

Abstract
NMDA receptor (NMDAR)-dependent long-term depression (LTD) in the hippocampus is mediated primarily by the calcium-dependent removal of AMPA receptors (AMPARs) from the postsynaptic density. The AMPAR-binding, PDZ (PSD-95/Dlg/ZO1) and BAR (Bin/amphiphysin/Rvs) domain-containing protein PICK1 has been implicated in the regulation of AMPAR trafficking underlying several forms of synaptic plasticity. Using a strategy involving small hairpin RNA-mediated knockdown of PICK1 and its replacement with recombinant PICK1, we performed a detailed structure–function analysis of the role of PICK1 in hippocampal synaptic plasticity and the underlying NMDAR-induced AMPAR trafficking. We found that PICK1 is not necessary for maintenance of the basal synaptic complement of AMPARs or expression of either metabotropic glutamate receptor-dependent LTD or NMDAR-dependent LTP. Rather, PICK1 function is specific to NMDAR-dependent LTD and the underlying AMPAR trafficking. Furthermore, although PICK1 does not regulate the initial phase of NMDAR-induced AMPAR endocytosis, it is required for intracellular retention of internalized AMPARs. Detailed biophysical analysis of an N-terminal acidic motif indicated that it is involved in intramolecular electrostatic interactions that are disrupted by calcium. Mutations that interfered with the calcium-induced structural changes in PICK1 precluded LTD and the underlying NMDAR-induced intracellular retention of AMPARs. These findings support a model whereby calcium-induced modification of PICK1 structure is critical for its function in the retention of internalized AMPARs that underlies the expression of hippocampal NMDAR-dependent LTD.
Introduction
Long-term potentiation (LTP) and long-term depression (LTD) triggered by the activation of NMDA receptors (NMDARs) are extensively studied forms of synaptic plasticity that are thought to play key roles in adaptive and pathological experience-dependent plasticity (Malenka and Bear, 2004; Citri and Malenka, 2008). Compelling evidence suggests that they result, at least in part, from activity-dependent regulated trafficking of AMPA receptors (AMPARs) to and away from synapses (Bredt and Nicoll, 2003; Kennedy and Ehlers, 2006; Shepherd and Huganir, 2007).
Protein interacting with C kinase 1 (PICK1), a small protein (416 aa) with a unique domain structure comprising an N-terminal PSD-95/Dlg/ZO1 (PDZ) domain and a C-terminal Bin/amphiphysin/Rvs (BAR) domain (see Fig. 1 A), has been implicated as a major component of the machinery that regulates AMPAR trafficking during LTD at hippocampal CA1 pyramidal cell synapses (Kim et al., 2001; Jin et al., 2006; Terashima et al., 2008). PICK1 interacts with protein kinase C as well as receptor ligands such as the AMPAR subunits GluA2/3 through its PDZ domain and is thought to self-associate through its BAR domain (Staudinger et al., 1997; Xia et al., 1999; Daw et al., 2000; Perez et al., 2001). It also interacts with components of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)-dependent membrane fusion machinery (Hanley et al., 2002), actin and the actin-nucleating Arp2/3 complex (Rocca et al., 2008), as well as with GRIP1 and charged membrane lipids, all through its BAR domain (Lu and Ziff, 2005; Jin et al., 2006). In addition, PICK1 contains two acidic stretches, one adjacent to its N terminus and another adjacent to its C terminus, which have been suggested to bind calcium (Hanley and Henley, 2005).
Figure 1.
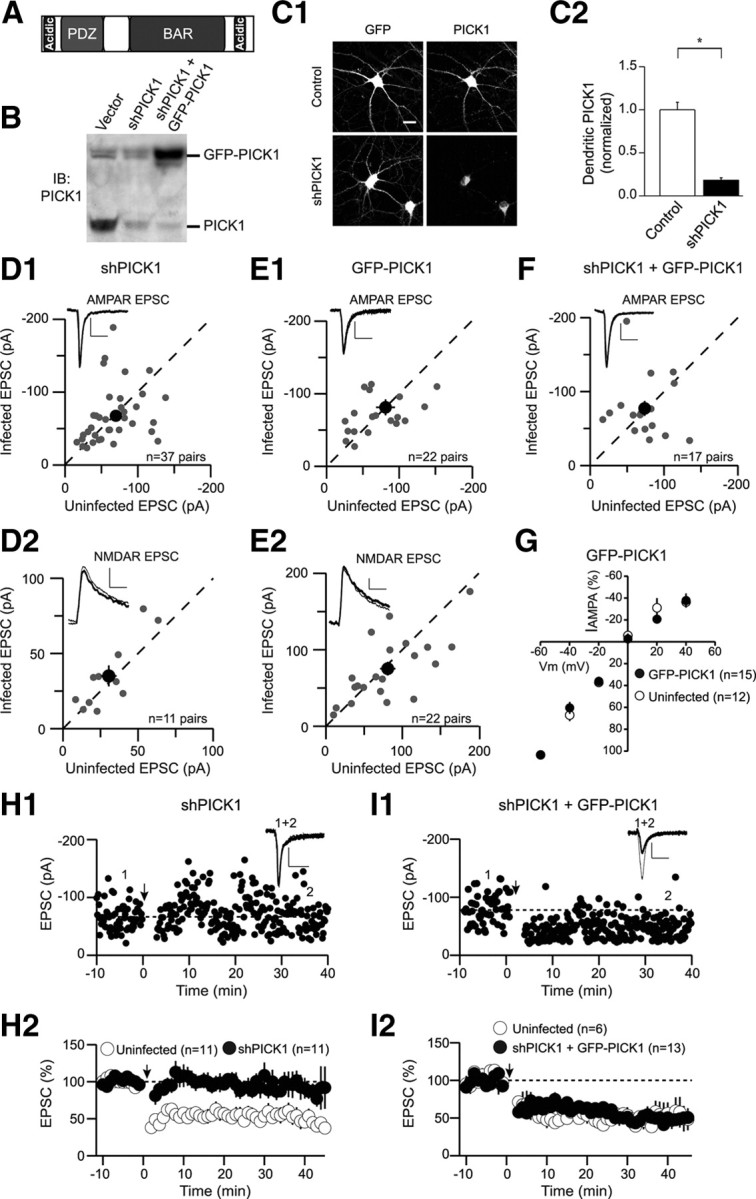
Knockdown of PICK1 does not affect basal synaptic transmission but blocks LTD. A, Schematic depicting the domain structure of PICK1. B, Western blot demonstrating shRNA-mediated knockdown of endogenous PICK1 in dissociated hippocampal cultures. C1, Representative cells showing that endogenous PICK1 staining is reduced by shPICK1 compared with control cells. The cell body staining remaining in the PICK1 knockdown was not abolished by a competitive peptide suggesting it is nonspecific background. Scale bar, 20 μm (for this and all subsequent figures). C2, Quantitation of dendritic PICK1 levels in control and PICK1 knockdown cells (control, 1 ± 0.09, n = 26; shPICK1, 0.18 ± 0.03, n = 29). In this and all subsequent figures, the asterisk (*) indicates p < 0.05, and error bars indicate SEM. D, Amplitude of AMPAR EPSCs (D1, shPICK1, −67.6 ± 6.3 pA; uninfected, −69.7 ± 6.7 pA, n = 37 pairs) and NMDAR EPSCs (D2, shPICK1, 35.1 ± 6.9 pA; uninfected, 30.5 ± 5.2 pA, n = 11 pairs) in neurons expressing shPICK1 are plotted against those of simultaneously recorded uninfected neighboring neurons. In this and subsequent figures, the gray symbols represent single neuron pairs; the black symbol represents mean ± SEM; sample traces appear in the top left corner of each graph. Calibration: 25 pA/50 ms (for this and all subsequent figures). E, Amplitude of AMPAR EPSCs (E1) (GFP-PICK1, −81.3 ± 11 pA; uninfected, −85.5 ± 13 pA, n = 22 pairs) and NMDAR EPSCs (E2) (GFP-PICK1, 75.3 ± 8.9 pA; uninfected, 80.9 ± 10 pA, n = 22 pairs) in neurons expressing GFP-PICK1 are plotted against those of simultaneously recorded uninfected neighboring neurons. F, Amplitude of AMPAR EPSCs in neurons expressing the PICK1 replacement construct (shPICK1 plus GFP-PICK1) are plotted against those of simultaneously recorded uninfected neighboring neurons (shPICK1 plus GFP-PICK1, −77.3 ± 10 pA; uninfected, −73.7 ± 7.6 pA, n = 17 pairs). G, Current–voltage relationship of the pure AMPAR EPSC in control cells (open circles) or cells expressing GFP-PICK1 (closed symbols). H, Knockdown of PICK1 inhibits LTD. H1, Sample experiment demonstrating lack of LTD in a cell expressing shPICK1. Downward arrow in this and all subsequent figures indicates time of LTD induction. In this and all subsequent figures, sample traces correspond to averaged EPSCs recorded at the indicated times (thin traces, averaged EPSCs from baseline; thick traces, averaged EPSCs after LTD induction). H2, Summary graph demonstrating block of LTD in shPICK1 infected neurons compared with interleaved uninfected controls (shPICK1, 87 ± 13%, n = 11; uninfected, 55 ± 10%, n = 11). In this and all subsequent summary graphs, points represent mean ± SEM. I, Replacement of endogenous PICK1 with GFP-PICK1 rescues LTD. I1, Sample experiment demonstrating LTD in a cell expressing shPICK1 plus GFP-PICK1. I2, Summary graph of LTD in shPICK1 plus GFP-PICK1-infected neurons and interleaved controls (shPICK1 plus GFP-PICK1, 50 ± 4% of baseline, n = 13; uninfected, 50 ± 12%, n = 6).
Because of its binding partners and potential function as a calcium sensor, PICK1 is well positioned to translate calcium influx through the NMDARs to a modification of the AMPAR trafficking underlying NMDAR-dependent LTD and perhaps other forms of synaptic plasticity (Chung et al., 2000; Daw et al., 2000; Kim et al., 2001; Perez et al., 2001; Terashima et al., 2004, 2008; Hanley and Henley, 2005; Jin et al., 2006). Here, we describe a comprehensive structure–function analysis of the function of PICK1 in CA1 pyramidal cells, using the novel and well controlled approach of lentivirus-mediated molecular replacement (Schlüter et al., 2006; Xu et al., 2008; Bhattacharyya et al., 2009) for an investigation of the roles of the different domains of PICK1 in LTD and the underlying AMPAR trafficking. We focus primarily on the role of calcium binding by PICK1 and complement multiple functional assays in slice preparations and dissociated hippocampal cultures with biophysical measurements of the effect of calcium binding on PICK1 structure. Our results suggest a simple unified model for the function of PICK1 during NMDAR-dependent LTD, whereby PICK1 is held inactive in its basal state through intramolecular electrostatic interactions involving the N-terminal acidic motif. Calcium influx through NMDARs during the induction of LTD produces a structural change in PICK1, potentially promoting its association with endocytosed AMPAR complexes and thereby delaying their recycling back to the synapse.
