

Abstract
Recent genome-wide screens of host genetic requirements for viral infection have reemphasized the critical role of host metabolism in enabling the production of viral particles. In this review, we highlight the metabolic aspects of viral infection found in these studies, and focus on the opportunities these requirements present for metabolic engineers. In particular, the objectives and approaches that metabolic engineers use are readily comparable to the behaviors exhibited by viruses during infection. As a result, metabolic engineers have a unique perspective that could lead to novel and effective methods to combat viral infection.
Keywords: virology, metabolic engineering, systems biology
Introduction
Viral infection is a significant challenge to human health, local and national economies, and the bioprocess industry. Human immunodeficiency virus (HIV) infection alone causes 3.5% of annual deaths worldwide [1]. Furthermore, viral contamination of cell-based production of biological and chemical goods has been responsible for up to $200 million in lost revenue per year per contamination [2]. Current methods for combating viruses have been met with limited success or have substantial drawbacks. For example, vaccines are very effective at fighting viral infection, but often a working vaccine is not easy to develop and is reliant on a host immune system. Antibody- [3] and RNAi-based therapies [4], while promising, have had relatively little success to date. And for industrial processes, the typical response to contamination is massive discard and disinfection, incurring huge monetary and time losses. Thus, there is a great need for new anti-viral strategies.
Viral proliferation requires a significant commitment of host resources to reproduce viral particles, including nucleic acids, proteins and membrane as well as energy to enable viral synthesis. Much of the early work in virology focused on the impact of viral infection on cellular metabolism and recently this aspect of infection is being re-appreciated. Large-scale forward-genetic screens are identifying many metabolic host-dependencies and metabolomic profiling studies reveal metabolic shifts in infected cells. The ability of the virus to alter metabolism of its host in order to produce new viral products has several parallels with metabolic engineering (Figure 1). Metabolic engineering is the effort to understand, perturb and design metabolic networks for the production of pharmaceuticals, food and specialty chemicals [5]. The virus has a similar objective to the engineer: increased production of a desired by-product – in this case, itself. In pursuing their objectives, the viral resources, like the engineer’s, include the host background and a set of proteins that may be introduced. The virus and engineer also face similar constraints. One constraint is the trade-off between maximum yield of the desired product and the viability of the host. In particular, increasing production rate might mean a lower growth rate and less overall product. Additionally, the engineer and virus must determine ways to avoid the defense mechanisms of the host. In response to these and other constraints, viruses have evolved complex methods to reproduce, including for example: (1) control of gene expression; (2) hypermutation to create previously unknown proteins which can be introduced into the host, or to modify existing proteins to evade an immune response; or (3) temporal control of the infection to produce virus and exit the host only under the most optimal circumstances. Similarly – and often inspired by or using viral methods [6–7] – the metabolic engineer is able to control gene expression, evolve proteins and employ inducible promoters for temporal control.
Figure 1. A comparison between viruses and metabolic engineers in terms of objectives, resources, constraints and methods.
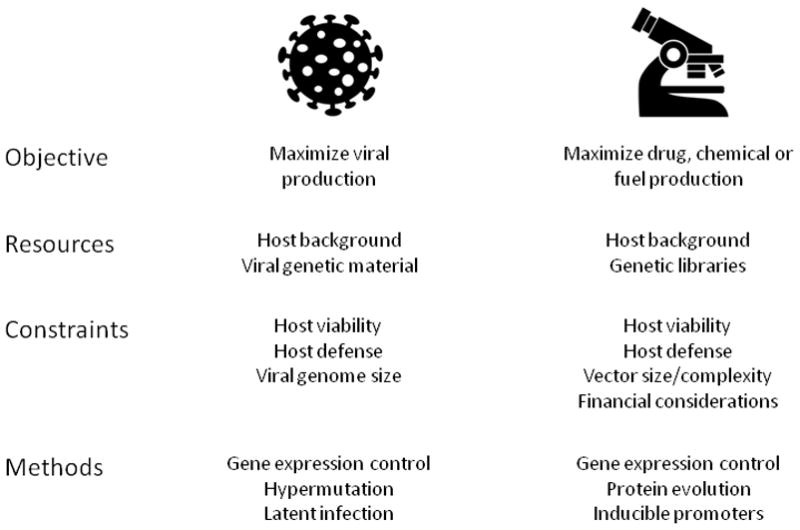
The viral properties are shown in the middle column and the metabolic engineer in the right column.
Of course there are also significant caveats to this comparison. For example, thanks to advances in DNA sequencing, gene annotation, and DNA synthesis, metabolic engineers have a much broader resource library, including vast existing cDNA libraries as well as the ability to locate desirable proteins in virtually any sequenced organism and synthesize the corresponding DNA de novo. Another resource specific to engineers is the availability of rational and computational approaches, which can accelerate the process of strain design. Finally, engineers have a significant financial constraint in their work, both in terms of development cost but also in scale-up to industrial production rates of the desired product.
