

Abstract
The human genome is epigenetically organized through a series of modifications to the histone proteins that interact with the DNA. In cancer, many of the proteins that regulate these modifications can be altered in both function and expression. One example of this is the family of histone deacetylases (HDACs), which as their name implies remove acetyl groups from the histone proteins, allowing for more condensed nucleosomal structure. HDACs have increased expression in cancer and are also believed to promote carcinogenesis through the acetylation and interaction with key transcriptional regulators. Given this, small molecule histone deacetylases inhibitors have been identified and developed, which not only inhibit HDACs, but can also lead to growth arrest, differentiation, and/or apoptosis in tumors both in vitro and in vivo. Here, we will discuss some of the recent developments in clinical trials utilizing HDACs inhibitors for the treatment of both hematological malignancies as well as solid tumors.
1. Introduction
DNA is woven together with proteins into an intricate organization of both extended euchromatin and condensed heterochromatin. The posttranslational modifications of the histone proteins involved in this structure regulate the epigenetic organization of the genome. This genomic organization is often altered on an epigenetic level, including the phosphorylation, acetylation, methylation, ubiquitination, sumoylation, and ADP-ribosylation of the eight histones within the nucleosome (H2A, H2B, H3, and H4).
In 1964, Mirsky and Allfrey published the first reports of histone acetylation and methylation being involved in RNA synthesis in a reversible fashion and being highly associated with open chromatin [1, 2]. Today, it is known that histone acetyltransferases transfer the acetyl group from acetyl-CoA forming ε-N-acetyl lysine on conserved lysines of the N-terminal tails of histones H3 and H4 (and to a lesser extent H2A and H2B), resulting in an open nucleosomal structure. This can be reversed by histone deacetylases (HDACs) of which, in mammals, there are currently 18 identified and have been divided into four classes based on cellular localization and function [3]. Class I includes HDACs 1, 2, 3, and 8 which are all nuclear and ubiquitously expressed. Class II, being able to shuttle back and forth between the nucleus and the cytoplasm and believed to be tissue restricted, includes HDACs 4, 5, 6, 7, 9, and 10; within this class, HDACs 6 and 10 (class IIb) have two catalytic sites, are expressed only in the cytoplasm, and are involved in a variety of biological processes. Class III contains the structurally diverse NAD+-dependent sirtuin family, which does not act primarily on histones [4]. Finally, the ubiquitously expressed HDAC11 represents Class IV, which has previously been characterized as being part of both Class I and Class II (Figure 1). Nonhistone targets of HDACs include p53, E2F, GATA-1, YY1, RelA, Mad-Max, c-Myc, NF-κB, HIF-1α, Ku70, α-tubulin, STAT3, Hsp90, TFIIE, TFIIF, and hormone receptors explaining the diverse biological effects that HDACs can impart to the cell ([5–17] for review, see [18, 19]).
Figure 1.

The histone deacetylase, family. Schematic representations of class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), class III (SIRT1), and class IV (HDAC11). Structure and Length of HDACs are shown. The total number of amino acid residues (aa) is depicted on the right, next to each HDAC. The enzymatic domains and the nucleus localization sequences are highlighted in brown and black, respectively.
Knockout mice for HDACs 1 and 2 display embryonic or perinatal lethality and class II HDACs knockouts, while viable and fertile (except for HDAC7) have significant developmental abnormalities [20–22]. HDACs expression, and activity can be altered in many cancers and in both lymphoma and leukemia HDACs is associated with the function of oncogenic-translocation products, such as PML-RARα in acute promyelocytic leukemia [23–25]. Furthermore, with the discovery of specific pan-HDACs inhibitors, it has been shown that blocking HDACs function can cause cell-cycle arrest and differentiation through the increased expression of p21WAF1/CIP1 [26, 27], affect tumor survival by blocking angiogenesis through the increased acetylation of HIF-1α [9], affect protein degradation through the acetylation of Hsp90 [13], and increase the expression of pro-apoptotic factors [28–31], making HDACs inhibitors a good candidate for single-agent cancer therapy and even combination therapy with conventional chemotherapeutics and radiation. Here, we will discuss the latest clinical advances in HDACs inhibitors.
2. HDACs Inhibitor Classifications
Riggs and colleagues identified the HDACs inhibitor prototype sodium butyrate to be an effective inhibitor of deacetylase activity [32, 33]. This was found to be non-competitive, reversible and specific for HDACs activity [34–36]. Sodium butyrate was also found to induce differentiation, RNA synthesis and strongly inhibit cell growth in the G1 phase of the cell cycle [37]. These findings paved the road for development of more specific and effective HDACs inhibitors to use in the clinic. HDACs inhibitors can be divided into four major structural classes: (1) small molecular weight carboxylates; (2) hydroxamic acids; (3) benzamides; and (4) cyclic peptides [19, 38, 39]. Pan-HDACs inhibitors include vorinostat, panobinostat, belinostat and isotype/class-specific HDACs inhibitors include romidepsin, mecetinostat (MGCD0103) and entinostat [39]. Vorinostat (Zolinza) and Romidepsin (Istodax) are the only HDACs inhibitors currently approved by the U.S. Food and Drug Adminitration (FDA) for the treatment of refractory cutaneous T-cell lymphoma (CTCL) [40, 41].
All HDACs inhibitors available or in development target the zinc molecule found in the active site of Class I, II, and IV HDACs and are characterized by their ability to inhibit the proliferation of transformed cells in culture and tumor growth in animal models by inducing cell-cycle arrest, differentiation, and/or apoptosis (Figure 2). It has been shown that HDACs inhibitors can selectively induce the expression of less than 10% of genes, some of which are involved in the inhibition of tumor growth (e.g., p21WAF1, p27Kip and p16ink4a) [19, 26, 38]. Furthermore, evidence shows that more genes may be repressed after HDACs inhibitors treatment than activated, this could be due to a chromatin conformation in a hyperacetylated state that represses transcription, the release of transcriptional repressors from HDACs protein complexes, the activation or inactivation of nonhistone transcriptional repressors and many other plausible explanations. Unfortunately, the mechanism of action is not completely elucidated, and there are also no substantiated HDAC or HAT measurements that can predict tumor response to HDACs inhibitors treatment. Otherwise, HDACs inhibitors induce broad hyperacetylation in both tumor and normal tissues, which can be used as a biomarker for drug activity. However, steps will need to be taken to further characterize the molecular mechanisms behind HDACs inhibitors function as well as predictive markers of response to further implement them functionally in the clinic.
