

Abstract
An aldol-based ‘build/couple/pair’ (B/C/P) strategy was applied to generate a collection of stereochemically and skeletally diverse small molecules. In the build phase, a series of asymmetric syn- and anti- aldol reactions were performed to produce four stereoisomers of a Boc protected γ-amino acid. In addition both stereoisomers of O-PMB-protected alaninol were generated to provide a chiral amine coupling partner. In the couple step, eight stereoisomeric amides were synthesized by coupling the chiral acid and amine building blocks. The amides were subsequently reduced to generate the corresponding secondary amines. In the pair phase, three different reactions were employed to enable intramolecular ring-forming processes, namely: nucleophilic aromatic substitution (SNAr), Huisgen [3+2] cycloaddition and ring-closing metathesis (RCM). Despite some stereochemical dependencies, the ring-forming reactions were optimized to proceed with good to excellent yields providing a variety of skeletons ranging in size from 8- to 14-membered rings. Scaffolds resulting from the RCM pairing reaction were diversified on solid-phase to yield a 14,400-membered library of macrolactams. Screening of this library led to the discovery of a novel class of histone deacetylase inhibitors, which display mixed enzyme inhibition and led to increased levels of acetylation in a primary mouse neuron culture. The development of stereo-structure/activity relationships (SSAR) was made possible by screening all 16 stereoisomers of the macrolactams produced through the aldol-based B/C/P strategy.
Introduction
The shift to high-throughput synthetic practices over the past decade has given rise to small-molecule screening collections that are rich in achiral, aromatic compounds. This is perhaps due in part to the abundant methods available for sp2-couplings that are particularly amenable to parallel synthesis. An analysis of a publicly available screening collection, the NIH Molecular Libraries-Small Molecule Repository (MLSMR), reveals that the compounds on average are less complex (as measured by sp3-content) than both natural products and drugs (vide infra). Recent evidence demonstrates, however, the importance of structural complexity and the presence of stereogenic centers, in transitioning compounds from discovery to the clinic to drugs.1 Natural products have served to augment screening collections (including the MLSMR) as a source of structural complexity and have indeed led to the successful development of drugs.2 Despite their utility in drug discovery, a major limitation of natural products has been the challenges associated with their formation and an inability to readily and efficiently generate analogs. We sought to address this dichotomy by designing a small-molecule screening collection that combines the structural complexity of natural products and the efficiency of high-throughput synthesis. Indeed previous efforts in the diversity-oriented synthesis (DOS) of natural product-inspired libraries have yielded useful biological probes and potential leads for drug discovery.3
In addition to targeting compounds of increased structural complexity, we hypothesized that the ability to systematically access the complete matrix of stereoisomers for a collection of chiral, sp3-rich compounds would be especially advantageous in facilitating both upfront and downstream discovery efforts. In particular we reasoned that access to such a collection would enable the rapid development of stereo-structure/activity relationships (SSAR)4 providing valuable information for the prioritization and optimization of hit compounds. In order to create a compound collection with ‘built-in’ SSAR, we employed a three-phase DOS strategy called ‘build/couple/pair’ (B/C/P).5 The initial phase (the build phase) entails the asymmetric synthesis of chiral building blocks with functionality suitable for subsequent reactions. The chiral building blocks are then coupled together (couple phase) through a variety of intermolecular bond forming reactions to provide all possible stereochemical combinations. Intramolecular cyclizations (the pair phase) then join functional groups in many pairwise combinations to create a variety of ring systems. In this fashion, the complete stereochemical matrix of a diverse set of small molecules can be created in a modular fashion, lending itself to the generation of powerful SSAR.
In the present study we chose to focus on synthetic pathways that would provide access to medium-and large-sized ring systems, given the presence of these frameworks in biologically important natural products. Medium-sized rings in particular are often underrepresented in screening collections, perhaps due to the limited methods for their synthesis. While synthetic approaches to five- and six-membered rings are common via cyclization and cycloaddition reactions, strategies to form 7- to 9-membered rings are often inhibited due to entropic factors and transannular interactions, which can pose unique challenges for the synthesis of such molecular frameworks.6 Meanwhile, macrocycles continue to be of interest in drug discovery in light of their proven therapeutic value. Macrocycles are appealing for drug discovery as they provide diverse functionality in a conformationally pre-organized ring structure, often resulting in high affinity and selectivity for protein targets. The majority of current macrocyclic drugs are derived from natural sources, and several synthetic macrocycles are now in preclinical and clinical development.7 We believe the modular nature of the B/C/P strategy is ideally suited to the systematic asymmetric synthesis of medium- and large-ring systems.