Materials and Methods
Drugs and antibodies.
All drugs were made as concentrated stock solutions and diluted 100- to 10,000-fold before use. Drugs that could not be dissolved in H2O were dissolved in DMSO [except 2-[(1S,2S)-2-carboxycyclopropyl]-3-(9H-xanthen-9-yl)-d-alanine (LY-341495), which was prepared in 1.2N NaOH]. Drugs used in the study were d-AP5, LY-341495, TTX, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX), 6,7-dinitroquinoxaline-2,3-dione (DNQX), (R,S)-3,5-dihydroxyphenylglycine (R,S-DHPG), and NMDA (all from Tocris), and picrotoxin (Sigma-Aldrich).
Antibodies used were as follows: goat polyclonal anti-PICK1 (Santa Cruz Biotechnology), rabbit polyclonal anti-PICK1 (Affinity Bioreagents), mouse monoclonal anti-PSD-95 (Affinity Bioreagents), rabbit polyclonal anti-green fluorescent protein (GFP) (Invitrogen; Abcam), mouse monoclonal anti-GFP (Millipore Bioscience Research Reagents), mouse monoclonal anti-Myc (Cell Signaling), rat monoclonal anti-hemagglutinin (HA) (Roche), rabbit polyclonal anti-GluA1 (Calbiochem), mouse monoclonal anti-GluA2 (Millipore Bioscience Research Reagents), and rabbit polyclonal anti-Bassoon (Synaptic Systems).
Constructs.
The cDNA encoding rat PICK1 was obtained from Open Biosystems. Lentiviral constructs were based on FUGW, into which a linker encoding a HA tag was cloned following GFP. PICK1 was cloned following the HA tag. It should be noted that tagging PICK1 on its C terminus with GFP resulted in mislocalization of the molecule, which appeared diffuse throughout the cytoplasm. The expression pattern of the N-terminally GFP-tagged PICK1 used throughout the study was similar to that of the endogenous protein and was characterized by a punctate distribution in dendrites.
Virus preparation and infection.
All lentiviral constructs were modified from the original FUGW+H vector backbone (Schlüter et al., 2006). For small hairpin RNA (shRNA) expression, a hairpin (shPICK1) targeting the PICK1 sequence (CTATGAGTACCGCCTTATCCT) was cloned into the H1 promoter cassette. When the effect of shRNAs was tested, the ubiquitin promoter-driven enhanced green fluorescent protein (eGFP) expression was used to identify infected cells. For molecular replacement studies, eGFP was replaced by fusion proteins of N-terminally, eGFP-tagged PICK1 in which silent mutations were introduced in the PICK1 construct target region of shPICK1 (CTATGAGTATAGGTTAATCCT). For the production of lentiviral particles, the transfer vector, the HIV-1 packaging vector Δ8.9, and the VSVG envelope glycoprotein vector were cotransfected into HEK293T fibroblasts using FUGENE6 transfection reagent (Roche). Supernatants of culture media were collected 40 h after transfection and centrifuged at 50,000 × g to concentrate the viral vector. To infect hippocampal slice cultures, concentrated viral solutions were injected into the CA1 pyramidal cell layer using a Picospritzer II (General Valve). To infect dissociated hippocampal cultures, concentrated virus was used for both 12 mm wells and 35 mm wells.
Hippocampal slice culture and acute slice preparation.
Procedures for preparation of hippocampal slice cultures were essentially as described previously (Schlüter et al., 2006) with minor modifications. Briefly, hippocampi of 6-d-old rats were isolated, and 225 μm slices were cut using a Vibratome (Leica Microsystems) in ice-cold sucrose solution containing the following (in mm): 204 sucrose, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 2 MgSO4, 1 NaH2PO4, 0.5 CaCl2 saturated with 95% O2 and 5% CO2. Slices were transferred onto MilliCell culture plate inserts (Millipore) and cultured in Neurobasal A medium supplemented with 1 μg/ml insulin, 0.5 mm ascorbic acid, and 25% horse serum. Slices were infected 2 d after plating, after which serum concentration was reduced to 5%. Medium was changed every 2–3 d. All recordings were performed 11–14 d after infection.
Acute transverse hippocampal slices (300 μm) were prepared from 21- to 28-d-old Sprague Dawley rats in ice-cold sucrose solution. Slices were incubated at room temperature (20–22°) in artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 26.3 NaHCO3, 11 glucose, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, and 1 NaH2PO4 for at least 1.5 h before use.
Electrophysiology.
Whole-cell patch-clamp recordings were made from CA1 pyramidal neurons using standard techniques (Xu et al., 2008; Citri et al., 2009). Recordings were performed in a recording chamber constantly perfused (1.5–2 ml/min) with warm (30°) ACSF containing the following (in mm): 119 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, and 2.5 CaCl2, bubbled with 95% O2 and 5% CO2. Picrotoxin (50 μm) was included to block inhibitory transmission. For recordings from hippocampal slice cultures, 2-chloroadenosine (2 μm) was included and 4 mm extracellular Ca2+ and Mg2+ was used. A double-barreled glass pipette filled with ACSF was used as a bipolar stimulation electrode and was placed within 300 μm of the recording pipettes in stratum radiatum. Whole-cell voltage-clamp recordings were performed with 3–5 MΩ electrodes containing the following (in mm): 117.5 CsMeSO3, 10 HEPES, 10 TEA (tetraethylammonium)-Cl, 8 NaCl, 15.5 CsCl, 1 MgCl2, 0.25 EGTA, 4 MgATP, 0.3 NaGTP, and 5 QX-314 (lidocaine N-ethyl bromide), pH adjusted to 7.3 with CsOH; osmolarity, 290 ± 5 mOsm. Stimulation pulses were provided at 0.1 Hz while holding the cell at −60 mV. Basal synaptic strength was measured by simultaneous whole-cell recordings from a visually identified infected cell and an adjacent uninfected cell (Schlüter et al., 2006; Xu et al., 2008). AMPAR EPSCs were recorded at −60 mV; NMDAR EPSCs were recorded at +40 mV and measured 50 ms after the stimulation, a time point at which AMPAR-mediated currents are absent or minimal. Average EPSCs for each cell were obtained by averaging the values of 20–40 consecutive stimuli. For I–V experiments, spermine (100 μm) was included in the patch solution and d-APV (50 μm) was included in the bath solution. Whole-cell voltage-clamp LTD experiments in hippocampal slice cultures were performed by recording baseline EPSCs at −60 mV while stimulating afferents at 0.1 Hz. After a 10 min baseline, LTD was induced in one of two ways: NMDAR-dependent LTD was elicited by delivering 300 stimuli over the course of 3 min at −10 mV [similar to the protocol used in the study by Terashima et al. (2008)], whereas metabotropic glutamate receptor (mGluR)-dependent LTD was induced by a 5 min application of R,S-DHPG (25 μm). As the synaptically evoked LTD was inhibited in the presence of d-APV (50 μm), it was defined as NMDAR dependent (data not shown). In the recordings from acute slices after in vivo virus injection (see Fig. 3), baseline EPSCs were recorded at −65 mV. LTD was induced by delivering two trains of 5 Hz stimulation for 3 min, separated by 4 min, while holding the cell at −40 mV. LTP was induced by delivering two trains of 100 Hz for 1 s, separated by 10 s, while holding the cell at 0 mV.
Figure 3.
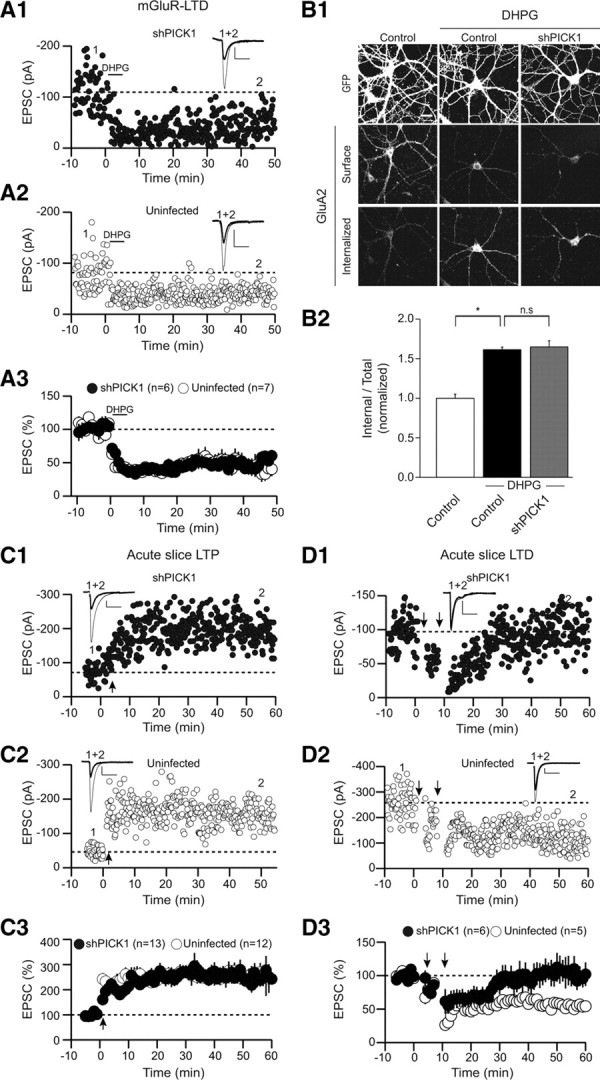
PICK1 knockdown does not affect mGluR-dependent LTD nor LTP. A, mGluR-dependent LTD is not affected by knockdown of PICK1. A1, A2, Sample experiments demonstrating mGluR-LTD in a shPICK1 expressing cell (A1) and an uninfected cell (A2). A3, Summary graphs of mGluR-dependent LTD in shPICK1-infected cells and interleaved control cells (shPICK1, 49 ± 9% of baseline, n = 6; uninfected, 49 ± 8%, n = 7). B, mGluR-induced intracellular accumulation of AMPARs is not affected by PICK1 knockdown. Representative images (B1) and quantitation (B2) (control, 1 ± 0.05, n = 23; control plus DHPG, 1.6 ± 0.03, n = 21; shPICK1 plus DHPG, 1.6 ± 0.08, n = 32). C, LTP is not affected by PICK1 knockdown in vivo. C1, C2, Sample experiments demonstrating LTP in shPICK1 expressing cell (C1) and an uninfected cell (C2). C3, Summary graphs of LTP in shPICK1-infected neurons and interleaved uninfected cells (shPICK1, 258 ± 24%, n = 13; uninfected, 252 ± 24%, n = 12). D, NMDAR-dependent LTD is inhibited by PICK1 knockdown in vivo. D1, D2, Sample experiments demonstrating block of LTD in a shPICK1-expressing cell (D1) compared with an uninfected, control cell (D2). D3, Summary graphs demonstrating block of LTD in shPICK1-infected neurons compared with interleaved uninfected control cells (shPICK1, 94 ± 17%, n = 6; uninfected, 63 ± 3%, n = 5).