Figure 2.

Mechanism of action of histone deacetylases inhibitors. It has been proposed that there are specific sites in the promoter region of a subset of genes that recruit the transcription factor complex (TFC) with histone deacetylases (HDACs). With inhibition of HDACs by HDACs inhibitors, histones are acetylated, and the DNA that is tightly wrapped around a deacetylated histone core relaxes. The accumulation of acetylated histones in nucleosomes leads to increased transcription of this subset of genes, which, in turn leads to downstream effects that result in cell-growth arrest, differentiation, and/or apoptosis.
3. HDACs Inhibitors in Clinical Trials
From the initial discovery of sodium butyrate, there has been tremendous interest and investigation in HDACs inhibitors, today there are at least 15 HDACs inhibitors that are currently under clinical investigation for both hematological malignancies and solid tumors, both for single-agent and combination therapy [42]. Initial molecules included valproic acid, phenyl-butyrate, SAHA (vorinostat), trapoxin A, oxamflatin, depudepsin, depsipeptide (romidepsin, Istodax) and trichostatin A [38, 43], which have paved the way to the second-generation HDACs inhibitors such as the hydroxamic acids: belinostat (PDX101), LAQ824, and panobinostat (LBH589), and the benzamides: entinostat (MS-27-275), CI994, and MGCD0103 (mocetinostat) [44]. Here, we will discuss some of the recent clinical trials regarding several of the most promising HDACs inhibitors (Table 1).
Table 1.
Table of HDACs inhibitors discussed in this paper, organized by class (refer to text for references).
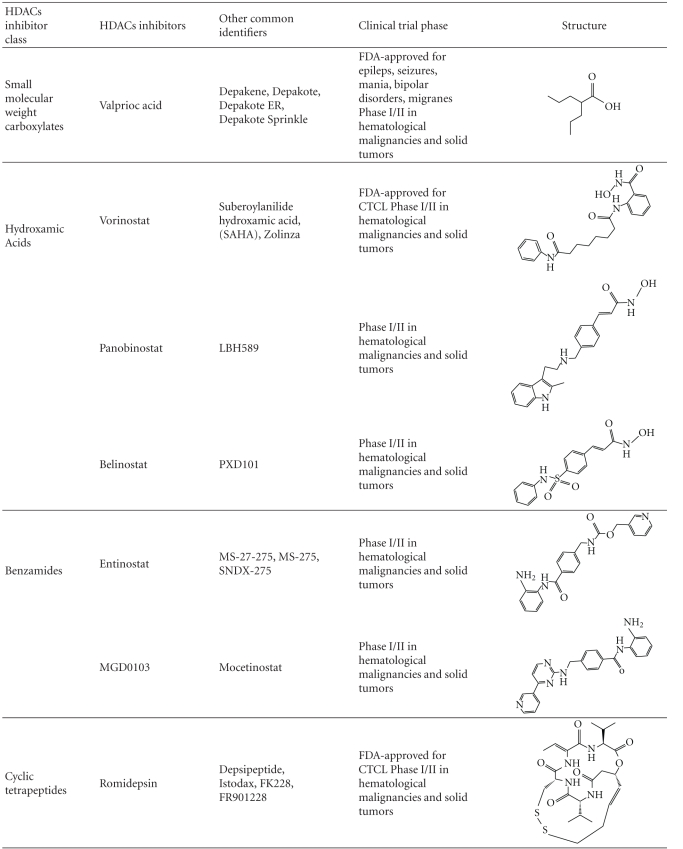 |
4. Vorinostat
In 2006, two phase II trials led vorinostat (Zolinza) to be approved by the U.S. FDA for the treatment of refractory cutaneous T-cell lymphoma CTCL [40]. A multicenter phase IIB trial enrolled a total of 74 patients for progressive, persistent, or recurrent CTCL who had received at least two prior therapies. Patients were treated daily with 400 mg of orally administered vorinostat and showed an overall response rate of 29.7%, a 6.1 month median duration of response, and a 9.8 month median time to progression [45]. Similar findings were published in a phase II study with a similar patient population [46]. When considering all patients from these trials together, 26% of patients experienced thrombocytopenia, 14% anemia, and only 5% of patients experienced grade 3 to 5 adverse events, including thrombocytopenia, pulmonary embolism, fatigue, and nausea. The most common adverse events were diarrhea, fatigue, and nausea. From the larger multicenter trial, 6 patients continued treatment with vorinostat for 2 years or longer with continued clinical effect (one complete remission (CR), four partial remission (PR), and one stable disease (SD)) [47].
A phase II clinical trial tested the use of vorinostat in other hematological malignancies, including relapsed diffuse large B-cell lymphoma (DLBCL), where out of 18 patients, one resulted in a CR and one in SD with grade 1 and 2 toxicities, but was concluded to have an overall minimal effect in treating DLBCL [48]. A second trial tested vorinostat in patients with lymphoma showing promising results. Out of 17 patients with relapsed indolent non-Hodgkin's lymphoma four patients achieved CR, two had PRs and four patients remained with SD [49].
A dose-escalation phase I trial was also performed for oral vorinostat as a single-agent therapy in acute myeloid leukemia (AML). Out of 41 total patients enrolled, 31 with AML, three with myelodysplastic syndrome (MDS), four with chronic lymphocytic leukemia (CLL), two with acute lymphoblastic leukemia, and one with chronic myeloid leukemia (CML). The maximum tolerated dose (MTD) was 200 mg when given twice daily and 250 mg when given three times daily, each given for 14 days in a 21-day cycle. The dose limiting toxicities (DLT) were again nausea, vomiting, and diarrhea. Seven of the patients with AML showed hematologic responses, including two CRs and two CRs with incomplete recovery [50].