Nevertheless, given the similarities between metabolic engineering and viral infection, can engineers leverage their tools and unique perspective to devise novel antiviral strategies? Others have noted the value of using engineering approaches to adapt viruses for biological applications [8]. The specific focus of this review is to highlight the common goal of engineers and viruses in manipulating cellular metabolism, and to discuss how metabolic engineers might use these similarities to combat viral infection in health and cellular culture. We begin with a discussion of the metabolic aspects of viral infection. This is followed by a brief review of the accomplishments and approaches of metabolic engineering, intended primarily for virologists new to the field. We then discuss current engineering efforts to bring these fields together, as well as possible future directions.
Metabolic Aspects of Viral Infection
Viruses are obligate parasites completely dependent on their host’s cellular metabolism to reproduce. Viral infection has been shown to inhibit other cellular processes such as replication and cellular macromolecular synthesis in order to redirect valuable resources to their own mass production [9]. Studies primarily from mid-1950’s to the early 1960’s established that uptake of a broad range of viruses, including Rous sarcoma virus [10], feline leukemia virus [11], and poliomyelitis virus [12], increase the rate of glycolysis of infected cells, in some cases by as much as 370% [13]. As a notable exception, influenza was found to inhibit glycolysis by blocking the glucose-6-phosphate isomerase (GPI) reaction [14]. However, later studies found that influenza could also increase glycolysis in early stages of infection [15].
Other aspects of metabolism have likewise been altered by viral infection. These early studies further showed that in addition to increasing ATP production through glycolysis, viral infections redirected cellular resources to viral replication. In particular, poliomyelitis virus infection was shown to alter cellular metabolism by increasing the rate of RNA breakdown and, thereby, expanding the available nucleotide pool by the possible release of a ribonuclease [16]. Similarly, during Rubella infection, the quantity of available ATP was observed to rapidly diminish [17]. Other viruses have been shown to penetrate mitochondria, hijacking mitochondrial production to virus-specific products [18].
Although the results from these studies were compelling, the molecular revolution soon led to a shift away from a metabolic focus to a genetic approach to virology, with very few major viral metabolic studies in the last four decades. However, recent development of new techniques in metabolic profiling, as well as forward-genetic screens have produced new data which emphasize the critical role of metabolism in infection. These results have inspired a renewed focus on the impact of host metabolism on viral infectivity. Studies based on metabolite profiling have been published about the impact of infection by viruses including Reovirus [19], Dengue 2 virus [20], and Mayaro virus [21]. As before, the results often highlight central metabolism. Metabolite profiling of human cytomegalo virus (HCMV) dynamics confirmed increases in glycolysis and pyrimidine nucleotide biosynthesis seen in earlier studies, and found increased rates in the citric acid cycle [22]. Further studies have also shown that both HCMV and influenza A additionally hijack the fatty acid biosynthesis pathway, and identified this pathway as a target for antiviral therapy [23].
The development of certain knockout libraries [24–25] and RNAi screening technologies have allowed for the rapid survey for viral host dependencies. Within the last few years, many genome-wide screens looking at host dependencies for HIV [26–29], Influenza [30–34], West Nile Virus [35], Dengue Virus [36] and HCV [37–39] have been reported. This data has only begun to be interpreted, with recent reviews comparing the results of the HIV [40–44] and influenza screens [45]. In addition, these screens have emphasized the importance of experimental design and analysis in RNAi-based screens [46–49].
Recently, we performed a screen for host dependencies in bacteriophage lambda and Escherichia coli. We performed plaque assays for 3,985 knockout strains contained in the ”Keio Collection” [24] and found a surprising 57 host dependencies. Forty-nine genes had previously been shown to play a role in lambda infection (Figure 2). Of these, twenty were identified by our screen. An additional eighteen genes were not in the Keio Collection, presumably because the knockout strains were unviable. Of the remaining 11 genes not identified, several of these strains grew too slow to be properly assessed using our methodology and were excluded from the analysis (e. g. ΔdnaK). Others, like recA, play a specific role in lambda infection which was not assayed in our experimental conditions [50]. On the other hand, 37 of the 57 genes we identified had not previously been directly linked to lambda replication. These 37 additional genes expand the list of potential host-dependencies from the approximately49 that have been previously established to 86genes — an increase of approximately 76%.
Figure 2. Host genes in central metabolism that play an important role in viral infection.