The B/C/P strategy implemented in this work is depicted in Figure 1. In the build phase we chose asymmetric propionate aldol reactions to selectively generate four stereoisomers of a chiral γ-amino acid building block (1). We employed both D- and L-alaninol for the second chiral building block (2). In the couple step the aldol-derived acids (1) and the PMB-protected alaninol (2) are reacted to give an amide, which in turn was reduced to render a secondary amine. The complete matrix of all possible stereoisomers of a linear amine (3) was achieved using this approach. In the pair phase the linear amine template arising from the previous coupling step was further derivatized and paired through nucleophilic aromatic substitution (SNAr), Huisgen [3+2] cycloaddition or ring-closing metathesis (RCM), resulting in a diverse collection of skeletal frameworks (4-8), each with a complete set of stereoisomers. The end products, ranging in size from 8- to 14-membered rings, retain functional handles that can be utilized for solid-phase library synthesis and analog development, thus maximizing the relevance of this pathway for expansion to a large screening collection.
Figure 1.

An aldol-based B/C/P strategy for generating macrocycles and medium-sized rings.
In order to visualize the structural diversity obtained by the three pairing strategies, a molecular shape based diversity analysis derived from normalized principal moments of inertia (PMI) ratios8 was applied (Figure 2). This simplistic representation categorizes molecular shape into three distinct topologies: rod-, disc- and sphere-like character. The minimum energy conformer required for the analysis was generated using stochastic search, a variant of Random Incremental Pulse Search (RIPS) method9 as implemented in MOE molecular modeling software program.10 The parameters for stochastic search were optimized with respect to convergence of the lowest energy conformer of every enantiomeric pair.11 The three principal moments of inertia were calculated in MOE and normalized PMI ratios for the triangular scatter plot12 were generated by an in-house Pipeline Pilot script13 as described by Sauer.7 The distinct shape space occupied by each ring system's conformational ensemble, with energy ≤3 kcal/mol from the global minimum, shows a clear preference for each scaffold type within the triangle. Since compounds do not necessarily bind to proteins in their lowest energy conformation14 we chose to plot conformations ≤3 kcal/mol for each diastereomer. Overall the RCM pairing strategy leads to scaffolds with the most sphere-like molecular shape, while the SNAr-derived scaffolds are more disc-like in nature. The appreciable difference in shape between the 1,4- and 1,5-macrocyclic triazoles is especially intriguing, as it is the result of a simple change in atom connectivity. As evident by this qualitative analysis a wide coverage of molecular shape space can be accessed from this aldol-based B/C/P strategy.
Figure 2.

Scatter plot representing molecular shape diversity of the bare library scaffolds. Normalized PMI ratios are plotted in a triangular graph to compare shape space covered by skeletons derived from the three pairing reactions: SNAr (8- and 9-membered rings), Click (1,4- and 1,5-triazoles) and RCM. The triangle is defined by its three corners, which are represented as rod-, disc- and sphere-like shapes. Representative examples of molecules exhibiting these extremes are shown. Minimum energy conformers ≤3 kcal/mol from the global minimum are plotted on the graph for all diastereomers.
Results and Discussion
1. Build phase
The asymmetric aldol reaction has been of enormous utility in the synthesis of complex natural products.15 We chose to employ complementary syn- and anti-aldol technologies16,17 for the preparation of the four stereoisomers of γ-amino acid 1 containing three useful handles (an acid, alcohol and amine) for generating skeletal and appendage diversity. The TBS- and Boc-protecting groups were selected based on their compatibility with a variety of downstream transformations. As we required substantial quantities (~500 g) of the aldol intermediates for library production and future follow-up studies, significant effort went into developing a practical and robust method for their large-scale preparation.18 As outlined in Scheme 1, the syn-aldol reaction of aldehyde 9 with Evans oxazolidinone (−)-10a was successful for the generation of (−)-11a using dibutylboron triflate as a solution in toluene along with dichloromethane as a co-solvent (1:1). The reaction proceeded at 0 °C to provide aldol adduct (−)-11a with high diastereoselectivity (>99:1) as measured by chiral SFC. Direct treatment of (−)-11a with LiOH and hydrogen peroxide at ambient temperature in MeOH/THF provided amino acid (−)-12a in ‘one-pot’ as a single stereoisomer in excellent yield (96%).19 This protocol has been successfully applied to routinely access >90 g20 batches of either (+)-12a or (−)-12a as crystalline white solids. Protection of the secondary alcohol as the TBS-ether was achieved via treatment with TBSOTf to afford (−)-1a.