Vorinostat has also been tested for use in treating several solid tumors, including platinum-resistant epithelial ovarian cancer, primary peritoneal carcinoma, and nonsmall cell lung carcinoma (NSCLC). After encouraging results from a phase I dose-escalation trial of vorinostat combined with carboplatin and paclitaxel in advanced solid malignancies, resulting in 11 out of 25 patients (10 of 19 with NSCLC and 1 of 4 with head and neck cancer) achieving a PR [51], a phase II National Cancer Institute-sponsored study has been carried out and results recently published [52]. This phase II randomized, double-blinded, placebo-controlled trial enrolled 94 patients with previously untreated stage IIIB or IV NSCLC to receive Carboplatin and Paclitaxel with either Vorinostat (400 mg daily on days 1 through 14 of each treatment cycle) or placebo. In the Vorinostat arm, a favorable trend toward improvement in median PFS (6 months versus 4,1 months in the placebo arm) and OS (13 months in the Vorinostat arm versus 9,7 months in the placebo arm) was clearly shown although at the price of an increased toxicity. Grade 4 thrombocytopenia was more frequent in the Vorinostat arm (18% versus 3% in the placebo arm) as well as grade 2-3 nausea, diarrhea, and fatigue. Moreover, 26% of patients in the Vorinostat arm discontinued therapy after the first cycle in comparison to 16% of the ones enrolled in the placebo arm. Comparably, the proportions of patients who completed all 6 cycles scheduled were 41% and 29%, respectively, for the placebo and Vorinostat arm.
Several trials also tested the efficacy of Vorinostat as single agent in different solid tumor sites (head and neck, breast, colorectal, and prostate cancer) and all reported a considerably high rate of adverse effects limiting the possibility of a reliable efficacy assessment. The most common adverse event reported in those trials were: fatigue (from 62% to 81%), nausea (from 58% to 74%), anorexia (from 58% to 81%), vomiting (from 33% to 56%), and thrombocytopenia (from 17% to 50%) [53–55].
Vorinostat is potentially also an attractive candidate for association with radiation since HDACs inhibition decreases cellular ability to repair DNA double-strand breaks both by Homologous Repair (HR) and Non-Homologous End Joining (NHEJ) [56, 57], thus resulting in a potent in vivo radiosensitizing effect [58]. A Phase I trial recently tested Vorinostat in combination with pelvic palliative radiotherapy (3 Gy per fraction up to 30 Gy) for gastrointestinal tumors. Vorinostat was administered orally once daily 3 hours before each radiotherapy fraction at doses ranging from 100 mg to 400 mg. The most common, any grade, adverse effects reported were fatigue, nausea, anorexia, and vomiting, respectively, in 94%, 65%, 59%, and 47% of patients [59].
5. Romidepsin
Romidepsin (Istodax, Gloucester Pharmaceuticals) is a natural compound isolated from Chromobacterium violaceum. It is a bicyclic tetrapeptide and is sometimes referred to as depsipeptide after the class of molecules to which it belongs. It was first tested for antibacterial activity, but it was found to have strong cytotoxic activity against different tumor cell lines, and later on mice. Romidepsin is mainly targeting class I HDACs, and it has also been recently approved by the FDA for treatment of CTCL. Two phase II multicentric single arm trials collected cumulatively 167 patients with refractory CTCL (mostly in advanced stages) treated with Romidepsin at a starting dose of 14 mg/m2 infused over 4 hours on days 1, 8, and 15 every 28 days. The endpoint for both studies was the overall response rate (ORR). Median time to first response was 2 months in both studies and ORR was 34% and 35%, respectively. The median duration of response was 15 and 13.7 months, respectively. Adverse effects observed in both studies were similar to the toxicities observed in phase I trials. Common (any grade) adverse effects included nausea (56% and 86%, resp.), fatigue (53% and 77%), vomiting (34% and 52%), and anorexia (23% and 54%). Furthermore, consistently with the toxicity pattern shown by Romidepsin in Phase I studies [60], ECG changes were also noted in a large proportion of patients of the study (up to 50%) consisting of T-wave flattening, ST tract depression, and QT interval prolongation. Cardiotoxicity, which has not been frequently found after Vorinostat treatments, seems to be a more specific side effect of Romidepsin and has been explained as being dependent upon the interaction of the drug with the HERG K+ channels [3]. Romidepsin has also been initially tested clinical conditions other than CTCL. In some Phase I/II trials, single-agent Depsipeptide has shown a limited clinical benefit in treating refractory neoplasms, including AML/MDS, CLL, lung cancer, hormone refractory prostate cancer, and renal cell cancer [61–66].
6. Mecetinostat (MGCD0103)
Mecetinostat (MGCD0103) is a class I isotype-selective orally available benzamide HDACs inhibitor. Early clinical trials have demonstrated activity in hematological malignancies, including myeloid leukemia and lymphoma and was well tolerated with DLTs of fatigue, nausea, vomiting, and diarrhea. A phase I trial resulted in a bone marrow CR in three of 29 patients with AML at a MTD of 60 mg/m2 administered three times weekly [67]. A phase II study in adults with relapsed or refractory DLBCL (33 patients) or follicular lymphoma (FL—17 patients) also demonstrated significant anticancer activity. Most of the 17 patients with DLBCL that were reassessed by CT after treatment showed a decrease in tumor volume, as well as one CR and 3 PRs. Out of ten patients with FL, one achieved PR. Grade 3 toxicities or greater included fatigue, neutropenia, thrombocytopenia, and anemia [68]. A phase II trial was also conducted in patients with relapsed or refractory Hodgkin's lymphoma. A treatment schedule of 110 mg or 85 mg three times per week in a 4-week cycle were given to 23 and 10 patients, respectively. From the 21 patients evaluated from the 110 mg cohort, there was an ORR of 38% (2 had CRs, and 6 had PRs). The patients who had CRs remained with progression free survival for >270 and >420 days, respectively. From the 10 patients in the 85 mg cohort, all 5 that were evaluated demonstrated tumor reductions of ≥30%, with one PR and 2 SDs [69].