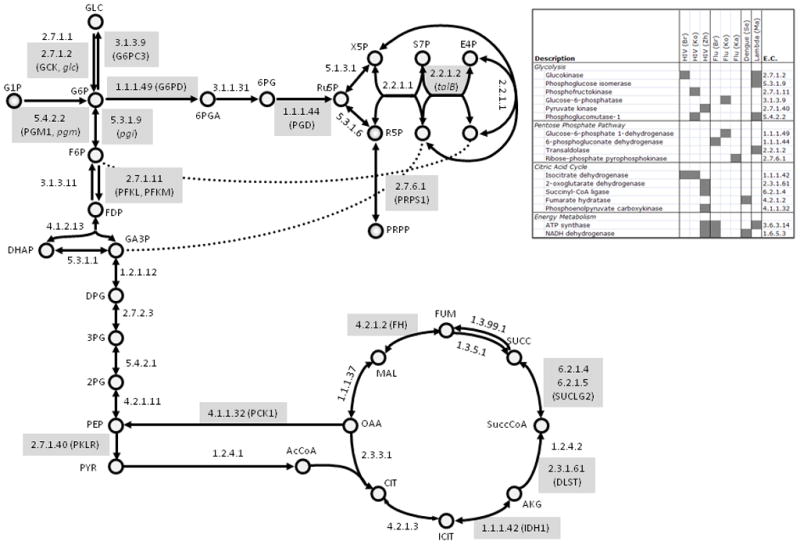
A central metabolic network is shown, where each arrow represents a reaction that is denoted by the corresponding Enzyme Commission (E.C.) number. Reactions that were found in one of several screens for host genetic requirements in viral infection are highlighted with a grey box, and the genes are noted in parentheses (human genes are all caps, and E. coli genes are italicized). A table is inset which provides more information about the highlighted genes, including which screen identified them as essential. HIV (Br) refers to [26], HIV (Ko) refers to[27], HIV (Zh) refers to [29], Flu (Br) refers to [30], Flu (Ko) refers to [33], Flu (Ka) refers to [32], Dengue (Se) refers to [36], Lambda (MA) refers to [74].
One of the key findings of this study was the pervasive role of metabolism in E. coli infection by phage lambda. Among the 86 E. coli genes that function in lambda replication, 31 have functions related to metabolism. These included several genes for enzymes in central metabolism (glk, pgm, pgi, and talB), carbon transport into glycolysis (lamB, crr, and manZ) and regulators of central metabolism (cyaA, malT, malI, fruR, and bglG). Additionally, in our study, we identify a new potential regulator of lamB expression in the unannotated gene yneJ. One potentially important aspect about these knockout strains is disproportionate affect on lambda phage replication in rich media. E. coli knockouts for most of these genes continue to grow relatively unaffected, but that lambda phage replication is significantly inhibited.
Given the critical role of metabolism found in lambda infection we suspected that the same might be true for other viral-host systems. Furthermore, metabolic genes are typically conserved in a broad range of organisms and so we wondered whether some of the genes identified in our study would also be found in the large-scale human studies. In fact, fifteen genes involved in central metabolism were found within a compiled gene list from the RNAi studies (Figure 2). The two viruses with the most host dependencies within central metabolism (HIV and influenza) have very different dependency lists. In agreement with early work on influenza — showing influenza to reduce glycolysis by inhibiting GPI [15] —all host dependencies for influenza lie upstream of GPI and within the pentose-phosphate pathway. Nearly reciprocal to influenza, the HIV host dependencies lie within glycolysis and the citric acid cycle, while no genes in the pentose-phosphate pathway were identified. This observation highlights a dramatic divergence in metabolic requirements between the two viruses.
Metabolic Engineering Methods
Over the years metabolic engineering has innovated and optimized a number of important experimental and computational methods to achieve its goals. One experimental focus of the field is the measurement of metabolite concentrations and subsequent inference of flux through specific pathways, using liquid-chromatography-tandem mass spectrometry with or without isotope labeling [51–52]. Flux and metabolome data, together with other approaches such as transcriptomics and proteomics, can lead to a deeper understanding of cellular responses to perturbation [53–54].
Beyond measurement, metabolic engineers are focused on cellular manipulation, from the deliberate modification of a few specific network nodes to the implementation of random mutagenesis under selection pressure [55]. Increasing production of a particular metabolite or other desired product often involves adding new genes to and/or removing competing pathways from a host. If the metabolic enzymes for a desired pathway are not known or are insufficiently active, cycles of random point mutation or sequence shuffling can be used to generate mutants. Under selection, these mutants can be screened to identify strains that are able to produce the desired product, produce it more efficiently, or better survive the potentially harsh conditions foreseen for implementation. To ensure that the enzymes are expressed adequately, constitutive or inducible promoters may be included to control the imported genes.
A great challenge in strain design lies in the fact that perturbations of individual genes or enzymes are typically introduced into a network context, and therefore the resulting production strain may exhibit unexpected behaviors. To address this issue, computational modeling has been particularly developed by metabolic engineers. Computational models are used to simulate and predict metabolic behaviors as a vehicle to better understanding metabolite data, and efficiently selecting manipulations to direct the production of a desired product. Beyond the modeling of metabolic networks as sets of ordinary differential equations [56], methods such as metabolic flux analysis has been used to predict cellular fluxes using experimental metabolite concentration measurements [57], and significant effort has been made to quantify the complex control of metabolic flux [58]. One method, called flux-balance analysis (FBA)[59] is based on conservation of mass and linear optimization and has the great advantage of enabling genome-scale analysis of metabolic network behaviors [60].