Scheme 1.

Syn- and Anti-Aldol Reaction to Provide Four Stereoisomers of γ-amino Acid 1
For the preparation of the γ-amino acid (1b) we adapted the ephedrine-based protocols reported by Abiko and co-workers for the synthesis of 1,2-anti-selective aldol products.17 Enolization of (−)-10b was carried out using freshly prepared dicyclohexylboron triflate in dichloromethane at °78 °C to provide (−)-11b upon reaction with aldehyde 9 with a diastereomeric ratio of 90:10. Hydrolysis of the ephedrine-based auxiliary was achieved using sodium hydroxide/hydrogen peroxide in a mixed tert-butanol/methanol solvent system to provide (−)-12b in 99% yield over two steps.21 This procedure has been successfully implemented for the preparation of >150 g20 batches of either (−)-12b or (+)-12b as colorless oils. Similar to the syn-aldol products, protection of the secondary alcohol as the TBS-ether was accomplished via treatment with TBSOTf to give compound (−)-1b in excellent yield.
Finally, completing the build portion of the B/C/P approach required the synthesis of both enantiomers of protected alaninol 2 (Figure 2). This could be accomplished through a chemoselective O-PMB-protection of D- and L-alaninol.22 Analogous to the aldol reaction, this procedure was executed on large scale (>100 g) providing both enantiomers of 2.
2. Couple phase
We next focused our attention on the union of chiral synthons 1 and 2 en route to the formation of the eight stereoisomers of linear template 3 (Table 1). To achieve this we developed a two-step reaction sequence, involving a straightforward amide coupling followed by borane-mediated reduction.23 The coupling of PMB-protected alaninol (−)-2 or (+)-2 with the four TBS-protected chiral γ-amino acids 1a-1b was performed with PyBOP to provide all stereochemical permutations of amide 13 in good to excellent yield (71-96%).24 Subsequent reduction of amide 13 to amine 3 could be achieved using borane-dimethylsulfide (BH3-DMS) at 65 °C,25 followed by quenching with Rochelle's salt in MeOH at 80 °C.26,27 This general procedure was successfully applied on large scale to provide up to 150 grams of amine 3 after simple aqueous work-up with good reproducibility, yield and purity for all stereoisomers (88-98%).
Table 1.
Preparation of the Complete Matrix of Stereoisomers of Amine 3
 | |||||||
|---|---|---|---|---|---|---|---|
| coupling partners | product | % yielda | coupling partners | product | % yielda | ||
| (−)-1a | (−)-2 |
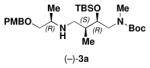
|
95 | (+)-1a | (+)-2 |
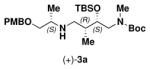
|
98 |
| ″ | (+)-2 |

|
91 | ″ | (−)-2 |
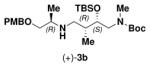
|
88 |
| (−)-1b | (−)-2 |
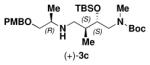
|
88 | (+)-1b | (+)-2 |

|
92 |
| ″ | (+)-2 |
|
98 | ″ | (−)-2 |

|
97 |
Isolated yield of amine 3 after aqueous work-up. Amide 13 was purified by silica gel chromatography prior to borane reduction.
3. Pair phase
Having developed a robust process for the synthesis of all eight stereoisomers of linear amine 3, we turned our attention to investigating suitable functional group pairing reactions28 for generating skeletal diversity. We aimed to develop methods for the synthesis of medium-sized rings and macrocycles from linear amine 3 by employing robust, high yielding reactions that were operationally simple, as well as regioselective. We describe below the development of three pairing reactions: (1) SNAr, (2) Huisgen [3+2] cycloaddition and (3) RCM, to access a variety of skeletons ranging in size from 8- to 14-membered rings.
3.1 Nucleophilic aromatic substitution
The intramolecular SNAr reaction has been a powerful tool for the synthesis of complex molecules involving the formation of biaryl ethers29 and to a lesser degree aryl-alkyl ethers.30 We hoped to utilize this cycloetherification strategy to construct eight- and nine-membered ring systems by forming an aryl-alkyl ether using the C-6 secondary alcohol of linear amine 3 as an internal nucleophile.