Data were collected using a Multiclamp 700B amplifier (Molecular Devices) and acquired with custom software written in Igor Pro (Wavemetrics). Recordings were filtered at 2 kHz and digitized at 5–10 kHz. EPSC amplitude, holding current, input resistance, and series resistance were continuously monitored on-line. For analysis, EPSC amplitudes were calculated, binned in 1 min intervals, and normalized to the EPSC amplitude during the baseline period. Percentage LTD (or LTP) was defined by the average of EPSC amplitude during minutes 30–40 after induction. In the figures, for presentation purposes, the stimulus artifacts of sample EPSCs have been removed. Recordings from uninfected control cells were interleaved throughout the course of all experiments. For each molecular manipulation shown in the figures, the control LTD graphs were generated from the uninfected neurons that were recorded from the same set of slices. During the course of this project, the LTD induction protocol was applied to 69 uninfected control cells. Approximately 80% of these yielded a depression >30%. Approximately 40% of the recordings from uninfected cells during the LTD experiments involved simultaneous paired recordings during which recordings were simultaneously made from adjacent uninfected and infected cells.
Statistical analysis.
All data are presented as mean ± SEM unless otherwise stated. For statistical comparisons, two-tailed Student's t test was used. Differences were considered significant if p < 0.05.
Dissociated hippocampal cultures.
Primary hippocampal neuronal cultures were prepared from postnatal day 0 (P0) Sprague Dawley rat pups as previously described (Bhattacharyya et al., 2009). Briefly, hippocampi were dissected from P0 rat pups, and tissue was dissociated by papain treatment followed by trituration with glass pipettes. Cells were plated on poly d-lysine-coated coverslips at a density of ∼75,000 cells per 12 mm well for immunocytochemistry or 5 × 105 cells per 35 mm plate for immunoblotting. Cells were grown in minimum essential medium (MEM) (Invitrogen) with 0.5 mm glutamine and N2 supplement. Glial growth was inhibited by FUDR (fluorodeoxyuridine) after 6 d in culture. Cells were infected at 10 d in vitro (DIV) and assayed 13–15 d later (23–25 DIV).
Immunocytochemistry.
The effect of PICK1 knockdown and replacement on surface expression of AMPARs was assayed by labeling surface AMPARs in live neurons by 15 min incubation at 37°C with either a rabbit polyclonal antibody directed against the N terminus of the GluA1 subunit (5 μg/ml) or mouse monoclonal antibody against the N terminus of the GluA2 subunit (10 μg/ml) (both in MEM). After washout of the antibody, cells were chilled on ice and fixed in 4% paraformaldehyde (PFA) on ice for 15 min without permeabilization. Surface receptors were then stained with goat anti-rabbit or goat anti-mouse (depending on the primary antibody used) Alexa 568 (Invitrogen) secondary antibody for visualization.
To measure synaptic GluA2 puncta, we quantified colocalization of surface GluA2 clusters and presynaptic Bassoon clusters. After staining the live cell with the mouse monoclonal antibody against the N terminus of GluA2, cells were fixed in 4% PFA on ice for 15 min without permeabilization. Subsequently, cells were incubated with goat anti-mouse Alexa 568 secondary antibody. Cells were then permeabilized in 0.1% Triton X-100 for 30 min at room temperature and subsequently were incubated with a polyclonal antibody against Bassoon overnight at 4°C followed by staining with donkey anti-rabbit Cy5 secondary antibody (Jackson ImmunoResearch) for 1 h at 37°C. To determine the efficiency of shPICK1 in reducing endogenous PICK1 levels, cells were infected with the shPICK1-containing lentivirus and 12–14 d after infection were fixed with 4% PFA, permeabilized using 0.1% Triton X-100, and stained for PICK1 with a rabbit polyclonal antibody against PICK1 followed by staining with goat anti-rabbit Alexa 568 secondary antibody.
As described by Bhattacharyya et al. (2009), to assay the endocytosis of AMPARs, surface receptors were first labeled with either rabbit polyclonal antibody directed against the N terminus of the GluA1 subunit or mouse monoclonal antibody directed against the N terminus of the GluA2 subunit in live cells as described above. After washout of the antibody, cells were treated with the appropriate mixtures of antagonists (1 μm TTX plus 100 μm LY-341495 plus 20 μm DNQX for NMDA application; 1 μm TTX, 20 μm DNQX, and 50 μm APV for DHPG application) for 5 min. Subsequently, 100 μm NMDA or 100 μm R,S-DHPG were applied for 3 or 5 min, respectively, in the presence of the appropriate antagonists as just described. Cells were washed and returned to the incubator for a total of 15 min. For the time course of endocytosis assay, cells were incubated for a total of 5, 10, and 15 min after the NMDA application. Cells were then fixed in 4% PFA for 15 min on ice without permeabilization. Surface receptors were stained with a saturating concentration of goat anti-rabbit or goat anti-mouse Alexa 568-conjugated secondary antibody (Invitrogen), followed by permeabilization of cells with 0.1% Triton X-100 for 30 min at room temperature and staining of internalized receptors with donkey anti-rabbit or donkey anti-mouse Cy5-conjugated secondary antibody (Jackson ImmunoResearch). Control experiments indicated that application of the first secondary antibody was effective in staining all remaining surface receptors as application of the second secondary antibody yielded no detectable Cy5 signal in nonpermeabilized cells (Bhattacharyya et al., 2009). We used a Cy5-conjugated secondary antibody to label the internalized receptors since Cy5 is not detectable in the visible range, enabling an unbiased sampling of cells in the different conditions. For eGFP staining, either an anti-GFP rabbit polyclonal antibody or anti-GFP monoclonal antibody was used. Depending on the species of the primary antibody against GFP, either goat anti-mouse or goat anti-rabbit Alexa 488 was used to visualize GFP.
Image acquisition and analysis.
Coverslips were mounted in Fluoromount (Electron Microscopy Sciences), and cells were imaged with a 63× oil-immersion objective mounted on a Zeiss LSM 510 laser-scanning confocal microscope. Images for all conditions in a particular experiment were obtained using identical acquisition parameters (gain, offset, laser power, pinhole size, scan speed, etc.) and were analyzed with MetaMorph software using identical parameters (Meta Imaging series 6.1; Universal Imaging). Untreated and treated cells from the same culture preparation were always compared with one another. Images from each experiment were thresholded using identical values between different conditions and the total thresholded area of fluorescently labeled surface and internalized receptors was measured using MetaMorph software. To measure the surface receptors in our assay, surface fluorescence was divided by total cell area, which was determined by measuring background fluorescence of the cell using a low threshold level. These values were then normalized to the average surface fluorescence of untreated control cells. For calculation of the proportion of surface AMPARs that were endocytosed in the dendrite (10 μm away from the soma), dendritic intracellular fluorescence was divided by total dendritic fluorescence (intracellular plus surface) for each cell. These values were then normalized to those of untreated control cells from the same experiment. Each experimental treatment and analysis was performed on a minimum of two coverslips with most experiments including six to eight coverslips. In all experiments except for those in which colocalization was addressed, collapsed Z-stacks were analyzed. For presentation, images were processed using Adobe Photoshop software (Adobe Systems) by adjusting brightness and contrast levels to the same degree for all conditions illustrated in each experiment. The n value given for each experiment refers to the number of cells analyzed. Data are presented as mean ± SEM. Group results were compared by using Student's t test. A value of p > 0.05 was considered not significant (n.s.).
Immunoblotting for PICK1 knockdown.
Dissociated hippocampal neurons plated in 35 mm wells were infected at 10 DIV and assayed 14 d later. Cells were harvested in RIPA buffer [50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxycholic acid, 50 mm NaF, 200 mm Na3VO4, and a protease inhibitor mixture (Roche)]. SDS-PAGE sample buffer was added to cleared lysates, which were separated on 4–12% gradient Bis-Tris gels (Invitrogen) and transferred to PVDF (polyvinylidene difluoride) membranes. Immunoblotting was performed using primary antibodies to PICK1 and PSD-95, detected with horseradish peroxidase-conjugated secondary antibodies and developed with the ECL Western blotting detection system (GE Healthcare). Prolonged expression of the shRNA is necessary to achieve a significant reduction in endogenous PICK1 probably because of the remarkable stability of PICK1 protein (Joch et al., 2007).
Coimmunoprecipitation studies.
HEK293 cells were transfected at 60–80% confluency in 10 cm plates using Fugene 6 (Roche). Cells were harvested and lysed 24–48 h after transfection in 500 μl of RIPA buffer, extracted at 4°C for 1 h, and spun for 10 min at >30,000 × g. Cell extracts were subjected to immunoprecipitation using 2 μg of anti-Myc antibody complexed with 15 μl of protein A-agarose (Roche) overnight at 4°C followed by four washes with HNTG buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 0.1% Triton X-100, and 10% glycerol) before solubilizing the bound proteins in SDS sample buffer. Samples were then run on SDS-PAGE gels for standard Western blotting with the indicated antibodies.
Protein expression and purification.
Recombinant plasmids encoding glutathione S-transferase (GST)-fusion proteins of wild-type (WT) or mutant PICK1 were transformed into Escherichia coli BL21 (DE3) competent cells, and the proteins were expressed at 16°C for 20 h with induction by 0.4 mm IPTG (isopropyl β-d-thiogalactoside). The harvested cell pellets were resuspended in 30 ml of lysis buffer [50 mm Tris, pH 8.5, 500 mm NaCl, 5 mm DTT, 10% (v/v) glycerol, 0.5 mm AEBSF [4-(2-aminoethyl)benzenesulfonyl fluoride], and a protease inhibitor mixture (Sigma-Aldrich)] and frozen at −80°C until lysis. GST-fusion proteins were purified by affinity chromatography on glutathione–agarose beads (Sigma-Aldrich). Isolated soluble proteins were obtained by cleavage overnight at 4°C with thrombin (0.25 mg/ml resin) in 50 mm Tris, pH 8.0, 150 mm NaCl, and 2.5 mm CaCl2. Proteins were further purified by ion exchange chromatography on a SourceQ column (GE Healthcare) in 20 mm Tris, pH 8.5, 5 mm DTT with a NaCl gradient. Each protein was subsequently run on a Superdex 200 gel filtration column (GE Healthcare) in 20 mm Tris, pH 8.5, 250 mm NaCl, 5 mm DTT. The fractions containing >95% pure proteins were pooled and used for circular dichroism (CD) experiments.