Systems metabolic engineering is the relatively new effort to combine these experimental and computational tools into an integrated development platform [61]. Such integration enables engineers to consider the network consequences of different design scenarios [62]. One example of systems metabolic engineering is the recent break through in producing the biodegradable plastic alternative polylactic acid (PLA) using a one-step fermentation in engineered E. coli [63]. The engineering effort began experimentally, as proteins which could convert lactic acid to PLA or a lactate copolymer were not previously known and had to be evolved [63]. The resulting strain was able to produce PLA, but at low efficiency, and so rational and computational design was used to optimize PLA production [64]. First, rational design was used to route carbon to lactate production, by deleting two genes and overexpressing a third. The overall effect of these modifications was a strain producing a copolymer with an increased fraction of lactate. Next, computational methods based on flux-balance analysis were used to guide further optimization of PLA and copolymer production. Possible gene deletions were considered in silico using FBA to identify gene deletions that would significantly enhance PLA production with minimal impact on growth rate. FBA was also used to determine further genes whose promoters could be exchanged with constitutive high expression promoters to increase PLA production. The analysis pointed to two further strain modifications which resulted in a significant increase of specific PLA copolymer production. The less intuitive nature of the modifications — particularly the synergy of mutation combinations and ability to predict mutations deleterious to growth rate — strongly supports a systems-level approach like flux-balance analysis in designing metabolic engineering strategies.
Taken together, these experimental and computational tools have had a dramatic impact on our ability to produce important substances biologically, from drugs to fuels to chemicals [65–67].
Using Metabolic Engineering to Understand Viral Infection
The metabolic aspects of viral infection, as well as the significant advances made by metabolic engineers over the years (described above), suggest that metabolic engineering approaches may be particularly useful in combating viral infection. For example, several groups have begun to explore metabolic host dependencies for the development of antiviral therapies with the rationale that by targeting the host, the virus is less likely to mutate and continue to proliferate. For example, HCV was found to use host lipid metabolism in its infection cycle [68], and several anti-HCV therapies directed specifically at host metabolic pathways have been developed [69]. These results support the use of metabolomics and fluxomics to gain a deeper understanding of host metabolism in viral infections. As many other viruses also require host metabolic proteins to replicate, this strategy should be broadly applied to other virues. Additionally, these techniques could improve target selection by predicting outcomes including possible harmful side-effects.
One critical asset for metabolic engineers in developing antiviral strategies is expertise in computationally modeling viral propagation. Viral infection models fall into two categories: unstructured and structured models [70]. Unstructured models are essentially variations on a core three-compartment model which keeps track of virus as well as the uninfected and infected host populations [71]. Such models have been used to model viral-host interaction over a broad range of infections, from HIV [72] to bacteriophage [73].
The results generated by these unstructured models can be useful in interpreting large data sets and identifying useful new experiments to perform, as we found in our work with E. coli and bacteriophage lambda [74]. Four of the genes identified by our initial screen for E. coli genes required for lambda infection had not been previously annotated. To gain insight into lambda infection dynamics, we monitored E. coli growth, and in some cases viral concentration, over the full course of infection. We then built an unstructured model of lambda infection, and fit the model parameters to our experimental time courses. Based on these results, we were able to predict the function of yneJ as a transcriptional regulator of lamB. This prediction was confirmed experimentally [74].
On the other hand, structured models of viral replication which include information about gene expression, protein interaction and other molecular details, can be useful to capture the underlying mechanism of infection. Some of the most detailed models involve bacteriophage, including a detailed structured model of the bacteriophage T7 life cycle [75] and a stochastic model of the lambda phage lysis/lysogeny decision switch [76]. However, structured models have also been used to describe the development of energy and substrate limitation during viral growth in mammalian cells [70], as well as representation of the viral transcription, translation, replication and assembly processes by vesicular stomatitis virus [77].
In addition to the structured virus models that have been created, in many cases a structured model of the host metabolism also already exists. For example, 29.9%of the E. coli genes identified as lambda dependencies are found in an FBA model [78], and five more genes encoding metabolic transcription factors, are found in a regulated FBA model which includes transcriptional regulation [79]. In total, 35.6% of the host dependency genes could be included if both models were combined (Figure 3). The question then becomes how to adapt these flux-balance models to the context of viral infection.
Figure 3. A Venn diagram categorizing E. coli genes required for optimal lambda phage infection as determined using forward or reverse genetics.