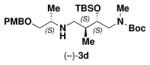
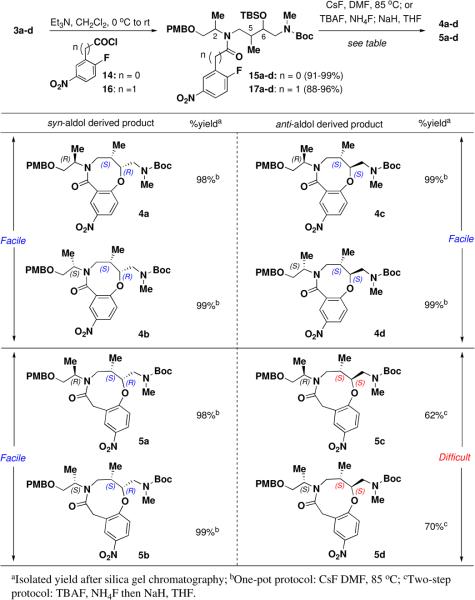
We first chose to explore the eight-membered ring formation (Table 2, 4a-d) using a ‘one-pot’ TBS deprotection/cyclization sequence with various sources of basic fluoride (e.g., TBAF, CsF). Linear amine 3a-d was first acylated with 2-fluoro-5-nitrobenzoyl chloride 1431 to afford tertiary amides 15a-d. Upon treatment with CsF in DMF at 85 °C, substrates 15a-d cyclized smoothly to afford the desired 8-membered rings 4a-d in nearly quantitative yield (>98%). Although we had anticipated encountering dimerization issues during the cycloetherification step,30d the reaction was extremely facile and could be run at concentrations as high as 0.3 M with no apparent dimer formation. TBAF was also successfully employed for this reaction but the use of CsF was preferred given the ease of purification involving only aqueous work-up with no need for chromatography. Notably, the success of the 8-membered ring cyclization was not influenced by the stereochemistry of the linear substrate and could be performed on >20 g scale for all stereoisomers (only one enantiomeric series shown).
Table 2.
Synthesis of 8- and 9-Membered Rings via SNAr
|
Unlike the eight-membered ring cyclizations, we did observe a stereochemical dependence for the formation of nine-membered rings (5a-5d). Linear amine 3a-d was first acylated with 2-fluoro-5-nitrophenyl acetyl chloride 16.31 The ‘one-pot’ CsF-promoted SNAr reaction proceeded smoothly for the syn-aldol derived substrates (2R,5S,6R)-17a and (2S,5S,6R)-17b to afford compounds 5a and 5b in high yield (>98%). In contrast, ring closure for the anti-aldol derived substrates (2R,5S,6S)-17c and (2S,5S,6S)-17d required a two-step protocol. Removal of the TBS group with buffered TBAF, followed by treatment with sodium hydride in THF afforded the desired anti-aldol derived ring systems (5c and 5d) in ~60-70% yield. Similar to the eight-membered ring synthesis, these one- or two-step cyclizations were executed on >20 g scale. The structure of one diastereomer (5c) in the nine-membered ring scaffold series was established by single crystal x-ray diffraction. (See Supporting Information.)
3.2 Huisgen [3+2] cycloaddition
Catalyst-controlled regioselective ligation of azides and alkynes to yield triazoles has emerged as an extremely useful reaction in both organic synthesis and chemical biology.32 Recent development of Ru-catalyzed azide-alkyne 1,3-dipolar cycloaddition (RuAAC) to form 1,5-triazoles33 complements the reliable Cu-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) to form 1,4-triazoles,32c further enhancing the utility of this cycloaddition reaction. Encouraged by our recent success in effecting regioselective macrocyclization in a divergent fashion under catalyst control,34 we undertook a study to evaluate azide-alkyne cycloaddition as a pairing reaction using a more structurally and stereochemically complex system. Preliminary studies indicated azido-acid 18 (Table 3) could be a suitable azide component, while terminal propargyl ether could serve as the alkyne unit for our macrocyclic pairing reaction to afford either 12- or 13-membered rings.
Table 3.
Regioselective Synthesis of Macrocyclic Triazoles via Catalyst-control

|
Acylation of amines 3a-d with azido acid 18 (Table 3) and subsequent TBAF-mediated TBS deprotection gave rise to the desired alcohols in good yield (72-93%). Initially, introduction of the alkyne component for the cycloaddition proved difficult. Propargylation under standard conditions using NaH in DMF resulted in incomplete conversion to alkyne 19a-d, even with a large excess of propargyl bromide. Formation of an oxazolidinone by-product (resulting from the reaction of the alkoxide intermediate with the Boc group) further complicated matters.35 Biphasic propargylation conditions using aqueous NaOH in DCM with a phase transfer catalyst also proved ineffective.36 Further optimization led to the use of NaHMDS in a mixed solvent system of THF:DMF (6:1) to affect the propargylation in high yield (>91%) for all stereoisomers.