Aside from the beneficial effects demonstrated in hematological malignancies, MGCD0103 also demonstrated clinical benefits in solid tumor treatment. A phase I trial in patients with advanced solid tumors given MGCD0103 three times per week for 2 of every 3 weeks showed tolerable DLTs of fatigue, nausea, vomiting, anorexia, and dehydration. After four or more cycles, SD was observed in five of 32 patients. A phase II dose of 45 mg/m2/day was recommended [70]. Phase I/II studies in solid tumors were also conducted in combination with gemcitabine. Phase I included patients with refractory solid tumors. Phase II was limited to gemcitabine naive patients with locally advanced or metastatic pancreatic cancer. During a 28-day cycle patients received MGCD0103 three times per week in a dose ascending 3 + 3 design targeting a DLT of <33%. Gemcitabine was administered three times per cycle weekly at 1000 mg/m2. Out of the 14 patients evaluated, there were 2 PRs in patients with pancreatic carcinoma, one PR in a patient with nasopharyngeal cancer, and one PR in a patient with cutaneous T-cell lymphoma. The phase II trial is ongoing at a dose of 90 mg for patients with pancreatic cancer [71].
7. Panobinostat (LBH589)
Panobinostat is a hydroxamate that has shown potential in early phase I and II clinical trials. In an initial trial, 15 patients with AML, ALL, or MDS were treated with 4.8 to 14 mg/m2 panobinostat administered intravenously as a 30-minute infusion. Transient blast cell reductions occurred in 8 of 11 patients with peripheral blasts. Four patients exhibited a DLT of grade 3 QTcF prolongation at 14 mg/m2, which were asymptomatic and cleared after treatment ended. Common toxicities included nausea, diarrhea, vomiting, hypokalemia, loss of appetite, and thrombocytopenia [72]. CTCL patients (stage IB-IVA), including Mycosis Fungoides (MF) and Sezary Syndrome (SS), who have failed two or more previous therapies were enrolled in a phase II clinical trail. Panobinostat was administered at 20 mg orally on days 1, 3, and 5 weekly until disease progression or intolerance to two groups of patients, one who had received prior treatment with oral bexorotene and a second without. The best overall responses were 3 PRs and 4 SDs. ECG monitoring of QTcF prolongation was performed, without any >500 ms [73].
8. Belinostat (PXD101)
Belinostat has shown promising anticancer activity in both hematologic malignancies as well as solid tumors. In a trial enrolling 16 patients with advanced hematological neoplasms, belinostat was administered intravenously at one of three dose levels: 600, 900, and 1000 mg/m2/d. While no CRs or PRs were noted, intravenous administration was well tolerated, and five patients (including two with DLBCL) achieved SDs after 2–9 treatment cycles. There were no grade 3 or 4 hematological toxicities (except one case of grade 3 lymphopenia), and the most common adverse effects were nausea, vomiting, fatigue and flushing. There were two grade 4 renal failures in patients with multiple myeloma (MM). The recommended dose for phase II studies was 1000 mg/m2/d, intravenously administered on days 1–5 of a 21-day cycle for patients with hematological neoplasia [74].
For solid tumors, Belinostat was tested in a phase I study of patients with advanced refractory cancers. The 46 patients received six dose levels, ranging from 150 to 1200 mg/m2/d over a 5-day cycle. DLTs were fatigue, diarrhea, atrial fibrillation, and grade 2 nausea/vomiting, which led to inability to complete the full cycle. 39% of patients resulted in SD. Of the 24 patients treated at the MTD, which was determined to be 1000 mg/m2/d, 50% achieved SD [75]. Patients with platinum resistant epithelial ovarian cancer (EOC) are resistant to conventional chemotherapy. Belinostat was administered intravenously at 1000 mg/m2/d on days 1–5 of a 21-day cycle to metastatic or recurrent platinum resistant EOC and low malignant potential (LMP) ovarian tumors. Of the 18 patients with LMP, 1 had PR, 10 had SDs. Median PFS in LMP was 13.4 months. Patients with EOC 9 had SD with a median PFS of 2.3 months [76].
9. Entinostat (MS-27-275)
Clinical trials of Entinostat, a benzamide derivative, initiated in 2005 with a Phase I study enrolling patients with advanced solid tumors or lymphoma. Entinostat was administered to a total of 22 patients once a week for 4 weeks during a 6-week cycle. The MTD was determined to be 6 mg/m2, and the common DLTs were hypophophatemia, hyponatremia, and hypoalbuminemia, which were all reversible [77]. After the analysis of three different dose schedules, 4 mg/m2 weekly or 2 to 6 mg/m2 every other week, for three weeks in a 28-day cycle; the biologically relevant plasma concentrations and antitumor activity were determined [78].
In solid tumors, a phase I combination therapy trial was performed on ten patients with an advanced NSCLC. Patients were treated with 5-azacitidine (AZA), a DNA methyltransferase inhibitor, subcutaneously on days 1–6 and 8–10 along with a fixed dose (7 mg/m2) on day 3 and 10 of a 28-day cycle of entinostat. The dose of AZA was varied by cohort using a standard 3 + 3 dose assessment. No DLTs were observed in the 30 mg/m2 dose cohort. However, in the 40 mg/m2 cohort, after one week, a patient was replaced due to rapidly progressing disease, and another patient experienced a grade 3 neutropenia and thrombocytopenia. The common toxicities included injection site reactions, nausea/vomiting, constipation, fatigue, and cytopenias. One patient had a PR, which continued longer than 8 months. Two patients had SDs and the remaining patients had PODs [79].
10. Valproic Acid
Valproic acid (VPA) has been increasingly studied in clinical trials for a variety of cancer types as a single agent or in combination with other therapies. In solid tumors, VPA was analyzed for activity in 12 patients with cervical cancer. Three four-patient dose cohorts were formed, for 20 mg/kg, 30 mg/kg, and 40 mg/kg administered orally for five days over a six-day protocol. Tumor-deacetylase activity decreased in eight patients in a statistically significant manner. A grade 2 depression in level of consciousness was registered in 9 patients [80]. Another phase I study in 26 patients revealed neurocognitive impairment, with grade 3 or 4 neurological side effects in 8 of the 26 patients. When administered intravenously the MTD was determined to be 60 mg/kg/d [81]. A phase II study for the treatment of advanced solid tumors with hydralazine and VPA revealed clinical benefit in 80% (12) of patients with cervix, breast, lung, testis, and ovarian carcinomas. Four patients had PRs and eight SDs, and the most common toxicity was hematological [82].