CD spectroscopy.
CD spectra were recorded on an Aviv 62DS model spectropolarimeter using a 1 mm pathlength cell. CD wavelength spectra were recorded from 200 to 260 nm in 1 nm steps on 8 μm PICK1 samples in 20 mm Tris, pH 8.5, 250 mm NaCl, 1 mm DTT. Thermal denaturation experiments were performed at the rate of 1°C per minute on analogous samples monitoring the CD absorption at 220 nm between 25 and 95°C. Calcium binding was tested by recording spectra in the presence of 1 mm EDTA or 1 mm calcium. All CD experiments were repeated at least three times with comparable results.
Results
Acute shRNA-mediated knockdown of PICK1 impairs LTD
To molecularly manipulate PICK1 in individual cells, we used a dual promoter lentiviral vector (Schlüter et al., 2006) in which the human H1 promoter drives expression of a shRNA targeting PICK1, while a ubiquitin promoter simultaneously drives expression of GFP or an N-terminal GFP-fusion protein of PICK1. This “molecular replacement strategy” enables strong temporal and spatial control of the knockdown, minimizing the developmental compensatory adaptations that can occur after genetic deletion of a protein (Elias et al., 2006), as well as confounds attributable to potential presynaptic effects of the shRNA. Furthermore, a successful rescue experiment with the wild-type protein effectively controls for possible off-target effects of the shRNA. Because mutant forms of the protein being studied are expressed at modest levels on the background of knockdown of the endogenous protein, it also minimizes the complications inherent to a dominant overexpression approach (Xu et al., 2008). Finally, we use the same lentiviruses to molecularly manipulate individual cells in vivo, in slice cultures, and in dissociated cultured neurons, enabling complementary and detailed analysis of the function of PICK1 in AMPAR trafficking and long-term plasticity in the most appropriate preparations.
We first identified a highly effective shRNA to PICK1 (shPICK1) that robustly reduced endogenous PICK1 levels in dissociated hippocampal neuronal cultures when assessed by Western blots (Fig. 1 B) and immunocytochemistry (Fig. 1 C) 11–14 d after infection. To investigate the role of PICK1 in regulating basal synaptic transmission, we injected shPICK1-expressing lentivirus into the CA1 cell body layer of hippocampal slice cultures. Robust GFP expression in infected cells allowed us to perform simultaneous whole-cell voltage-clamp recordings from an infected cell and a neighboring uninfected cell while stimulating the same Schaffer collateral axons (Schlüter et al., 2006; Xu et al., 2008). Knockdown of PICK1 had no detectable effects on AMPAR EPSCs (Fig. 1 D1) or NMDAR EPSCs (Fig. 1 D2). Similarly, overexpression of PICK1, which was driven by the ubiquitin promoter, had no effect on either basal AMPAR EPSCs (Fig. 1 E1) or NMDAR EPSCs (Fig. 1 E2). Furthermore, simultaneous knockdown of endogenous PICK1 with shPICK1 and its replacement with recombinant, shRNA-resistant GFP-PICK1 did not affect the size of AMPAR EPSCs (Fig. 1 F). Finally, the voltage dependence of AMPAR EPSCs was not affected by overexpression of GFP-PICK1 (Fig. 1 G). These results were surprising given that previous work in which either PICK1 or a peptide inhibitor of PICK1-GluA2 interactions was overexpressed using Sindbis viruses reported dramatic effects on AMPAR EPSCs (Daw et al., 2000; Kim et al., 2001; Terashima et al., 2004, 2008). A potential explanation for this discrepancy stems from the difference between ubiquitin promoter driven lentivirus-mediated expression and the much more excessive expression levels achieved by Sindbis viruses.
Importantly, knockdown of PICK1 with shPICK1 essentially blocked LTD, which was reliably elicited in uninfected control cells (Fig. 1 H). Furthermore, replacement of endogenous PICK1 with recombinant, GFP-tagged, shRNA-resistant PICK1 (shPICK1 plus GFP-PICK1) restored LTD to control levels (Fig. 1 I). These results indicate that shPICK1 was effective in inhibiting the synaptic function of endogenous PICK1 in hippocampal NMDAR-dependent LTD. Furthermore, the specificity of the knockdown and the functionality of the GFP-PICK1 construct are ensured by the rescue of LTD with recombinant full-length shRNA-insensitive GFP-PICK1.
PICK1 knockdown impairs NMDAR-triggered intracellular accumulation of surface AMPARs
Substantial evidence suggests that NMDAR-dependent LTD is primarily attributable to the removal of synaptic AMPARs through a process involving regulated clathrin and dynamin-dependent endocytosis (Carroll et al., 2001; Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Shepherd and Huganir, 2007). A valuable model system for examining the mechanisms underlying the NMDAR-triggered endocytosis of AMPARs and their subsequent processing is dissociated cultured neurons in which it is possible to directly monitor the trafficking of endogenous receptors (Carroll et al., 1999; Beattie et al., 2000; Ehlers, 2000; Lin et al., 2000; Bhattacharyya et al., 2009). Therefore, in addition to assaying the functional consequences of molecular manipulation of PICK1 using electrophysiological measurements in hippocampal slice preparations, we also examined the effects of the same molecular manipulations on AMPAR trafficking in dissociated hippocampal neurons. The two assays complement each other and provide a cross-check of our results, thus increasing confidence in the conclusions we reach. Knockdown of PICK1 in cultured neurons had no effect on surface levels of dendritic GluA1-containing AMPARs and neither did replacing endogenous PICK1 with recombinant GFP-PICK1 (Fig. 2 A). Consistent with a previous report (Sossa et al., 2006), an increase in the surface levels of GluA2 was observed after PICK1 knockdown, and this increase was reversed by replacement of endogenous PICK1 with GFP-PICK1 (Fig. 2 B). To address the synaptic localization of these GluA2-containing AMPARs, we quantified the proportion of synapses [as measured by Bassoon staining; (Regalado et al., 2006)] containing GluA2 clusters. Although the number of GluA2 clusters per 50 μm of dendritic length was increased after PICK1 knockdown (Fig. 2 C2), no significant change was observed in the density of synapses (Fig. 2 C3), and most importantly, no change occurred in the fraction of synapses that contained detectable GluA2 (Fig. 2 C4). These results suggest that the increase in GluA2 surface levels caused by PICK1 knockdown occurs at extrasynaptic sites, a conclusion consistent with electron microscopy analysis of the PICK1 knock-out mouse (Steinberg et al., 2006) in which increased extrasynaptic levels of GluA2 were also observed.
Figure 2.
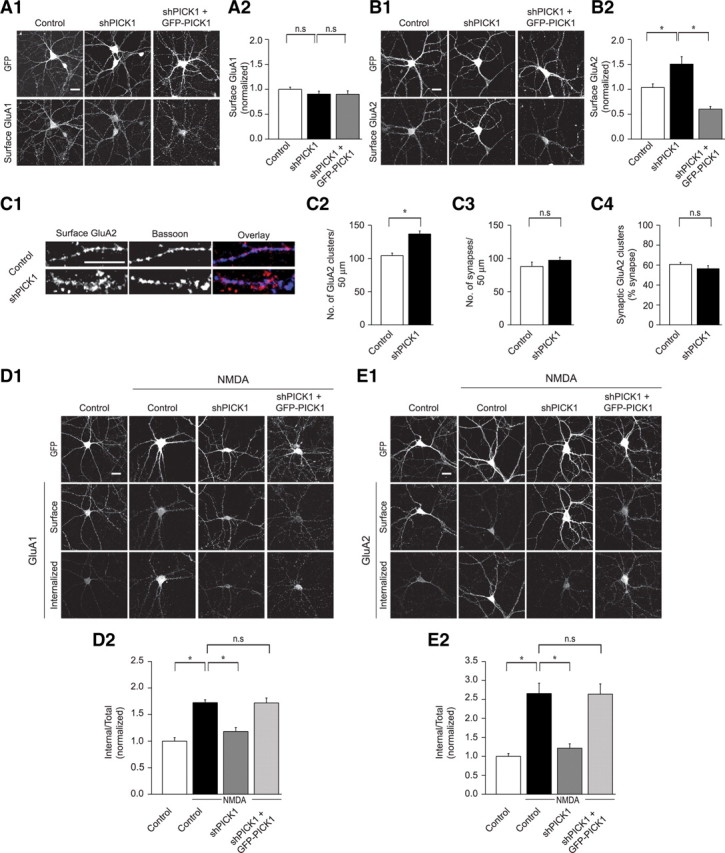
Knockdown of PICK1 abolishes NMDAR-induced intracellular accumulation of AMPARs. A, Knockdown of PICK1 does not affect the surface expression of GluA1 as shown in images of representative cells (A1), and after quantitation (A2) (control, 1 ± 0.04, n = 37; shPICK1, 0.9 ± 0.06, n = 44; shPICK1 plus GFP-PICK1, 0.9 ± 0.07, n = 20). B, shPICK1-infected cells exhibit an increase in surface expression of GluA2, an effect that is reversed by replacement of endogenous receptors with GFP-PICK1 (shPICK1 plus GFP-PICK1). B1, Representative cells. B2, Quantitation (control, 1 ± 0.07, n = 38; shPICK1, 1.5 ± 0.15, n = 47; shPICK1 plus GFP-PICK1, 0.6 ± 0.05, n = 24). C, PICK1 knockdown causes an increase in extrasynaptic, but not synaptic GluA2. C1, Examples of dendritic staining for surface GluA2 (red) and Bassoon (blue) in control or shPICK1-infected cells. Scale bar, 10 μm. C2, Quantitation of number of GluA2 clusters/50 μm dendrite (control, 104.4 ± 3.7, n = 15; shPICK1, 136.8 ± 4.6, n = 12). C3, Quantitation of number of synapses/50 μm dendrite (control, 88.1 ± 6.5, n = 15; shPICK1, 97.3 ± 4.5, n = 12). C4, Quantitation of percentage of synapses (defined by Bassoon staining) that express detectable surface GluA2 puncta (control, 60.7 ± 1.9%, n = 15; shPICK1, 56.4 ± 3.1%, n = 12). D, E, PICK1 knockdown inhibits NMDAR-induced intracellular accumulation of surface GluA1-containing AMPARs (D) and surface GluA2-containing AMPARs (E). Representative images (D1, E1) and quantitation (D2, E2) of NMDAR-triggered intracellular accumulation of surface AMPARs in cells expressing GFP, shPICK1, or shPICK1 plus GFP-PICK1. (D2, Control, 1.0 ± 0.07, n = 24; control plus NMDA, 1.7 ± 0.06, n = 21; shPICK1 plus NMDA, 1.2 ± 0.08, n = 24; shPICK1 plus GFP-PICK1 plus NMDA, 1.7 ± 0.09, n = 21.) (E2, Control, 1.0 ± 0.07, n = 32; control plus NMDA, 2.7 ± 0.27, n = 31; shPICK1 plus NMDA, 1.2 ± 0.12, n = 27; shPICK1 plus GFP-PICK1 plus NMDA, 2.6 ± 0.27, n = 24.)