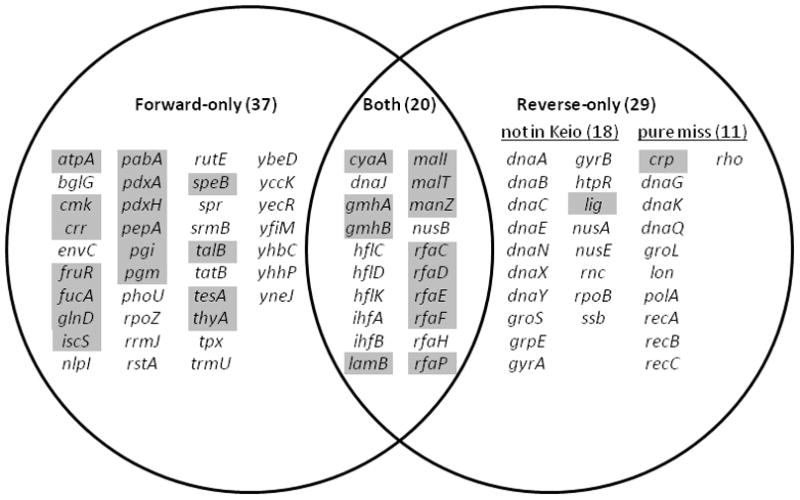
The genes in the right circle were identified by several groups over many years using reverse genetics. The genes in the left circle were identified in a forward genetic screen recently performed by our lab [74]. Genes highlighted in grey are found in either the Feist flux balance model [78]or the Covert regulated flux balance model of E. coli [79].
One approach might be to implement creation of viral biomass into flux balance models as a pseudoflux. A first step was taken in this direction by modeling the impact of viral production on E. coli metabolism, by bacteriophage MS2, a single-stranded RNA E. coli virus with a genome of only 3,569 nucleotides [80]. Beginning with an FBA model of E. coli metabolism [78], a new objective was introduced to represent replication of the MS2 RNA viral particles. The objective included production of the four viral proteins as well as the positive and negative strand RNA required for genome replication. This model predicted that infection by MS2 would result in an increase in pentose phosphate pathway flux concomitant with decreased flux through the citric acid cycle and no significant change in glycolytic flux [80].
FBA depends on the articulation of an objective function, such as the maximization of cellular biomass or of ATP production [52, 81]. In the above case with MS2, the authors used FBA to capture a time period during which they assumed that all of the host machinery was being used only to produce viral particles. This assumption enabled them to substitute a purely viral objective for the cellular objective function. In other cases, it might seem counter-intuitive that viruses are able to completely subvert the cellular objective. Often, the cellular and viral objectives are in competition to one another, similar to the trade-off between PLA production and cell growth described earlier.
This conflict between the host’s and hijacker’s objectives is well known to metabolic engineers, and therefore an innovation from engineering might be useful in better understanding this aspect of viral infection. Specifically, bilevel optimization approaches such as OptKnock [82] and RobustKnock [83] have been developed to determine a set of gene knockouts that parallelizes competing objectives. For example, these methods might be used to determine an E. coli strain that has coupled production of a desirable by-product such as ethanol to its own production of biomass. Similarly, viral infection entails the effective knockout of many genes by transcriptional repression, and it may be that in certain cases this serves to parallelize the viral objective of virion production with the cellular objective of biomass production. In such cases the host cell would be driven to produce virus until lysis is induced, and the OptKnock and RobustKnock algorithms would be well suited to understand this process.
Another significant new direction in this area could be to integrate the viral models with the existing host metabolic models – from E. coli to human [84]. The challenge in this case is to somehow incorporate very different simulation methods, such as linear optimization and differential equation-based models. We recently developed a framework, called “integrated flux balance analysis” that incorporates dynamic flux-balance analysis [59, 85] with Boolean logic (as in regulated FBA, see [86–87], as well as ordinary differential equations [88] into one simulation. The model is able to outperform both the ODE-and FBA-based models, and is available open source [89]. With this approach, existing FBA models could be incorporated with the viral models to obtain a comprehensive model of viral infection and its effect on host metabolism.
Concluding Remarks
In summary, we find some overlap between the goals and methods of viruses and the goals of metabolic engineers, and therefore believe that a metabolic engineering approach to fighting viral infection may be useful. Significant effort at this interface could lead to better understanding of the important metabolite concentrations and host pathways in this and other viral infections, as well as methods for diverting flux away from critical pathways to create super-resistant hosts. In this way the same tools that currently enable metabolic engineers to produce desired products might be adapted to prevent the viral products from forming.
Of course, these approaches will not apply equally to all viral infections, or even to all stages of viral infection. Comparison of the required E. coli genes for lambda versus T7 infection, for example, reveals a relatively minor role for core metabolism in the T7 life cycle [74]. Additionally, the large-scale RNAi screens to identify host gene dependencies for West Nile and Hepatitis C viruses did not identify a significant number of metabolic genes [30, 35, 38]. Nevertheless, in cases where viral infection depends significantly on metabolic factors, the metabolic engineering approach to virology could lead to critical new insights with applications to human health and biotechnology.
Acknowledgments
The authors are grateful for support of this work through a Bio-X Graduate Student Fellowship to EWB and an NIH Director’s Pioneer Award (DP1OD006413) to MWC.