VPA has been more frequently studied in the use of combination therapies, specifically with all transretinoic acid (ATRA). From a study of 75 patients with AML/MDS, 66 were initially treated with VPA monotherapy followed by ATRA in nonresponsive or relapsed patients. VPA was administered for a median treatment duration of 4 months and ATRA, 2 months. 24% of patients showed hematological improvement with a median response duration of 4 months. Four out of 10 relapsed patients, when administered ATRA had a second response and both treatments were well tolerated [83]. VPA was also combined with both AZA as well as ATRA in patients with AML or high-risk MDS. A total of 53 patients were treated with AZA at the fixed dose of 75 mg/m2 daily for 7 days, ATRA at 45 mg/m2 orally daily for 5 days starting on day 3, and VPA, which was dose escalated and administered orally daily for 7 days concomitantly. The ORR was found to be 42%, the median remission duration was 26 weeks, the MTD for VPA was 50 mg/kg daily for 7 days and the DLT was reversible neurotoxicity [84]. In another study of patients with AML/MDS, increasing doses of VPA administered orally and concomitantly with a fixed dose of decitabine (15 mg/m2 by intravenous daily infusion) for 10 days revealed a safe daily dose of 50 mg/kg. 22% (12) of patients had an objective response, this included 10 CRs and 2 CRs with incomplete platelet recovery [85].
11. Associations of HDACs Inhibitors with Other Target Drugs
Despite the very high number of gene products potentially deregulated in solid tumors, high throughput screening analyses suggest that mutations often occur in genes that collaborate in a relatively limited pool of common cell signaling pathways [86]. This hypothesis may have a great relevance in the clinic. In fact, having at hand several classes of effective “pathway-oriented” target drugs, and admitting that a tumor may be driven by a limited number of deregulated pathways, it possible that the concomitant use of a combination of drugs directed against different pathways functionally related may result in an improved antineoplastic effect or in the overcoming of drug resistance.
Recent studies on multiple myeloma (MM) models suggest that HDACs inhibitors may synergize with proteasome inhibitors. Although the molecular mechanism underlying this effect is not completely understood several means have been proposed [87] and encouraging data has come from the early clinical experimentation, including a phase I trial [88] of randomized patients with relapsed/refractory MM to receive Vorinostat (200 mg twice daily or 400 mg once daily for 14 days) in combination with bortezomib (0.7 or 0.9 mg/m2on days 4, 8, 11, and 15 or 0.9 or 1.1 or 1.3 mg/m2 on days 1, 4, 8, and 11). Among 34 evaluable patients, the best response to vorinostat plus bortezomib was a partial response (PR) in 9 (26%) patients, minimal response (MR) in 7 (21%) patients, and stable disease (SD) in 18 (53%) patients. Mean duration of SD was 89 days, range 9–369 days. Of the 13 evaluable patients who had previously been treated with bortezomib, 5 achieved a PR, 1 had an MR, and 7 had SD. Eleven of the 34 patients enrolled (32.4%) discontinued treatment due to adverse effects (AEs). Most common AEs were fatigue, nausea, diarrhea, and hematological toxicities. A phase II open label study from the same group is currently ongoing. Another Phase I trial accrued 23 heavily pretreated (median of 7 previous regimens) patients with relapsed/refractory MM to receiving escalating doses of Bortezomib (1 or 1.3 mg/m2 on days 1, 4, 8, and 11 and Vorinosat 100 mg twice daily, 200 mg twice daily, and 400 mg once daily, or 500 mg once daily for 8 days each 21-day cycle). Overall response rate was 42%, two patients receiving 500 mg vorinostat had prolonged QT interval and fatigue as dose-limiting toxicities. The most common grade >3 toxicities were myelosuppression (n = 13), fatigue (n = 11), and diarrhea (n = 5). In the same setting of patients with relapsed/refractory MM, the combination of Romidepsin and Bortezomib and Dexamethasone has also shown promising results. In a Phase I/II trial, of 18 evaluable patients, this schedule resulted in a overall response rate of 67%. The most common drug related grade 3 toxicities included fatigue (2 pts.), neutropenia (1 pts.), sepsis (2 pts.), and peripheral neuropathy (1 pts). Preclinical data seems to confirm a synergic effect of Panobinostat and Bortezomib, and a Phase I trial is currently ongoing (NCT00532389). These encouraging results are paving the way to a relevant number of trials testing the association of different HDAC and Proteasome inhibitors, and results are expected in a relatively short time.
12. HDACs Inhibitor-Related Toxicity
The relationship between the toxicity of HDACs inhibitors and their pharmacodynamic/pharmacokinetic properties is still largely unknown. This makes it difficult to optimize HDACs inhibitors treatment. Studies in preclinical models have shown that HDACs inhibitors are a class of agents that has been generally well tolerated and proved a very good toxicity profile in comparison with other chemotherapeutic drugs used in cancer therapy. The main adverse effect is fatigue, which is generally mild and tolerable in most patients, but in 30% of patients, it can be severe enough to cause drug discontinuation. Gastrointestinal toxicities are also common side effects and include anorexia, nausea, vomiting, and diarrhea. Overall, they are mild and controllable with symptomatic treatment. Biochemical disorders such as hypokalemia, hyponatremia, hypocalcemia, hyperglycemia, hypophosphatemia, and hypoalbuminemia are common with various HDACs inhibitors, while neurocortical disturbances including somnolence, confusion, and tremor are observed mainly with phenylbutyrate and valproic acid. All these side effects are generally reversible upon cessation of administration of the drug.
Another side effect of histone deacetylase inhibitors is transient thrombocytopenia that is relatively common with most HDACs inhibitors [89], it is generally mild, although has been dose limiting in some studies.
A significant adverse reaction regards the cardiotoxicity. Early studies in preclinical animal models have shown that various HDACs inhibitors such as Romidepsin are able to cause myocardial inflammation and cardiac enzyme elevation. These studies represent a controversial issue since high doses of HDACs inhibitors were used [90, 91] compared to the doses that were confirmed appropriate for use in Phase I trials. Specifically, the effect of Romidepsin on cardiac function was assessed in 42 patients with T-cell lymphoma. They received a total of 736 doses of Romidepsin and an intensive cardiac monitoring was evaluated [92]. Grade I (T-wave flattening) and grade II (ST segment depression) ECG changes occurred in more than half of the ECGs obtained post treatment; however, these changes were reversible and of short duration, with no elevation in cardiac enzymes and no significant changes in left ventricular ejection fraction.
