. 2006. Rapid and simple detection of IFN-neutralizing antibodies in chronic hepatitis C non-responsive to IFN-alpha. J. Med. Virol. 78:74-82. [DOI] [PubMed] [Google Scholar]
- 26.Kochs, G., A. Garcia-Sastre, and L. Martinez-Sobrido. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81:7011-7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochs, G., I. Koerner, L. Thiel, S. Kothlow, B. Kaspers, N. Ruggli, A. Summerfield, J. Pavlovic, J. Stech, and P. Staeheli. 2007. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J. Gen. Virol. 88:1403-1409. [DOI] [PubMed] [Google Scholar]
- 28.Kochs, G., L. Martinez-Sobrido, S. Lienenklaus, S. Weiss, A. Garcia-Sastre, and P. Staeheli. 2009. Strong interferon-inducing capacity of a highly virulent variant of influenza A virus strain PR8 with deletions in the NS1 gene. J. Gen. Virol. 90:2990-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krug, R. M., W. Yuan, D. L. Noah, and A. G. Latham. 2003. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology 309:181-189. [DOI] [PubMed] [Google Scholar]
- 30.Kuo, R. L., and R. M. Krug. 2009. Influenza A virus polymerase is an integral component of the CPSF30-NS1A protein complex in infected cells. J. Virol. 83:1611-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, S., J. Y. Min, R. M. Krug, and G. C. Sen. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13-21. [DOI] [PubMed] [Google Scholar]
- 32.Lienenklaus, S., M. Cornitescu, N. Zietara, M. Lyszkiewicz, N. Gekara, J. Jablonska, F. Edenhofer, K. Rajewsky, D. Bruder, M. Hafner, P. Staeheli, and S. Weiss. 2009. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol. 183:3229-3236. [DOI] [PubMed] [Google Scholar]
- 33.Marion, R. M., T. Zurcher, S. de la Luna, and J. Ortin. 1997. Influenza virus NS1 protein interacts with viral transcription-replication complexes in vivo. J. Gen. Virol. 78:2447-2451. [DOI] [PubMed] [Google Scholar]
- 34.Melen, K., L. Kinnunen, R. Fagerlund, N. Ikonen, K. Y. Twu, R. M. Krug, and I. Julkunen. 2007. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J. Virol. 81:5995-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mibayashi, M., L. Martinez-Sobrido, Y. M. Loo, W. B. Cardenas, M. Gale, Jr., and A. Garcia-Sastre. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81:514-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min, J. Y., and R. M. Krug. 2006. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U. S. A. 103:7100-7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min, J. Y., S. Li, G. C. Sen, and R. M. Krug. 2007. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 363:236-243. [DOI] [PubMed] [Google Scholar]
- 38.Mordstein, M., G. Kochs, L. Dumoutier, J. C. Renauld, S. R. Paludan, K. Klucher, and P. Staeheli. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4:e1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nemeroff, M. E., X. Y. Qian, and R. M. Krug. 1995. The influenza virus NS1 protein forms multimers in vitro and in vivo. Virology 212:422-428. [DOI] [PubMed] [Google Scholar]
- 40.Neumann, G., T. Noda, and Y. Kawaoka. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noah, D. L., K. Y. Twu, and R. M. Krug. 2003. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAs. Virology 307:386-395. [DOI] [PubMed] [Google Scholar]
- 42.Palese, P. 2004. Influenza: old and new threats. Nat. Med. 10:S82-87. [DOI] [PubMed] [Google Scholar]
- 43.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 44.Qiu, Y., and R. M. Krug. 1994. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J. Virol. 68:2425-2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinlivan, M., D. Zamarin, A. Garcia-Sastre, A. Cullinane, T. Chambers, and P. Palese. 2005. Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol. 79:8431-8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reed, L. J., and H. Muench. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. (Lond.) 27:493-497. [Google Scholar]
- 47.Rehwinkel, J., C. P. Tan, D. Goubau, O. Schulz, A. Pichlmair, K. Bier, N. Robb, F. Vreede, W. Barclay, E. Fodor, and E. S. C. Reis. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397-408. [DOI] [PubMed] [Google Scholar]
- 48.Rolling, T., I. Koerner, P. Zimmermann, K. Holz, O. Haller, P. Staeheli, and G. Kochs. 2009. Adaptive mutations resulting in enhanced polymerase activity contribute to high virulence of influenza A virus in mice. J. Virol. 83:6673-6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadler, A. J., and B. R. Williams. 2007. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 316:253-292. [DOI] [PubMed] [Google Scholar]
- 50.Salvatore, M., C. F. Basler, J. P. Parisien, C. M. Horvath, S. Bourmakina, H. Zheng, T. Muster, P. Palese, and A. Garcia-Sastre. 2002. Effects of influenza A virus NS1 protein on protein expression: the NS1 protein enhances translation and is not required for shutoff of host protein synthesis. J. Virol. 76:1206-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Satterly, N., P. L. Tsai, J. van Deursen, D. R. Nussenzveig, Y. Wang, P. A. Faria, A. Levay, D. E. Levy, and B. M. Fontoura. 2007. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. U. S. A. 104:1853-1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seo, S. H., E. Hoffmann, and R. G. Webster. 2002. Lethal H5N1 influenza viruses escape host anti-viral cytokine response. Nat. Med. 8:950-954. [DOI] [PubMed] [Google Scholar]
- 53.Shin, Y. K., Q. Liu, S. K. Tikoo, L. A. Babiuk, and Y. Zhou. 2007. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 88:13-18. [DOI] [PubMed] [Google Scholar]
- 54.Solorzano, A., R. J. Webby, K. M. Lager, B. H. Janke, A. Garcia-Sastre, and J. A. Richt. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J. Virol. 79:7535-7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Staeheli, P., P. Dreiding, O. Haller, and J. Lindenmann. 1985. Polyclonal and monoclonal antibodies to the interferon-inducible protein Mx of influenza virus-resistant mice. J. Biol. Chem. 260:1821-1825. [PubMed] [Google Scholar]
- 56.Staeheli, P., O. Haller, W. Boll, J. Lindenmann, and C. Weissmann. 1986. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 44:147-158. [DOI] [PubMed] [Google Scholar]
- 57.Talon, J., C. M. Horvath, R. Polley, C. F. Basler, T. Muster, P. Palese, and A. Garcia-Sastre. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 74:7989-7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tumpey, T. M., K. J. Szretter, N. Van Hoeven, G. Kochs, O. Haller, A. Garcia-Sastre, and P. Staeheli. 2007. The Mx1 gene protects mice against pandemic 1918 and highly lethal human H5N1 influenza viruses. J. Virol. 81:10818-10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Twu, K. Y., R. L. Kuo, J. Marklund, and R. M. Krug. 2007. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 81:8112-8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang, W., K. Riedel, P. Lynch, C. Y. Chien, G. T. Montelione, and R. M. Krug. 1999. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA. 5:195-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang, X., C. F. Basler, B. R. Williams, R. H. Silverman, P. Palese, and A. Garcia-Sastre. 2002. Functional replacement of the carboxy-terminal two-thirds of the influenza A virus NS1 protein with short heterologous dimerization domains. J. Virol. 76:12951-12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. Garcia-Sastre. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 74:11566-11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang, Y. L., L. F. Reis, J. Pavlovic, A. Aguzzi, R. Schafer, A. Kumar, B. R. Williams, M. Aguet, and C. Weissmann. 1995. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 14:6095-6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.