We next addressed the effect of PICK1 knockdown on NMDAR-induced AMPAR trafficking using well established procedures that allow staining of both internalized and remaining surface AMPARs (Bhattacharyya et al., 2009). To induce internalization primarily of synaptic AMPARs, NMDA was applied in the presence of TTX and antagonists of both AMPARs and mGluRs (Bhattacharyya et al., 2009). PICK1 knockdown almost completely abolished the NMDAR-induced intracellular accumulation of surface GluA1-containing receptors when measured 15 min after NMDA application. This effect was completely rescued by simultaneous expression of recombinant shRNA-resistant GFP-PICK1, indicating that the inhibition was attributable to the specific knockdown of PICK1 (Fig. 2 D). Essentially identical results were obtained when the NMDAR-induced intracellular accumulation of surface GluA2-containing AMPARs was examined after PICK1 knockdown and its replacement with GFP-PICK1 (Fig. 2 E). These results demonstrate that PICK1 plays an essential role in the intracellular accumulation of surface GluA1- and GluA2-containing AMPARs after NMDAR activation in cultured neurons, and are consistent with an essential role for PICK1 in NMDAR-dependent LTD (Fig. 1).
PICK1 functions specifically in hippocampal NMDAR-dependent LTD
Activation of either NMDARs or mGluRs can induce LTD in several cell types, including CA1 pyramidal cells (Malenka and Bear, 2004; Citri and Malenka, 2008). There is growing evidence that despite the fact that both forms of LTD involve loss of synaptic AMPARs, they use different molecular mechanisms (Oliet et al., 1997; Carroll et al., 1998; Daw et al., 2000; Kim et al., 2001; Jo et al., 2008; Citri et al., 2009; Lüscher and Huber, 2010). To address whether PICK1 is required for mGluR-LTD in CA1 pyramidal cells, we examined the effects of shPICK1 on mGluR-LTD induced by application of the group I mGluR agonist DHPG (25 μm). In contrast with its clear effects on NMDAR-dependent LTD, knockdown of PICK1 had no effect on DHPG-induced LTD (Fig. 3 A). Similarly, PICK1 knockdown had no effect on the internalization of AMPARs caused by DHPG application in cultured neurons (Fig. 3 B).
PICK1 has been suggested to be required for LTP (Terashima et al., 2008). To address the effect of shRNA-mediated PICK1 knockdown on LTP, we prepared acute hippocampal slices from rats that received intrahippocampal stereotaxic injections of lentiviruses expressing shPICK1 14 d earlier (at PND 14). Robust LTP of the same magnitude was induced in both uninfected cells and cells expressing shPICK1 (Fig. 3 C). We also examined NMDAR-dependent LTD in acute slices after identical in vivo injections of the same batch of lentivirus that was used for the LTP studies. Although LTD was reliably elicited in uninfected neurons, LTD was essentially abolished in cells in which PICK1 had been knocked down by expression of shPICK1 (Fig. 3 D). These results demonstrate that the PICK1 knockdown was effective when expressed in vivo and that, in CA1 pyramidal cells, PICK1 functions specifically in NMDAR-dependent LTD and does not appear to be required for either LTP or mGluR-LTD. Although we have no satisfactory explanation for the discrepancy between our results and those previously published (Terashima et al., 2008), it should be noted that no rescue experiments were performed in the previous study to control for off-target effects of the PICK1 shRNA or developmental confounds attributable to the knock-out of PICK1 (Rocca et al., 2008).
PICK1 delays AMPAR recycling to the plasma membrane
After NMDAR activation, AMPARs are internalized, followed by recycling of a proportion of the receptors back to the plasma membrane (Ehlers, 2000; Biou et al., 2008). PICK1 could be required for either the initial internalization of the AMPARs and/or function to retain AMPARs in intracellular compartments, thus preventing their recycling back to the surface. An involvement in either process could account for the dramatic decrease in internalized AMPARs 15 min after NMDA treatment in cells in which PICK1 was knocked down (Fig. 2 D,E). To distinguish between these possibilities, we performed an analysis of the time course of NMDAR-induced AMPAR internalization (Fig. 4). In control cells, AMPARs internalized within 5 min after NMDAR stimulation and were maintained intracellularly over the ensuing 10 min (Fig. 4 A,B). In cells in which PICK1 was knocked down, the same proportion of surface AMPARs were internalized when measured 5 min after NMDAR activation (Fig. 4 A,B). However, at later time points, the proportion of surface AMPARs that remained in intracellular compartments was significantly reduced in PICK1 knockdown cells (Fig. 4 A,B). To address the possibility that the internalized AMPARs were being recycled to the dendritic membrane, we quantified the surface levels of AMPARs at these same time points. In both the PICK1 knockdown cells and control cells, AMPARs were initially removed from the surface to the same degree (Fig. 4 C). However, at later time points, surface levels of AMPARs remained reduced in control cells, whereas in cells in which PICK1 was knocked down surface AMPARs recovered to baseline levels (Fig. 4 C). Together, these results show that PICK1 knockdown has no detectable effect on the initial endocytosis of AMPARs, but does dramatically enhance their recycling, strongly suggesting that PICK1 functions to prolong intracellular retention of AMPARs after their NMDAR-triggered internalization.
Figure 4.

PICK1 delays recycling of internalized AMPARs to the dendritic plasma membrane. A, Representative images of intracellular and surface AMPARs after NMDAR activation at various time points in control cells or shPICK1 expressing cells. (t = 0 min indicates cells not treated with NMDA). B, Quantitation of internalized AMPARs at different time points after NMDA application in control and shPICK1-infected cells [control, 1 ± 0.08, n = 37; control plus NMDA (5 min), 1.8 ± 0.11, n = 41; control plus NMDA (10 min), 2.2 ± 0.19, n = 21; control plus NMDA (15 min), 2.1 ± 0.14, n = 37] [shPICK1, 1 ± 0.05, n = 43; shPICK1 plus NMDA (5 min), 2 ± 0.10, n = 30; shPICK1 plus NMDA (10 min), 1.6 ± 0.19, n = 18; shPICK1 plus NMDA (15 min), 1.1 ± 0.10, n = 35]. C, Quantitation of surface levels of GluA2 at various time points in control and shPICK1-infected cells [control, 1 ± 0.06, n = 37; control plus NMDA (5 min), 0.7 ± 0.04, n = 41; control plus NMDA (10 min), 0.5 ± 0.04, n = 21; control plus NMDA (15 min), 0.6 ± 0.04, n = 37] [shPICK1, 1 ± 0.15, n = 43; shPICK1 plus NMDA (5 min), 0.5 ± 0.07, n = 30; shPICK1 plus NMDA (10 min), 0.6 ± 0.16, n = 18; shPICK1 plus NMDA (15 min), 0.9 ± 0.12, n = 35].
The BAR domain is essential for PICK1 localization and function
PICK1 contains a BAR domain that is important for self-association (Perez et al., 2001) and that potentially creates a large crescent shape through which the molecule interacts with a defined membrane curvature and thereby may be targeted to specific organelles (Peter et al., 2004). Positively charged lysine residues that mediate binding to negatively charged head groups of membrane phosphoinositides have been identified in the concave face of the BAR domain (Lu and Ziff, 2005; Jin et al., 2006; Steinberg et al., 2006). To address the function of the BAR domain of PICK1 in LTD, we created a replacement construct in which five lysine residues within the BAR domain were mutated to glutamic acid (5KE; K251E, K252E, K257E, K266E, K268E) (Fig. 5 A), a mutant that has been shown to have minimal lipid binding capacity (Jin et al., 2006), as well as reduced actin binding (Rocca et al., 2008). Whereas endogenous PICK1 and the wild-type replacement protein are found localized in puncta along dendrites, the GFP-PICK1(5KE) mutant appeared to have a much more diffuse pattern of expression, both in dissociated neurons and in slice cultures. Replacement of endogenous PICK1 with this PICK1 mutant [GFP-PICK1(5KE)] had no effect on basal AMPAR EPSCs (Fig. 5 B). This mutant form of PICK1 also was incapable of supporting LTD (Fig. 5 C) or maintaining the pool of AMPARs that were internalized after NMDAR activation (Fig. 5 D). These results support an essential role for the BAR domain in the proper localization and function of PICK1.
Figure 5.

The PICK1 BAR domain is necessary for NMDAR-dependent LTD and AMPAR trafficking. A, Schematic depicting the BAR domain mutant PICK1(5KE). B, Basal AMPAR EPSCs are not affected by replacement of PICK1 with GFP-PICK1(5KE) [shPICK1 plus GFP-PICK1(5KE), −74.4 ± 9.1 pA; uninfected, −82.3 ± 10 pA, n = 17 pairs]. C, Summary graph demonstrating block of LTD in shPICK1 plus GFP-PICK1(5KE)-infected neurons compared with interleaved uninfected control cells [shPICK1 plus GFP-PICK1(5KE), 89 ± 11%, n = 15; uninfected, 59 ± 8%, n = 13]. D, GFP-PICK1(5KE) cannot maintain the pool of AMPARs that were internalized after NMDAR activation. Representative images (D1) and quantitation (D2) of NMDAR-induced AMPAR intracellular accumulation in GFP, shPICK1 or shPICK1 plus GFP-PICK1(5KE) expressing cells [control, 1 ± 0.07, n = 32; control plus NMDA, 1.7 ± 0.15, n = 26; shPICK1 plus NMDA, 1 ± 0.11, n = 24; shPICK1 plus GFP-PICK1(5KE) plus NMDA, 1.1 ± 0.13, n = 34].