With macrocyclization precursors 19a-d in hand, we began exploring the RuAAC to form a 12-membered ring. After screening all known catalysts,37 [Cp*RuCl]4 was found to best effect macrocyclization regioselectively on all four diastereoisomers. The macrocyclization of the syn-aldol derived substrates (2R,5S,6R)-19a and (2S,5S,6R)-19b cleanly afforded triazoles 6a and 6b in good yield (78 and 75%), while the anti-aldol derived substrates (2R,5S,6S)-19c and (2S,5S,6S)-19d yielded triazoles 6c and 6d in modest yields (59 and 56%) due to competing dimer formation. The Ru-catalyzed cyclization reactions were reliable and could be routinely performed on 5-10 g batches to yield the macrocyclic triazoles.
Next, we began to evaluate intramolecular CuAAC to generate the 13-membered macrocyclic triazoles (7). We selected a polystyrene bound copper catalyst, PS-CuPF6, to effect the macrocyclization based on our recent success using this reagent to achieve pseudodilution conditions.34,38 Use of the solid-phase catalyst limited the extent of dimer formation to achieve macrocyclization on all four diastereomers of azido-alkyne 19. The anti-aldol derived substrates (2R,5S,6S)-19c and (2S,5S,6S)-19d readily participated in the intramolecular CuAAC reaction to afford triazoles 7c and 7d in 76 and 78% yield, with no dimer formation observed. In contrast, the macrocyclization reaction of syn-aldol derived substrates (2R,5S,6R)-19a and (2S,5S,6R)-19b proved to be more challenging. The desired products (7a and 7b) were isolated in modest yields (57 and 58%) along with 5-10% dimer formation. With respect to stereochemistry, this difference in reactivity for the Cu-catalyzed cyclization is the reverse of what was observed for the Ru-catalyzed reaction in that the syn-derived substrates cyclize more readily than the anti-substrates. Over the course of our study, we were able to establish the structure of 7a by single crystal x-ray diffraction. (See Supporting Information.)
3.3 Ring-closing metathesis
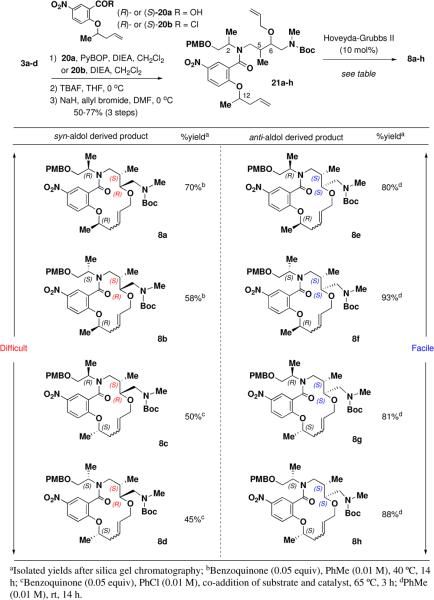
Ring-closing metathesis (RCM) is a reliable method for the construction of small, medium and large rings in both target- and diversity-oriented synthesis.39 Encouraged by recent reports that utilize macrocyclization by RCM on process scale,40 we decided to investigate the utility of RCM as a pairing reaction with a suitable derivative of linear amine 3. After preliminary evaluation of several different ring sizes, we found nitrobenzoic acid (chloride) 20 (Table 4) to be an appropriate fragment for the RCM pairing reaction to enable the formation of a 14-membered ring. Linear amines 3a-d were first acylated with either enantiomer of 20a or 20b41 and the resulting amides were subjected to TBS deprotection and allylation to give RCM precursors 21a-h in three steps and 50-77% overall yield. The choice of solvent proved to be critical for the success of the allylation. When THF was used as a solvent, we observed oxazolidinone formation similar to problems encountered during the preparation of alkyne 19.35 The formation of this by-product was significantly suppressed by switching to DMF and adding allyl bromide prior to the introduction of sodium hydride to the reaction mixture.
Table 4.