In addition, cardiac dysrhythmias were observed in a small number of patients but most of these patients had pretreatment documented dysrhythmias. Similar ECG changes and QT-interval prolongation have been reported in other Phase I/II Romidepsin studies [60, 66, 93–96]. In other Romidepsin studies, there have been reports of sudden death; however, the relationship to the drug remains unclear. In particular, a Phase II study of 15 patients with metastatic neuroendocrine tumors, administered with standard doses of Romidepsin reported one sudden death in a 48-year-old patient [66]. However, this patient had a history of hypertension, and a biventricular hypertrophy was revealed by postmortem examination, both are known risk factors for sudden death. Cardiotoxicity may be a class effect of HDACs inhibitors, being more frequent with Romidepsin and other class-I inhibitors rather than Vorinostat and other pan-HDAC inhibitors but it is unlikely that these side reactions are limited just to those HDACs inhibitors. Additional parallel cardiotoxicity studies with other various HDACs inhibitors are necessary.
Possible room for improvement could be in the development of isoform-selective HDACs inhibitors (extensively reviewed in [97]). It is known from knockout studies that the deletion of some specific HDACs isoforms can cause precise phenotypic defects. In particular, mice lacking some of the HDACs isoforms (namely, HDAC2, HDAC3, HDAC5, and HDAC9) show severe cardiac malformations and dysfunctions [98, 99], suggesting that HDACs inhibitors, specific for other HDACs could possibly have a better cardiotoxicity profile still retaining the full pro-apoptotic action. Furthermore the introduction of reliable sensitivity biomarkers in the design of trials will allow a better stratification of patients thus minimizing the risk of exposure of the unresponsive subjects to HDACs treatment and toxicity. Recently, a genome-wide loss-of-function screening was undertaken to reveal genes that govern tumor cell sensitivity to HDAC inhibitors in a sarcoma cell model, and HR23B, a protein involved in shuttling ubiquitinated proteins to the proteasome was identified as a potential biomarker [100]. HR23B expression was further investigated in 21 skin biopsies from 20 patients with CTCL enrolled in a Vorinostat Phase II trial [46] and analyzed by immunohistochemistry. The proportion of patients with a strong HR23B staining who had a clinical response was 69%, thus suggesting a pretty high positive predictive value (PPV). Similar PPV for HR23B were obtained when looking at patients treated with other HDACs inhibitors [101].
13. Conclusions
HDAC, inhibitors represent a promising new group of anticancer agents, even though the mechanisms of HDAC inhibitor-induced tumor cell death require further elucidation. While vorinostat and romidepsin are the only US FDA-approved HDACs inhibitors currently utilized in cancer therapy, as we have shown here, there are many HDACs inhibitors that are presently under intense clinical investigation, both as single agents and combination therapies. These will hopefully be able to further improve the range of treatment options available for hematologic malignancies as well as for solid tumors.
As we come closer to understanding the molecular mechanisms inherently responsible for tumorigenesis, as well as the full range of HDACs inhibitor cellular actions, we will be able to target in a more appropriate way and be able to pair cancer therapies for clinical use. In order to establish rigorous patient selection criteria and optimal drug combinations to properly design further trials and maximize the clinical gain, the bridge between the biological function and the therapeutic benefit of these drugs needs to be further elucidated.
Acknowledgment
This work was supported by grants from “Fondazione Banco di Sardegna” to L. Bagella.
References
- 1.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proceedings of the National Academy of Sciences of the United States of America. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allfrey VG, Mirsky AE. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science. 1964;144(3618):p. 559. doi: 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- 3.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. Journal of Clinical Oncology. 2009;27(32):5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 4.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annual Review of Biochemistry. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 5.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90(4):595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 6.Martínez-Balbás MA, Bauer U-M, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO Journal. 2000;19(4):662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel JH, Du Y, Ard PG, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Molecular and Cellular Biology. 2004;24(24):10826–10834. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L-F, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-κB action regulated by reversible acetylation. Science. 2001;293(5535):1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 9.Jeong J-W, Bae M-K, Ahn M-Y, et al. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell. 2002;111(5):709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Fu M, Angeletti RH, et al. Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. Journal of Biological Chemistry. 2001;276(21):18375–18383. doi: 10.1074/jbc.M100800200. [DOI] [PubMed] [Google Scholar]
- 11.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. Journal of Biological Chemistry. 2002;277(29):25904–25913. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 12.Cohen HY, Lavu S, Bitterman KJ, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Molecular Cell. 2004;13(5):627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 13.Kovacs JJ, Murphy PJM, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Molecular Cell. 2005;18(5):601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Yuan Z-L, Guan Y-J, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307(5707):269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Li N, Caron C, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO Journal. 2003;22(5):1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature Reviews Drug Discovery. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 17.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature Reviews Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 18.Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opinion on Investigational Drugs. 2005;14(12):1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 19.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annual Review of Pharmacology and Toxicology. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 20.Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO Journal. 2002;21(11):2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trivedi CM, Luo Y, Yin Z, et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity. Nature Medicine. 2007;13(3):324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 22.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3(1):28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 23.Richon VM, O’Brien JP. Histone deacetylase inhibitors: a new class of potential therapeutic agents for cancer treatment. Clinical Cancer Research. 2002;8(3):662–664. [PubMed] [Google Scholar]