PICK1 PDZ domain function is not required for LTD
We next addressed the function of the PDZ domain of PICK1, using a well characterized mutation (K27A, D28A; termed KD-AA) (Fig. 6 A) that has been reported to abolish the PICK1 PDZ domain interaction with both PKC and GluA2 (Staudinger et al., 1997; Dev et al., 1999; Xia et al., 1999; Hanley et al., 2002). Replacement of endogenous PICK1 with GFP-PICK1(KD-AA) had no effect on basal AMPAR-mediated transmission (Fig. 6 B). Surprisingly, this mutant form of PICK1 behaved like wild-type PICK1 in that it rescued the block of LTD caused by knockdown of endogenous PICK1 (Fig. 6 C). Furthermore, it rescued the block of maintained NMDAR-triggered AMPAR endocytosis because of shPICK1 expression (Fig. 6 D). These results are surprising, as a central tenet for the function of PICK1 has been that it mediates its effects on AMPAR trafficking through direct association with GluA2 (Xia et al., 1999; Kim et al., 2001; Perez et al., 2001; Duprat et al., 2003; Dev et al., 2004; Terashima et al., 2004, 2008; Hanley, 2008; Thorsen et al., 2010). Therefore, to further test the importance of the direct interaction between GluA2 and the PICK1 PDZ domain, we tested the effects of an additional PDZ domain mutant (K27E) (Fig. 6 E), which has been reported to specifically abolish the interactions of the PDZ domain with GluA2, while maintaining PICK1–PKC interactions (Dev et al., 2004). This mutant form of PICK1 behaved similarly to wild-type PICK1 and PICK1(KD-AA) in that it had no effect on basal AMPAR EPSCs (Fig. 6 F) and fully supported LTD (Fig. 6 G).
Figure 6.

PICK1 PDZ domain mutants support NMDAR-dependent LTD and AMPAR trafficking. A, Schematic depicting the PDZ domain mutant PICK1(KD-AA). B, Basal AMPAR EPSCs are not affected by replacement of PICK1 with GFP-PICK1(KD-AA) [shPICK1 plus GFP-PICK1(KD-AA), −61.9 ± 6.3 pA; uninfected, −69.4 ± 8.7 pA, n = 16 pairs]. C, LTD is not affected by replacement of endogenous PICK1 with GFP-PICK1(KD-AA). C1, Sample experiment demonstrating LTD in a cell expressing shPICK1 plus GFP-PICK1(KD-AA). C2, Summary graphs of LTD in shPICK1 plus GFP-PICK1(KD-AA)-infected neurons and interleaved uninfected control cells [shPICK1 plus GFP-PICK1(KD-AA), 66 ± 6%, n = 15; uninfected, 59 ± 8%, n = 13; the same uninfected control graph is shown for the PICK1(5KE) and PICK1(KD-AA) mutants as they were analyzed in parallel and therefore are compared with a common set of interleaved controls]. D, Replacement of endogenous PICK1 with GFP-PICK1(KD-AA) rescues the block of maintained NMDAR-triggered AMPAR endocytosis because of PICK1 knockdown. Representative images (D1) and quantitation (D2) of NMDAR-triggered intracellular accumulation of GluA2-containing AMPARs in cells expressing GFP, shPICK1, or shPICK1 plus GFP-PICK1(KD-AA) [control, 1 ± 0.05, n = 63; control plus NMDA, 1.9 ± 0.14, n = 60; shPICK1 plus NMDA, 1.13 ± 0.07, n = 49; shPICK1 plus GFP-PICK1(KD-AA), 1.9 ± 0.15, n = 50]. E, Schematic depicting the PDZ domain mutant PICK1(K27E). F, Basal AMPAR EPSCs are not affected by replacement of PICK1 with GFP-PICK1(K27E) [shPICK1 plus GFP-PICK1(K27E), −74.8 ± 13.3 pA; uninfected, −70.7 ± 8.2 pA, n = 15 pairs]. G, LTD is not affected by replacement of endogenous PICK1 with GFP-PICK1(K27E). G1, Sample experiment demonstrating LTD in a cell expressing shPICK1 plus GFP-PICK1(K27E). G2, Summary graphs of LTD in shPICK1 plus GFP-PICK1(K27E)-infected neurons and interleaved uninfected control cells [shPICK1 plus GFP-PICK1(K27E), 65 ± 12%, n = 7; uninfected, 57 ± 8, n = 6]. H, Coimmunoprecipitation experiment demonstrating a weakened interaction of PDZ domain mutants of PICK1 with GluA2. I, Coimmunoprecipitation experiment demonstrating that coexpression of GRIP1 can reinstate a complex of GluA2 with PDZ-domain mutants of PICK1. IP, Immunoprecipitation; IB, immunoblotting.
Because the effects of the PDZ domain mutants on LTD were surprising, we performed coimmunoprecipitation (co-IP) experiments in HEK293 cells, which expressed GFP-PICK1 and myc-tagged GluA2, to test whether, as expected, these PICK1 mutants disrupt the interactions of PICK1 with GluA2. Whereas a reliable co-IP of wild-type GFP-PICK1 with myc-GluA2 occurred, consistent with published studies (Xia et al., 1999; Perez et al., 2001; Dev et al., 2004), a dramatically reduced interaction was evident in the co-IP of either the KD-AA or the K27E mutants (Fig. 6 H). Although together our results suggest that direct interactions of GluA2 with the PDZ domain of PICK1 are dispensable for LTD, it is possible that an intermediary protein could provide a direct link between these proteins. Indeed, PICK1 has been reported to interact with GRIP1, in a complex that mediates a PDZ-independent interaction of PICK1 with GluA2 (Lu and Ziff, 2005). We therefore tested the capacity of the PDZ domain mutant proteins to interact with GluA2 in the presence of recombinant GRIP1. Coexpression of GRIP1 facilitated a reconstitution of the interaction of PICK1 with GluA2, such that the PDZ domain mutants of PICK1 were now able to immunoprecipitate well with myc-GluA2 (Fig. 6 I). We conclude that, although direct interactions between PICK1 and GluA2 are not essential for the function of PICK1 in NMDAR-induced AMPAR trafficking and LTD, formation of a GluA2–PICK1 complex, perhaps mediated by GRIP1, could be critical for the function of PICK1 in regulating synaptic AMPARs.
Calcium-induced changes in PICK1 structure are essential for LTD
Equilibrium dialysis assays have provided evidence that N-terminal and C-terminal acidic motifs are important for calcium binding by PICK1 (Hanley and Henley, 2005) and overexpression of PICK1 lacking the N-terminal motif abolished the enhanced NMDA-triggered AMPAR endocytosis observed after overexpression of wild-type PICK1 (Hanley and Henley, 2005). These results are consistent with the hypothesis that calcium binding promotes structural changes in PICK1, and these are critical for its function in regulating AMPAR trafficking during LTD (Hanley and Henley, 2005; Hanley, 2008). However, neither the effects of calcium on PICK1 structure nor the role of the N-terminal motif of PICK1 in LTD have been addressed in previous work. We therefore performed a series of experiments to directly address the role of calcium-induced structural modification of PICK1 in NMDAR-dependent LTD and the underlying NMDAR-induced AMPAR endocytosis.
To determine whether calcium induces structural changes in PICK1, we acquired CD spectra of purified bacterially expressed recombinant PICK1 in the absence or presence of calcium. In the absence of calcium, the CD spectrum of wild-type PICK1 exhibits double minima at 208 and 220 nm (Fig. 7 A, open circles), which is characteristic of an α-helical structure. This result is expected for proteins containing both α-helical and β-sheet structure, such as PICK1, because the α-helical CD signal dominates over the β-sheet signal. Importantly, the CD spectrum of PICK1 in the presence of 1 mm calcium (Fig. 7 A, solid circles) exhibited a distinct decrease in the CD signal around 208 nm. Although small, this change was reproducible and suggests that PICK1 indeed binds calcium. This conclusion was tested by performing thermal denaturation experiments, which were monitored through the CD signal at 220 nm (Fig. 7 B). In the absence of calcium, the thermal denaturation curve of PICK1 contained two transitions, one at ∼38°C and another at >90°C at which complete loss of the CD signal could not be reached (open circles). These findings imply that PICK1 denatures in two steps that may correspond to different domains and/or to global interactions between domains. Addition of calcium (solid circles) led to a shift in the second transition toward lower temperatures, indicating that calcium binding destabilizes tertiary interactions in PICK1.
Figure 7.

Calcium-induced modification of PICK1 structure requires the N-terminal acidic motif and is essential for LTD. A, CD spectra of recombinant PICK1 in the absence (open circles) or presence (solid circles) of 1 mm calcium. B, Thermal denaturation of recombinant PICK1 monitored through the CD signal at 220 nm in the absence (open circles) or presence (solid circles) of 1 mm calcium. C, CD spectra of recombinant PICK1(D3N) in the absence (open triangles) or presence (solid triangles) of 1 mm calcium. D, Thermal denaturation of recombinant PICK1(D3N) in the absence (open triangles) or presence (solid triangles) of 1 mm calcium. E, Superposition of the CD spectra of recombinant WT PICK1 in the presence of 1 mm calcium (solid circles) with that of PICK1(D3N) in the absence of calcium (open triangles). F, Thermal denaturation of recombinant PICK1(Δ9) in the absence (open diamonds) or presence (solid diamonds) of 1 mm calcium. G, H, Schematics depicting the N-terminal domain mutants PICK1(Δ9) (G) and PICK1(D3N) (H). I, J, AMPAR EPSCs are not affected by replacement of PICK1 with GFP-PICK1(Δ9) [I, shPICK1 plus GFP-PICK1(Δ9), −72.1 ± 8.3 pA; uninfected, −74.3 ± 12.4 pA, n = 16 pairs] or GFP-PICK1(D3N) [J, shPICK1 plus GFP-PICK1(D3N), −64.6 ± 8.3 pA; uninfected, −65.7 ± 8.4 pA, n = 13 pairs]. K, L, LTD is inhibited by replacement of endogenous PICK1 with GFP-PICK1(Δ9) (K) or GFP-PICK1(D3N) (L). K1, L1, Sample experiments demonstrating lack of LTD in a cell expressing shPICK1 plus GFP-PICK1(Δ9) (K1) or a cell expressing shPICK1 plus GFP-PICK1(D3N) (L1). K2, L2, Summary graphs demonstrating block of LTD in shPICK1 plus GFP-PICK1(Δ9)-infected neurons [K2, shPICK1 plus GFP-PICK1(Δ9), 93 ± 5%, n = 16; uninfected, 46 ± 7%, n = 14] or shPICK1 plus GFP-PICK1(D3N)-infected neurons [L2, shPICK1 plus GFP-PICK1(D3N), 102 ± 11%, n = 10; uninfected, 46 ± 7%, n = 14; the same uninfected control graph is shown in both panels because the PICK1(Δ9) and PICK1(D3N) mutants were analyzed in parallel and are therefore compared with a common set of interleaved controls].