Abbreviations
- FBA
flux balance analysis
- GPI
glucose-6-phosphate isomerase
Biography
Markus Covert’s main interests focus on integrating experimental and computational approaches to study large biological systems. His received his PhD at UC San Diego, where he worked with Bernhard Palsson in metabolic and transcriptional regulatory modeling of Escherichia coli. As a postdoctoral fellow with David Baltimore at Caltech, he used a combined experimental/computational approach to study the NF-kappa B signaling network. He started as an Assistant Professor in Stanford’s Bioengineering Department in January 2007, and recently won the NIH Director’s Pioneer Award for his efforts to build the first “gene-complete” computational model of a cell.
Footnotes
The authors have declared no conflict of interest.
References
- 1.Mathers C, et al. The global burden of disease : 2004 update. Geneva, Switzerland: World Health Organization; 2008. p. vii.p. 146. [Google Scholar]
- 2.Thayer A. Genzyme Signs Consent Decree With U.S. Chemical & Engineering News. 2010;88(22):33. [Google Scholar]
- 3.Sawyer LA. Antibodies for the prevention and treatment of viral diseases. Antiviral Res. 2000;47(2):57–77. doi: 10.1016/s0166-3542(00)00111-x. [DOI] [PubMed] [Google Scholar]
- 4.Dykxhoorn DM, Lieberman J. Silencing viral infection. PLoS Med. 2006;3(7):e242. doi: 10.1371/journal.pmed.0030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park SJ, Cochran Jennifer R. Protein Engineering and Design. Taylor and Francis; Boca Raton: 2009. [Google Scholar]
- 6.Sharan SK, et al. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc. 2009;4(2):206–23. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lois C, et al. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295(5556):868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 8.Yin J. Chemical engineering and virology: Challenges and opportunities at the interface. Aiche Journal. 2007;53(9):2202–2209. [Google Scholar]
- 9.Vaheri A, V, Cristofalo J. Metabolism of rubella virus-infected BHK 21 cells. Enhanced glycolysis and late cellular inhibition. Arch Gesamte Virus forsch. 1967;21(3):425–36. doi: 10.1007/BF01241741. [DOI] [PubMed] [Google Scholar]
- 10.Singh VN, et al. Alterations in glucose metabolism in chick-embryo cells transformed by Rous sarcoma virus: intracellular levels of glycolytic intermediates. Proc Natl Acad Sci U S A. 1974;71(10):4129–32. doi: 10.1073/pnas.71.10.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardell D, Essex M. Glycolysis during early infection of feline and human cells with feline leukemia virus. Infect Immun. 1974;9(5):824–7. doi: 10.1128/iai.9.5.824-827.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy HB, Baron S. The effect of animal viruses on host cell metabolism. II. Effect of poliomyelitis virus on glycolysis and uptake of glycine by monkey kidney tissue cultures. J Infect Dis. 1957;100(2):109–18. doi: 10.1093/infdis/100.2.109. [DOI] [PubMed] [Google Scholar]
- 13.Green M, Henle G, Deinhardt F. Respiration and glycolysis of human cells grown in tissue culture. Virology. 1958;5(2):206–19. doi: 10.1016/0042-6822(58)90019-9. [DOI] [PubMed] [Google Scholar]
- 14.Fisher TN, Ginsberg HS. The reaction of influenza viruses with guinea pig polymorphonuclear leucocytes. II. The reduction of white blood cell glycolysis by influenza viruses and receptor-destroying enzyme (RDE) Virology. 1956;2(5):637–55. doi: 10.1016/0042-6822(56)90044-7. [DOI] [PubMed] [Google Scholar]
- 15.Klemperer H. Glucose breakdown in chick embryo cells infected with influenza virus. Virology. 1961;13:68–77. doi: 10.1016/0042-6822(61)90033-2. [DOI] [PubMed] [Google Scholar]
- 16.Salzman NP, Lockart RZ, Jr, Sebring ED. Alterations in HeLa cell metabolism resulting from poliovirus infection. Virology. 1959;9:244–59. doi: 10.1016/0042-6822(59)90118-7. [DOI] [PubMed] [Google Scholar]
- 17.Bardeletti G. Respiration and ATP level in BHK21/13S cells during the earlist stages of rubella virus replication. Intervirology. 1977;8(2):100–9. doi: 10.1159/000148884. [DOI] [PubMed] [Google Scholar]
- 18.Gaitskhoki VS, et al. Reconstruction of autonomic genetic and protein-synthesizing systems from viral RNA and isolated mitochondria. Dokl Akad Nauk SSSR. 1971;201(1):220–3. [PubMed] [Google Scholar]
- 19.Burgener A, Coombs K, Butler M. Intracellular ATP and total adenylate concentrations are critical predictors of reovirus productivity from Vero cells. Biotechnol Bioeng. 2006;94(4):667–79. doi: 10.1002/bit.20873. [DOI] [PubMed] [Google Scholar]
- 20.El-Bacha T, et al. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim Biophys Acta. 2007;1772(10):1158–66. doi: 10.1016/j.bbadis.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 21.El-Bacha T, et al. Mayaro virus infection alters glucose metabolism in cultured cells through activation of the enzyme 6-phosphofructo 1-kinase. Mol Cell Biochem. 2004;266(1–2):191–8. doi: 10.1023/b:mcbi.0000049154.17866.00. [DOI] [PubMed] [Google Scholar]