- 24.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nature Reviews Drug Discovery. 2006;5(1):37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa M, Oda Y, Eguchi T, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncology Reports. 2007;18(4):769–774. [PubMed] [Google Scholar]
- 26.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expressjon and gene-associated histone acetylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandor V, Senderowicz A, Mertins S, et al. P21-dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. British Journal of Cancer. 2000;83(6):817–825. doi: 10.1054/bjoc.2000.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(44):16090–16095. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nature Medicine. 2005;11(1):71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 30.Nebbioso A, Clarke N, Voltz E, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nature Medicine. 2005;11(1):77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Adachi M, Kawamura R, Imai K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death and Differentiation. 2006;13(1):129–140. doi: 10.1038/sj.cdd.4401686. [DOI] [PubMed] [Google Scholar]
- 32.Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268(5619):462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 33.Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Molecular and Cellular Biochemistry. 1982;42(2):65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- 34.Candido EPM, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14(1):105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 35.Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell. 1978;14(1):115–121. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- 36.Boffa LC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. Journal of Biological Chemistry. 1978;253(10):3364–3366. [PubMed] [Google Scholar]
- 37.Rastl E, Swetly P. Expression of poly(adenosine diphosphate-ribose) polymerase activity in erythroleukemic mouse cells during cell cycle and erythropoietic differentiation. Journal of Biological Chemistry. 1978;253(12):4333–4340. [PubMed] [Google Scholar]
- 38.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. Journal of the National Cancer Institute. 2000;92(15):1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 39.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clinical Cancer Research. 2009;15(12):3958–3969. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 40.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature Biotechnology. 2007;25(1):84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 41.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. Journal of Clinical Oncology. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marks PA, Xu W-S. Histone deacetylase inhibitors: potential in cancer therapy. Journal of Cellular Biochemistry. 2009;107(4):600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YB, Lee K-H, Sugita K, Yoshida M, Horinouchi S. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene. 1999;18(15):2461–2470. doi: 10.1038/sj.onc.1202564. [DOI] [PubMed] [Google Scholar]
- 44.Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. Journal of Hematology and Oncology. 2010;3, article 5 doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous t-cell lymphoma. Journal of Clinical Oncology. 2007;25(21):3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 46.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109(1):31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olsen E, Duvic M, Breneman D, et al. Vorinostat provides prolonged safety and clinical benefit to patients with advanced cutaneous t-cell lymphoma (CTCL) Journal of Clinical Oncology. 2008;26 [Google Scholar]
- 48.Crump M, Coiffier B, Jacobsen ED, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Annals of Oncology. 2008;19(5):964–969. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 49.Kirschbaum M, Zain J, Popplewell L, et al. Phase 2 study of suberoylanilide hydroxamic acid (SAHA) in relapsed or refractory indolent non-Hodgkin's lymphoma: a California Cancer Consortium study. Journal of Clinical Oncology. 2007;25 abstract no. 18515. [Google Scholar]
- 50.Garcia-Manero G, Yang H, Bueso-Ramos C, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 51.Ramalingam SS, Parise RA, Ramananthan RK, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clinical Cancer Research. 2007;13(12):3605–3610. doi: 10.1158/1078-0432.CCR-07-0162. [DOI] [PubMed] [Google Scholar]
- 52.Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2010;28(1):56–62. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bradley D, Rathkopf D, Dunn R, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago phase 2 consortium. Cancer. 2009;115(23):5541–5549. doi: 10.1002/cncr.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vansteenkiste J, Van Cutsem E, Dumez H, et al. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Investigational New Drugs. 2008;26(5):483–488. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 55.Blumenschein GR, Jr., Kies MS, Papadimitrakopoulou VA, et al. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza™, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Investigational New Drugs. 2008;26(1):81–87. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 56.Kachhap SK, Rosmus N, Collis SJ, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS ONE. 2010;5(6, article e11208):1–12. doi: 10.1371/journal.pone.0011208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nature Structural and Molecular Biology. 2010;17(9):1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Folkvord S, Ree AH, Furre T, Halvorsen T, Flatmark K. Radiosensitization by SAHA in experimental colorectal carcinoma models-in vivo effects and relevance of histone acetylation status. International Journal of Radiation Oncology, Biology, Physics. 2009;74(2):546–552. doi: 10.1016/j.ijrobp.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 59.Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. The Lancet Oncology. 2010;11(5):459–464. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 60.Sandor V, Bakke S, Robey RW, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clinical Cancer Research. 2002;8(3):718–728. [PubMed] [Google Scholar]
- 61.Byrd JC, Marcucci G, Parthun MR, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105(3):959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 62.Fouladi M, Furman WL, Chin T, et al. Phase I study of depsipeptide in pediatric patients with refractory solid tumors: a Children's Oncology Group report. Journal of Clinical Oncology. 2006;24(22):3678–3685. doi: 10.1200/JCO.2006.06.4964. [DOI] [PubMed] [Google Scholar]
- 63.Klimek VM, Fircanis S, Maslak P, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (Romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clinical Cancer Research. 2008;14(3):826–832. doi: 10.1158/1078-0432.CCR-07-0318. [DOI] [PubMed] [Google Scholar]
- 64.Schrump DS, Fischette MR, Nguyen DM, et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clinical Cancer Research. 2008;14(1):188–198. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 65.Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clinical Genitourinary Cancer. 2006;5(1):57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]
- 66.Parker C, Molife R, Karavasilis V, et al. Romidepsin (FK228), a histone heacetylase inhibitor: final results of a phase II study in metastatic hormone refractory prostate cancer (HRPC) Journal of Clinical Oncology. 2007;25 abstract no. 15507. [Google Scholar]