To investigate whether the N-terminal region plays a role in the calcium-mediated structural change in PICK1, we generated a mutant in which the first three acidic amino acids in the N-terminal acidic stretch (D4, D6, D8) were mutated to asparagine [termed PICK1(D3N)]. This mutation notably reduced the calcium-dependent shift in CD spectra (Fig. 7 C). Moreover, the denaturation curves obtained for the PICK1(D3N) mutant in the presence and absence of calcium were similar and exhibited only one transition at 38°C (Fig. 7 D), which corresponds to the first transition observed for wild-type PICK1 (Fig. 7 B). These observations suggest that the D3N mutation hinders calcium binding to PICK1 and mimics the structural effect of calcium binding to the wild-type protein. This conclusion is supported by the observation that the decrease in the CD signal at 208 nm observed for WT PICK1 in the presence of calcium was already observed for the PICK1(D3N) mutant even in the absence of calcium (Fig. 7 E). We also generated a mutant form of PICK1 in which the N-terminal acidic stretch was deleted [termed PICK1(Δ9)]. This mutant had a tendency to aggregate in solution, which hindered acquisition of high-quality CD data. However, we were able to obtain thermal denaturation curves that revealed a behavior similar to that of the PICK1(D3N) mutant (Fig. 7 F). Overall, these results support a model whereby the N terminus of PICK1 establishes tertiary interactions with another region in the protein and these interactions are weakened by calcium binding, possibly because of neutralization of the charge within the N terminus.
To determine whether this N-terminal motif is critical for synaptic function, we replaced endogenous PICK1 with either GFP-PICK1(Δ9) (Fig. 7 G) or GFP-PICK1(D3N) (Fig. 7 H). Neither PICK1 mutant had a significant effect on basal AMPAR EPSCs (Fig. 7 I,J). Furthermore, neither mutant was able to rescue the block of LTD caused by knockdown of endogenous PICK1 (Fig. 7 K,L). The effects of these same PICK1 N-terminal mutants on AMPAR trafficking in cultured neurons were also examined. Consistent with the findings that these mutants do not rescue LTD, the PICK1(Δ9) and PICK1(D3N) mutants could not support maintained NMDAR-induced internalization of AMPARs (Fig. 8 A,B).
Figure 8.

Calcium-induced modification of PICK1 structure is essential for NMDAR-induced intracellular accumulation of AMPARs. A, B, Replacement of endogenous PICK1 with GFP-PICK1(Δ9) (A) or GFP-PICK1(D3N) (B) does not rescue the inhibition of maintained NMDAR-induced AMPAR endocytosis because of the knockdown of endogenous PICK1. Representative images (A1, B1) and quantitation (A2, B2) of NMDAR-triggered internal accumulation of GluA2-containing AMPARs in cells expressing GFP, shPICK1, shPICK1 plus GFP-PICK1(Δ9) [A, control, 1 ± 0.06, n = 47; control plus NMDA, 1.7 ± 0.12, n = 46; shPICK1 plus NMDA, 1.09 ± 0.08, n = 39; shPICK1 plus GFP-PICK1(Δ9) plus NMDA, 1.14 ± 0.09, n = 48] or shPICK1 plus GFP-PICK1(D3N) [B, control, 1 ± 0.06, n = 47; control plus NMDA, 1.8 ± 0.11, n = 47; shPICK1 plus NMDA, 1 ± 0.08, n = 38; shPICK1 plus NMDA plus GFP-PICK1(D3N), 1.1 ± 0.08, n = 41].
Our results are consistent with a model whereby calcium influx during the induction of NMDAR-dependent LTD causes a structural change in PICK1 that is essential for its function in preventing internalized AMPARs from returning to the surface after their endocytosis. Mutations that cause loss of negative charge within the N-terminal acidic motif appear to abolish a constitutive electrostatic intramolecular interaction as well as the capacity of calcium to elicit a structural change in PICK1.
To further establish the necessity of calcium-mediated disruption of intramolecular electrostatic interactions for PICK1 function in LTD, we generated a PICK1 mutant in which the sequence of the N-terminal acidic motif was inverted [Fig. 9 A,B, PICK1(FLIP)]. In this mutant, the total charge of the N-terminal region of PICK1 is maintained, but the spatial arrangement of amino acids is disrupted. The CD spectrum of the PICK1(FLIP) mutant was completely insensitive to calcium (Fig. 9 C), as was the thermal denaturation curve of this mutant (Fig. 9 D). Furthermore, the thermal denaturation curve and the CD spectrum were very similar to that of wild-type PICK1 and remained in the wild-type conformation even in the presence of calcium (Fig. 9 E). These results suggest that inversion of the calcium-binding motif disrupts the spatial organization of amino acids necessary for calcium binding while maintaining electrostatic interactions important for PICK1 intramolecular folding.
Figure 9.

The orientation of the N-terminal acidic motif of PICK1 is essential for LTD. A, Schematic depicting the N-terminal mutant PICK1(FLIP). B, Amino acid sequence of the FLIP mutation, demonstrating the inversion within the acidic motif. C–E, CD spectroscopy demonstrates that PICK1(FLIP) is insensitive to calcium and maintains the conformation of the wild-type protein. C, CD spectra of PICK1(FLIP) in the absence (open squares) or presence (solid squares) of 1 mm calcium. D, Thermal denaturation of PICK1(FLIP) in the absence (open squares) or presence (solid squares) of 1 mm calcium. E, Superposition of the CD spectra of recombinant WT PICK1 (open circles) with that of PICK1(FLIP) in the presence of 1 mm calcium (solid squares). F, AMPAR EPSCs are not affected by replacement of PICK1 with GFP-PICK1(FLIP) [shPICK1 plus GFP-PICK1(FLIP), −78.2 ± 9.6 pA; uninfected, −85.1 ± 13.3 pA, n = 11 pairs]. G, NMDAR-dependent LTD is inhibited by replacement of endogenous PICK1 with GFP-PICK1(FLIP). G1, Sample experiment demonstrating lack of LTD in a cell expressing shPICK1 plus GFP-PICK1(FLIP). G2, Summary graphs demonstrating lack of LTD in shPICK1 plus GFP-PICK1(FLIP)-infected neurons compared with interleaved uninfected control neurons [shPICK1 plus GFP-PICK1(FLIP), 101 ± 13%, n = 13; uninfected, 53 ± 6%, n = 17]. H, Replacement of endogenous PICK1 with GFP-PICK1(FLIP) does not rescue the inhibition of maintained NMDAR-induced AMPAR endocytosis because of the knockdown of endogenous PICK1. H1, Representative images. H2, Quantitation of NMDAR-triggered intracellular accumulation of GluA2-containing AMPARs in cells expressing GFP, shPICK1, or shPICK1 plus GFP-PICK1(FLIP) [control, 1 ± 0.07, n = 52; control plus NMDA, 2 ± 0.17, n = 42; shPICK1 plus NMDA, 1.2 ± 0.09, n = 44; shPICK1 plus GFP-PICK1(FLIP), 1.4 ± 0.10, n = 41].
Having established that PICK1(FLIP) does not undergo calcium-dependent structural modifications, we tested its synaptic effects. Replacement of endogenous PICK1 with this mutant had no effect on basal AMPAR EPSCs (Fig. 9 F). Consistent with an essential role for calcium-induced relief of intramolecular tethering for the function of PICK1 in LTD, PICK1(FLIP) could not support NMDAR-dependent LTD (Fig. 9 G) nor could it support the intracellular retention of AMPARs after NMDAR activation (Fig. 9 H). These results provide evidence for a model whereby in the basal state PICK1 adopts an inactive structure containing intramolecular interactions that involve the N-terminal acidic motif. Calcium-mediated disruption of these intramolecular interactions is necessary for PICK1 to promote prolonged intracellular retention of AMPARs during LTD.
Discussion
The acceptance of the central role of activity-dependent trafficking of AMPARs in experience-dependent plasticity (Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Malenka and Bear, 2004; Shepherd and Huganir, 2007) has generated great interest in understanding the molecular mechanisms controlling AMPAR synaptic content during LTP and LTD. PICK1, a unique PDZ and BAR domain-containing protein, has been implicated as one of the key proteins playing an important role in the regulation of the synaptic content of AMPARs during several different forms of synaptic plasticity. Our results suggest that PICK1 is critical for the expression of NMDAR-dependent LTD, but not LTP nor mGluR-dependent LTD in CA1 pyramidal cells. Time course analyses of the trafficking of AMPARs after NMDAR activation revealed that PICK1 is not required for the initial endocytosis of AMPARs but instead plays a critical role in their intracellular retention after internalization. Examination of the effects of mutant forms of PICK1 provided insight into the roles of key PICK1 domains. Most importantly, we found that calcium-induced structural modifications of PICK1 involving its N-terminal acidic motif are required for the function of PICK1 in LTD.
A simplified view of PICK1 is that its BAR domain is critical for its interactions with curved membranes, such as those found in endocytic vesicles, whereas its PDZ domain is critical for direct interactions with AMPARs, in particular GluA2 and 3. To test the importance of the BAR domain, we mutated five BAR domain lysines to glutamic acids to disrupt binding to negatively charged head groups in membrane phosphoinositides (Lu and Ziff, 2005; Jin et al., 2006; Steinberg et al., 2006). Consistent with previous work (Jin et al., 2006), the BAR domain mutant was mislocalized and could not support LTD or the maintained internalization of AMPARs after NMDAR activation. These results indicate that the subcellular localization of PICK1 and its function in LTD are inextricably related and that both require an intact BAR domain.
Surprisingly, mutations in the PDZ domain of PICK1 that disrupt its direct binding to GluA2 (Hanley et al., 2002; Dev et al., 2004; Terashima et al., 2004; Jin et al., 2006; Rocca et al., 2008) did not prevent PICK1 from rescuing either LTD or the maintained NMDAR-triggered endocytosis of AMPARs. This finding suggests that, in contrast to a predominant model (Daw et al., 2000; Kim et al., 2001; Hanley, 2008), direct binding of PICK1 to AMPAR subunits is not required during LTD. Much of the previous work that suggested a critical role for direct PICK1–GluA2/3 interactions in LTD depended on the postsynaptic loading or expression of a peptide that disrupted this interaction. However, the results using such peptides have been inconsistent, with reports of either a block of LTD or a minimal effect on LTD in CA1 pyramidal cells (Daw et al., 2000; Xia et al., 2000; Kim et al., 2001; Duprat et al., 2003; Terashima et al., 2008; Thorsen et al., 2010). The reported effects of such peptides on basal synaptic transmission have been similarly inconsistent (Daw et al., 2000; Kim et al., 2001; Terashima et al., 2004). In the context of these conflicting results, it is important to note that a critical factor in interpreting the results of experiments in which short peptides are used is the difficulty in ruling out the possibility of off-target effects of the peptides on protein–protein interactions that do not involve PICK1.