- 22.Munger J, et al. Dynamics of the cellular metabolome during human cytomegalo virus infection. PLoS Pathog. 2006;2(12):e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munger J, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008;26(10):1179–86. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006–0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winzeler EA, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285(5429):901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 26.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319(5865):921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 27.Konig R, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135(1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rato S, et al. Novel HIV-1 knockdown targets identified by an enriched kinases/phosphatases shRNA library using a long-term iterative screen in Jurkat T-cells. PLoS One. 2010;5(2):e9276. doi: 10.1371/journal.pone.0009276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4(5):495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Brass AL, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139(7):1243–54. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao L, et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454(7206):890–3. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlas A, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463(7282):818–22. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 33.Konig R, et al. Human host factors required for influenza virus replication. Nature. 2010;463(7282):813–7. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapira SD, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139(7):1255–67. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan MN, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455(7210):242–5. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sessions OM, et al. Discovery of insect and human dengue virus host factors. Nature. 2009;458(7241):1047–50. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherry S, et al. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev. 2005;19(4):445–52. doi: 10.1101/gad.1267905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A. 2009;106(38):16410–5. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tai AW, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5(3):298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An P, Winkler CA. Host genes associated with HIV/AIDS: advances in gene discovery. Trends Genet. 2010;26(3):119–31. doi: 10.1016/j.tig.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arhel N, Kirchhoff F. Host proteins involved in HIV infection: new therapeutic targets. Biochim Biophys Acta. 2010;1802(3):313–21. doi: 10.1016/j.bbadis.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Bushman FD, et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5(5):e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goff SP. Knockdown screens to knockout HIV-1. Cell. 2008;135(3):417–20. doi: 10.1016/j.cell.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kok KH, Lei T, Jin DY. siRNA and shRNA screens advance key understanding of host factors required for HIV-1 replication. Retrovirology. 2009;6:78. doi: 10.1186/1742-4690-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Min JY, Subbarao K. Cellular targets for influenza drugs. Nat Biotechnol. 2010;28(3):239–40. doi: 10.1038/nbt0310-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borner K, et al. From experimental setup to bioinformatics: an RNAi screening platform to identify host factors involved in HIV-1 replication. Biotechnol J. 2010;5(1):39–49. doi: 10.1002/biot.200900226. [DOI] [PubMed] [Google Scholar]
- 47.Sharma S, Rao A. RNAi screening: tips and techniques. Nat Immunol. 2009;10(8):799–804. doi: 10.1038/ni0809-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang XD, Heyse JF. Determination of sample size in genome-scale RNAi screens. Bioinformatics. 2009;25(7):841–4. doi: 10.1093/bioinformatics/btp082. [DOI] [PubMed] [Google Scholar]
- 49.Zhang XD, Marine SD, Ferrer M. Error rates and powers in genome-scale RNAi screens. J Biomol Screen. 2009;14(3):230–8. doi: 10.1177/1087057109331475. [DOI] [PubMed] [Google Scholar]
- 50.Tomizawa J, Ogawa T. Effect of ultraviolet irradiation on bacteriophage lambda immunity. J Mol Biol. 1967;23(2):247–63. doi: 10.1016/s0022-2836(67)80031-7. [DOI] [PubMed] [Google Scholar]
- 51.Fischer E, Zamboni N, Sauer U. High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem. 2004;325(2):308–16. doi: 10.1016/j.ab.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 52.Sauer U. High-throughput phenomics: experimental methods for mapping fluxomes. Curr Opin Biotechnol. 2004;15(1):58–63. doi: 10.1016/j.copbio.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Yus E, et al. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009;326(5957):1263–8. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- 54.Ishii N, et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science. 2007;316(5824):593–7. doi: 10.1126/science.1132067. [DOI] [PubMed] [Google Scholar]
- 55.Keasling JD. Gene-expression tools for the metabolic engineering of bacteria. Trends Biotechnol. 1999;17(11):452–60. doi: 10.1016/s0167-7799(99)01376-1. [DOI] [PubMed] [Google Scholar]
- 56.Reich JG, Sel*kov EiEe. Energy metabolism of the cell : a theoretical treatise. London; New York: Academic Press; 1981. p. viii.p. 345. [Google Scholar]
- 57.Stephanopoulos G, Aristidou AA, Nielsen JH. Metabolic engineering : principles and methodologies. San Diego: Academic Press; 1998. p. xxi.p. 725. [Google Scholar]
- 58.Westerhoff HV, Palsson BO. The evolution of molecular biology into systems biology. Nat Biotechnol. 2004;22(10):1249–52. doi: 10.1038/nbt1020. [DOI] [PubMed] [Google Scholar]