- 67.Garcia-Manero G, Assouline S, Cortes J, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112(4):981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crump M, Andreadis C, Assouline S, Rizzieri D, Wedgwood A, Mclauglin P. Treatment of relapsed or refractory non-hodgkin lymphoma with the oral isotype-selective histone deacetylase inhibitor MGCD01013: interim results from a phase II study. Journal of Clinical Oncology. 2008;26 abstract no. 8528. [Google Scholar]
- 69.Bociek RG, Kuruvilla G, Pro B, Wedgwood A, Li Z. Isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 demonstrates clinical activity and safety in patients with relapsed/refractory classical Hodgkin Lymphoma (HL) Journal of Clinical Oncology. 2008;26 abstract no. 8507. [Google Scholar]
- 70.Siu LL, Pili R, Duran I, et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. Journal of Clinical Oncology. 2008;26(12):1940–1947. doi: 10.1200/JCO.2007.14.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hurwitz H, Nelson B, O'Dwyer PJ, Chiorean EG, Gabrail N, Li Z. Phase I/II: the oral isotype-selective HDAC inhibitor MGCD0103 in combination with gemcitabine (Gem) in patients (pts) with refractory solid tumors. Journal of Clinical Oncology. 2008;26 abstract no. 4625. [Google Scholar]
- 72.Giles F, Fischer T, Cortes J, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clinical Cancer Research. 2006;12(15):4628–4635. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 73.Duvic M, Vanaclocha F, Bernengo MG, et al. Phase II study of oral panobinostat (LBH589), a potent pan-deacetylase inhibitor, in patients with refractory Cutaneous T-Cell Lymphoma (CTCL) Journal of Clinical Oncology. 2008;26 abstract no. 8555. [Google Scholar]
- 74.Gimsing P, Hansen M, Knudsen LM, et al. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. European Journal of Haematology. 2008;81(3):170–176. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 75.Steele NL, Plumb JA, Vidal L, et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clinical Cancer Research. 2008;14(3):804–810. doi: 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- 76.Mackay HJ, Hirte H, Colgan T, et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. European Journal of Cancer. 2010;46(9):1573–1579. doi: 10.1016/j.ejca.2010.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kummar S, Gutierrez M, Gardner ER, et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clinical Cancer Research. 2007;13(18):5411–5417. doi: 10.1158/1078-0432.CCR-07-0791. [DOI] [PubMed] [Google Scholar]
- 78.Gore L, Rothenberg ML, O’Bryant CL, et al. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clinical Cancer Research. 2008;14(14):4517–4525. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Juergens RA, Vendetti F, Coleman B, Sebree RS, Rudek MA, Belinsky SA. Phase I trial of 5-azacitidine (%AC) and SNDX-275 in advanced lung cancer (NSCLC) Journal of Clinical Oncology. 2008;26 abstract no. 19036. [Google Scholar]
- 80.Chavez-Blanco A, Segura-Pacheco B, Perez-Cardenas E, et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Molecular Cancer. 2005;4, article 22 doi: 10.1186/1476-4598-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Atmaca A, Al-Batran S-E, Maurer A, et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. British Journal of Cancer. 2007;97(2):177–182. doi: 10.1038/sj.bjc.6603851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Candelaria M, Gallardo-Rincón D, Arce C, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Annals of Oncology. 2007;18(9):1529–1538. doi: 10.1093/annonc/mdm204. [DOI] [PubMed] [Google Scholar]
- 83.Kuendgen A, Knipp S, Fox F, et al. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Annals of Hematology. 2005;84(supplement 1):61–66. doi: 10.1007/s00277-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 84.Soriano AO, Yang H, Faderl S, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110(7):2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 85.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Swanton C, Caldas C. Molecular classification of solid tumours: towards pathway-driven therapeutics. British Journal of Cancer. 2009;100(10):1517–1522. doi: 10.1038/sj.bjc.6605031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jagannath S, Dimopoulos MA, Lonial S. Combined proteasome and histone deacetylase inhibition: a promising synergy for patients with relapsed/refractory multiple myeloma. Leukemia Research. 2010;34(9):1111–1118. doi: 10.1016/j.leukres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 88.Weber D, Badros A, Jagannath S, et al. Vorinostat plus Bortezomib for the treatment of relapsed/refractory multiple myeloma: early clinical experience. Blood. 2008;112 abstract no. 871. [Google Scholar]
- 89.Imanishi R, Ohtsuru A, Iwamatsu M, et al. A histone deacetylase inhibitor enhances killing of undifferentiated thyroid carcinoma cells by p53 gene therapy. Journal of Clinical Endocrinology and Metabolism. 2002;87(10):4821–4824. doi: 10.1210/jc.2002-020877. [DOI] [PubMed] [Google Scholar]
- 90.Piekarz RL, Sackett DL, Bates SE. Histone deacetylase inhibitors and demethylating agents: clinical development of histone deacetylase inhibitors for cancer therapy. Cancer Journal. 2007;13(1):30–39. doi: 10.1097/PPO.0b013e31803c73cc. [DOI] [PubMed] [Google Scholar]
- 91.Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opinion on Investigational Drugs. 2007;16(5):659–678. doi: 10.1517/13543784.16.5.659. [DOI] [PubMed] [Google Scholar]
- 92.Page JG, Rodman L, Heath JE, Tomaszeqski JE, Smith AC. Effect of infusion rate on the toxicity of depsipeptide (NCS-63-176) in Beagle Dogs. Proceedings of the American Association for Cancer Research. 1995;36 abstract no. 2193. [Google Scholar]
- 93.Page JG, Rodman L, Heath JE, Thomaszewski JE, Smith AC. Comparison of toxicity of depsipeptide (NSC-630176) in dogs and rats. Proceedings of the American Association for Cancer Research. 1996;37 abstract no. 2549. [Google Scholar]
- 94.Piekarz RL, Frye AR, Wright JJ, et al. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clinical Cancer Research. 2006;12(12):3762–3773. doi: 10.1158/1078-0432.CCR-05-2095. [DOI] [PubMed] [Google Scholar]
- 95.Marshall JL, Rizvi N, Kauh J, et al. A phase I trial of Depsipeptide (FR901228) in patients with advanced cancer. Journal of Experimental Therapeutics and Oncology. 2002;2(6):325–332. doi: 10.1046/j.1359-4117.2002.01039.x. [DOI] [PubMed] [Google Scholar]
- 96.Whittaker S, McCulloch W, Robak T, et al. International multicenter Phase II study of the HDAC inhibitor (HDACi) depsipeptide (FK228) in cutaneous T-cell lymphoma (CTCL): interim report. Journal of Clinical Oncology. 2006;24 abstract no. 3062. [Google Scholar]
- 97.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step? Cancer Letters. 2009;280(2):211–221. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 98.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Molecular and Cellular Biology. 2004;24(19):8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Montgomery RL, Potthoff MJ, Haberland M, et al. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. Journal of Clinical Investigation. 2008;118(11):3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fotheringham S, Epping MT, Stimson L, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15(1):57–66. doi: 10.1016/j.ccr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 101.Khan O, Fotheringham S, Wood V, et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(14):6532–6537. doi: 10.1073/pnas.0913912107. [DOI] [PMC free article] [PubMed] [Google Scholar]