A major goal of our study was to determine the importance of calcium in regulating the structure and function of PICK1. Previous work suggested that an N-terminal acidic motif was important for binding calcium and that calcium binding to this motif regulated the interaction between PICK1 and GluA2 (Hanley and Henley, 2005). Combined with other results suggesting an intramolecular interaction between the PDZ and BAR domains of PICK1 (Lu and Ziff, 2005; Rocca et al., 2008), these results led to the suggestion that calcium promotes structural changes in PICK1, which contribute to its function in regulating AMPAR trafficking and LTD (Hanley and Henley, 2005; Hanley, 2008). To address this model experimentally, we performed CD spectrometry and thermal denaturation studies on wild-type PICK1 and PICK1 containing mutations within its N-terminal acidic motif. The changes induced by calcium on the CD spectrum and thermal denaturation profile of wild-type PICK1 demonstrate that this protein indeed binds calcium. Moreover, the calcium-induced shift in the second transition of the denaturation curve suggests that calcium binding destabilizes tertiary interactions within PICK1. Mutations that remove the N-terminal acidic motif or decrease its negative charge abolish this second transition and the calcium-induced CD spectral changes, yielding a CD spectrum that resembles that observed for calcium-bound wild-type PICK1. Importantly, these N-terminal mutant forms of PICK1 were incapable of supporting either LTD or maintained NMDAR-dependent internalization of AMPARs. Together, these data suggest that the N-terminal acidic motif is critical for intramolecular electrostatic interactions that maintain the basal conformation of PICK1. An active disruption of this electrostatic interaction by calcium promotes a conformational change in PICK1 that is required for the normal expression of LTD. Our data also indicate that inverting the sequence of the N-terminal motif preserves the intramolecular interactions but abolishes calcium binding and the ability of PICK1 to support LTD, providing additional evidence for a model whereby calcium-mediated disruption of an intramolecular interaction is necessary for the function of PICK1 in LTD.
Previous work has led to several models for the function of PICK1 in the regulation of AMPAR trafficking during synaptic plasticity in hippocampal pyramidal cells. It has been proposed to be an endocytic adaptor that associates GluA2-containing AMPARs with endocytic vesicles, promoting removal of the receptors from the plasma membrane (Daw et al., 2000; Xia et al., 2000; Kim et al., 2001; Perez et al., 2001; Hanley et al., 2002; Hanley and Henley, 2005; Rocca et al., 2008). Alternatively, two different roles for PICK1 in the regulation of AMPAR recycling have been proposed. One model suggests a role for PICK1 in promoting AMPAR recycling and incorporation at the synapse (Lu and Ziff, 2005), whereas the other suggests that PICK1 inhibits the recycling of AMPARs after their endocytosis (Lin and Huganir, 2007). Our results are consistent with this latter model in that we find that PICK1 is not required for the initial endocytosis of AMPARs triggered by NMDAR activation but is required for their prolonged intracellular retention. In line with this idea, PICK1 colocalizes with the early endosome marker protein EEA1 and this colocalization is rapidly enhanced after NMDAR activation (Sossa et al., 2006).
Together, our results are consistent with a relatively simple model (Fig. 10). In the basal state, PICK1 exists in a closed conformation that requires electrostatic interactions involving the N-terminal acidic motif and perhaps the PDZ domain, which has been reported to interact with the BAR domain (Lu and Ziff, 2005; Hanley, 2008; Rocca et al., 2008). During the induction of LTD, calcium influx through activated NMDARs promotes endocytosis of AMPARs in a PICK1-independent fashion while also relieving inhibitory intramolecular interaction in PICK1. Calcium binding to PICK1 might promote an open configuration that exposes the BAR domain and perhaps other portions of the protein, which mediate the recruitment of PICK1 to endosomes. PICK1 might associate with the receptors directly, or through an intermediary, such as GRIP1. This association delays the recycling of recently endocytosed AMPARs to the postsynaptic membrane. Recently, actin polymerization and, specifically, the Arp2/3 complex have been implicated in the regulation of early endosome fission and maturation (Derivery et al., 2009). Furthermore, Arp2/3-dependent actin polymerization at the early endosome has been reported to be essential for recycling of the EGFR (epidermal growth factor receptor) (Lladó et al., 2008). As PICK1 has been implicated in inhibition of Arp2/3-mediated actin polymerization (Rocca et al., 2008), calcium-dependent recruitment of PICK1 to AMPAR-containing early endosomes could delay the actin polymerization-dependent maturation of the endosomes, resulting in prolonged intracellular retention of AMPARs.
Figure 10.

Model for the function of PICK1 in NMDAR-dependent LTD. In the basal state, PICK1 exists in a closed conformation (0) that requires electrostatic interactions involving the N-terminal acidic motif and perhaps the PDZ domain. During the induction of LTD, calcium influx through activated NMDARs promotes endocytosis of AMPARs in a PICK1-independent fashion (1, 2) while also relieving inhibitory intramolecular interaction in PICK1 (3). Calcium binding to PICK1 promotes an open configuration that exposes the BAR domain and perhaps other portions of the protein, which mediate the recruitment of PICK1 to endosomes (4). The association of PICK1 with recently endocytosed AMPARs delays or prevents their recycling to the postsynaptic membrane (5).
All of our conclusions about the role of PICK1 in synaptic plasticity are based on assays performed in hippocampal pyramidal neurons and all electrophysiological measurements were made from CA1 pyramidal cells in slice preparations. This is important to note since different roles have been ascribed to PICK1 in the regulation of AMPAR trafficking and various forms of synaptic plasticity at diverse synapses throughout the brain (Xia et al., 2000; Chung et al., 2003; Gardner et al., 2005; Liu and Cull-Candy, 2005; Steinberg et al., 2006; Jo et al., 2008). Thus, we must assume that the detailed synaptic functions of PICK1 are cell type specific and cannot be generalized. The differences in PICK1 function between distinct cell types points out the importance of elucidating the detailed molecular mechanisms of different forms of synaptic plasticity. The knowledge garnered from such studies should allow manipulation of plasticity at synapses within defined circuits, manipulations that in turn will facilitate our understanding of how activity-dependent neural circuit modifications mediate experience-dependent plasticity.
Footnotes
This work was supported by Welch Foundation Grant I-1304 (J.R.), National Institutes of Health Grants MH63394 (R.C.M.) and NS37200 (J.R.), and the Morby Charitable Fund for Alzheimer's Research (R.C.M.). A.C. gratefully acknowledges the support of the International Human Frontier Science Program Organization. We are grateful to Arundhati Sengupta Ghosh for technical assistance. We thank Tom Sudhof for helpful suggestions and members of the Malenka Laboratory for critical reading of this manuscript.
References
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biou V, Bhattacharyya S, Malenka RC. Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc Natl Acad Sci U S A. 2008;105:1038–1043. doi: 10.1073/pnas.0711412105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Nicoll RA, Malenka RC. Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol. 1998;80:2797–2800. doi: 10.1152/jn.1998.80.5.2797. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Lüscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, von Zastrow M, Malenka RC. Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci. 2001;2:315–324. doi: 10.1038/35072500. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Citri A, Soler-Llavina G, Bhattacharyya S, Malenka RC. N-methyl-d-aspartate receptor- and metabotropic glutamate receptor-dependent long-term depression are differentially regulated by the ubiquitin-proteasome system. Eur J Neurosci. 2009;30:1443–1450. doi: 10.1111/j.1460-9568.2009.06950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- Derivery E, Sousa C, Gautier JJ, Lombard B, Loew D, Gautreau A. The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev Cell. 2009;17:712–723. doi: 10.1016/j.devcel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Dev KK, Nishimune A, Henley JM, Nakanishi S. The protein kinase C alpha binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology. 1999;38:635–644. doi: 10.1016/s0028-3908(98)00230-5. [DOI] [PubMed] [Google Scholar]
- Dev KK, Nakanishi S, Henley JM. The PDZ domain of PICK1 differentially accepts protein kinase C-alpha and GluR2 as interacting ligands. J Biol Chem. 2004;279:41393–41397. doi: 10.1074/jbc.M404499200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprat F, Daw M, Lim W, Collingridge G, Isaac J. GluR2 protein-protein interactions and the regulation of AMPA receptors during synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:715–720. doi: 10.1098/rstb.2002.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG, Khatri L, Hanson PI, Ziff EB. NSF ATPase and alpha-/beta-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron. 2002;34:53–67. doi: 10.1016/s0896-6273(02)00638-4. [DOI] [PubMed] [Google Scholar]
- Jin W, Ge WP, Xu J, Cao M, Peng L, Yung W, Liao D, Duan S, Zhang M, Xia J. Lipid binding regulates synaptic targeting of PICK1, AMPA receptor trafficking, and synaptic plasticity. J Neurosci. 2006;26:2380–2390. doi: 10.1523/JNEUROSCI.3503-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Heon S, Kim MJ, Son GH, Park Y, Henley JM, Weiss JL, Sheng M, Collingridge GL, Cho K. Metabotropic glutamate receptor-mediated LTD involves two interacting Ca2+ sensors, NCS-1 and PICK1. Neuron. 2008;60:1095–1111. doi: 10.1016/j.neuron.2008.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joch M, Ase AR, Chen CX, MacDonald PA, Kontogiannea M, Corera AT, Brice A, Séguéla P, Fon EA. Parkin-mediated monoubiquitination of the PDZ protein PICK1 regulates the activity of acid-sensing ion channels. Mol Biol Cell. 2007;18:3105–3118. doi: 10.1091/mbc.E05-11-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci U S A. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG. Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat Neurosci. 2005;8:768–775. doi: 10.1038/nn1468. [DOI] [PubMed] [Google Scholar]
- Lladó A, Timpson P, Vilà de Muga S, Moretó J, Pol A, Grewal T, Daly RJ, Enrich C, Tebar F. Protein kinase Cdelta and calmodulin regulate epidermal growth factor receptor recycling from early endosomes through Arp2/3 complex and cortactin. Mol Biol Cell. 2008;19:17–29. doi: 10.1091/mbc.E07-05-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1 targets activated protein kinase Cα to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Regalado MP, Terry-Lorenzo RT, Waites CL, Garner CC, Malenka RC. Transsynaptic signaling by postsynaptic synapse-associated protein 97. J Neurosci. 2006;26:2343–2357. doi: 10.1523/JNEUROSCI.5247-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Sossa KG, Court BL, Carroll RC. NMDA receptors mediate calcium-dependent, bidirectional changes in dendritic PICK1 clustering. Mol Cell Neurosci. 2006;31:574–585. doi: 10.1016/j.mcn.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J Biol Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JT. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci. 2004;24:5381–5390. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsen TS, Madsen KL, Rebola N, Rathje M, Anggono V, Bach A, Moreira IS, Stuhr-Hansen N, Dyhring T, Peters D, Beuming T, Huganir R, Weinstein H, Mulle C, Strømgaard K, Rønn LCB, Gether U. Identification of a small-molecule inhibitor of the PICK1 PDZ domain that inhibits hippocampal LTP and LTD. Proc Natl Acad Sci U S A. 2010;107:413–418. doi: 10.1073/pnas.0902225107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]