- 59.Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol. 1994;60(10):3724–31. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Price ND, et al. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21(4):162–9. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 61.Blazeck J, Alper H. Systems metabolic engineering: Genome-scale models and beyond. Biotechnol J. 2010 doi: 10.1002/biot.200900247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park JH, et al. Application of systems biology for bioprocess development. Trends Biotechnol. 2008;26(8):404–12. doi: 10.1016/j.tibtech.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Yang TH, et al. Biosynthesis of polylactic acid and its copolymers using evolved propionate CoA transferase and PHA synthase. Biotechnol Bioeng. 2010;105(1):150–60. doi: 10.1002/bit.22547. [DOI] [PubMed] [Google Scholar]
- 64.Jung YK, et al. Metabolic engineering of Escherichia coli for the production of polylactic acid and its copolymers. Biotechnol Bioeng. 2010;105(1):161–71. doi: 10.1002/bit.22548. [DOI] [PubMed] [Google Scholar]
- 65.Carothers JM, Goler JA, Keasling JD. Chemical synthesis using synthetic biology. Curr Opin Biotechnol. 2009;20(4):498–503. doi: 10.1016/j.copbio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 66.Lee SK, et al. Metabolic engineering of microorganisms for biofuels production: from bugs to synthetic biology to fuels. Curr Opin Biotechnol. 2008;19(6):556–63. doi: 10.1016/j.copbio.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 67.Park JH, et al. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci U S A. 2007;104(19):7797–802. doi: 10.1073/pnas.0702609104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21(1):33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ikeda M, Kato N. Modulation of host metabolism as a target of new antivirals. Adv Drug Deliv Rev. 2007;59(12):1277–89. doi: 10.1016/j.addr.2007.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sidorenko Y, Reichl U. Structured model of influenza virus replication in MDCK cells. Biotechnol Bioeng. 2004;88(1):1–14. doi: 10.1002/bit.20096. [DOI] [PubMed] [Google Scholar]
- 71.Ellner SP, Guckenheimer J. Dynamic models in biology. Princeton, N.J: Princeton University Press; 2006. p. xxii.p. 329. [Google Scholar]
- 72.Knorr AL, Srivastava R. Evaluation of HIV-1 kinetic models using quantitative discrimination analysis. Bioinformatics. 2005;21(8):1668–77. doi: 10.1093/bioinformatics/bti230. [DOI] [PubMed] [Google Scholar]
- 73.Pearl S, et al. Nongenetic individuality in the host-phage interaction. PLoS Biol. 2008;6(5):e120. doi: 10.1371/journal.pbio.0060120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maynard A Forward-Genetic Screen and Dynamic Analysis of Lambda Phage Host-Dependencies Reveals an Extensive Interaction Network and a New Anti-Viral Strategy. Plos Genetics. doi: 10.1371/journal.pgen.1001017. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Endy D, Kong D, Yin J. Intracellular kinetics of a growing virus: A genetically structured simulation for bacteriophage T7. Biotechnol Bioeng. 1997;55(2):375–89. doi: 10.1002/(SICI)1097-0290(19970720)55:2<375::AID-BIT15>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 76.Arkin A, Ross J, McAdams HH. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics. 1998;149(4):1633–48. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hensel SC, Rawlings JB, Yin J. Stochastic kinetic modeling of vesicular stomatitis virus intracellular growth. Bull Math Biol. 2009;71(7):1671–92. doi: 10.1007/s11538-009-9419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Covert MW, et al. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429(6987):92–6. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 80.Jain R, Srivastava R. Metabolic investigation of host/pathogen interaction using MS2-infected Escherichia coli. BMC Syst Biol. 2009;3:121. doi: 10.1186/1752-0509-3-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5(1):93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84(6):647–57. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 83.Tepper N, Shlomi T. Predicting metabolic engineering knockout strategies for chemical production: accounting for competing pathways. Bioinformatics. 2010;26(4):536–43. doi: 10.1093/bioinformatics/btp704. [DOI] [PubMed] [Google Scholar]
- 84.Duarte NC, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104(6):1777–82. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mahadevan R, Edwards JS, Doyle FJ., 3rd Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J. 2002;83(3):1331–40. doi: 10.1016/S0006-3495(02)73903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomas R. Boolean formalization of genetic control circuits. J Theor Biol. 1973;42(3):563–85. doi: 10.1016/0022-5193(73)90247-6. [DOI] [PubMed] [Google Scholar]
- 87.Covert MW, Schilling CH, Palsson B. Regulation of gene expression in flux balance models of metabolism. J Theor Biol. 2001;213(1):73–88. doi: 10.1006/jtbi.2001.2405. [DOI] [PubMed] [Google Scholar]
- 88.Kremling A, Bettenbrock K, Gilles ED. Analysis of global control of Escherichia coli carbohydrate uptake. BMC Syst Biol. 2007;1:42. doi: 10.1186/1752-0509-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Covert MW, et al. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics. 2008;24(18):2044–50. doi: 10.1093/bioinformatics/btn352. [DOI] [PMC free article] [PubMed] [Google Scholar]