Synthesis of 14-Membered Macrolactams by Ring-Closing Metathesis
|
The outcome of the RCM macrocyclization reaction broadly falls into two categories and is highly dependent on the stereochemistry at C-5 and C-6 and to a lesser extent on C-12. Screening of several catalysts revealed the second-generation Hoveyda-Grubbs catalyst to be optimal for providing the desired 14-membered rings (8a-d, Table 4) as a mixture of E/Z isomers.42,43 The RCM reaction of the syn-aldol derived substrates (21a-d) proved challenging due to terminal olefin isomerization and dimerization. Cyclization of substrates 21a and 21b could be effected in 70 and 58% yield to provide 8a and 8b using 1,4-benzoquinone as an additive to prevent olefin isomerization.44,45 Ring closure of 21c and 21d was most difficult requiring slow co-addition of the substrate and catalyst at elevated temperatures (65 °C) to minimize dimerization in addition to the use of benzoquinone to prevent isomerization. Under these optimized reaction conditions the desired products (8c and 8d) were isolated in modest yield (50 and 45%). Meanwhile, the anti-aldol derived substrates (21e-h) participated in the RCM reaction with high efficiency. These four reactions were conducted at room temperature and the products (8e-h) were isolated in 80-93% yield with negligible dimerization and isomerization for all substrates. We found all the RCM reactions to be scalable up to 10-50 g without any loss in efficiency compared to milligram scale reactions. During the course of our studies we were able to confirm the structure for the E-isomer of ent-8b by single crystal X-ray diffraction. (See Supporting Information).
4. Synthesis of library scaffolds
Having demonstrated the successful cyclization of the linear template to yield three unique skeletons, we investigated the conversion of these products to compounds that are suitable for solid-phase library synthesis. Namely, PMB removal was required to provide a primary alcohol for loading onto solid-support, while adjustment of the nitrogen protecting groups was needed to ensure compatibility with our silicon-based linker once immobilized onto solid-support.46,47 To illustrate the necessary protecting group manipulations, we selected one diastereomer from each pairing reaction to advance to the final library scaffold. As shown in Scheme 2, the products of SNAr (4c) and RCM (8a) were both subjected to hydrogenation to provide anilines 22 and 23, which were protected as the Fmoc carbamates (24 and 25) under standard conditions. As expected, hydrogenation of the RCM E/Z product mixture resulted in concomitant reduction of the double bond to provide a single isomer suitable for NMR characterization at the aniline stage (23).
Scheme 2.

a Protecting Group Modifications to Yield Final Library Scaffolds
Initial attempts to remove the Boc and PMB protecting groups simultaneously under acidic conditions (HCl or TFA) led to inconsistent results among each scaffold series. While the desired SNAr product (29) could be obtained from 24 in moderate yields, deprotection of the RCM macrocycle (25) under acidic conditions led to a complex mixture of products. Meanwhile, t-butyl cation addition was observed during TFA-mediated Boc removal for the 1,4-macrocyclic triazole (7a). Surprisingly, this side reaction could not be suppressed by the addition of various scavengers, such as Et3SiH or thioanisole. In light of these difficulties, an alternate deprotection sequence was pursued for all cores. We discovered that a two-step protocol using TBSOTf followed by HF-pyridine could be employed for the chemoselective removal of the Boc group via a silyl carbamate intermediate to afford the desired amine.48 Protection of the resulting amines as the Alloc (26 and 27) or Fmoc (28) carbamates proceeded smoothly. Finally, PMB removal was achieved using DDQ to afford library scaffolds (29-31) in good yield and high purity.
6. Solid-phase library synthesis
The 48 scaffolds generated using the aldol-based B/C/P strategy have been utilized for the production of >30,000 compounds using SynPhase™ Lantern technology.49 As an example, the solid-phase diversification of the 16 RCM-based scaffolds (30a-h and ent-30a-h)50 to yield a library of 14,400 compounds is described below (Scheme 3). Loading onto solid-support was achieved for all stereoisomers of 30 via activation of silicon-functionalized Lanterns47 with TfOH followed by reaction with the scaffold (1.2 equiv) in the presence of 2,6-lutidine (target loading level = 15 μmol/Lantern). (Lanterns were equipped with radio frequency transponders to enable tracking and sorting of library members.) Macrolactam scaffold 30 contains two handles for the introduction of appendage diversity: a masked aniline and secondary amine moiety, both suitable for reaction with various electrophiles. The first diversity site, an aniline, was exposed under standard conditions required for Fmoc removal (20% piperidine in DMF) and then reacted with a total of 25 building blocks including sulfonyl chlorides, isocyanates, acids and formaldehyde.51 Next, the Alloc group was removed from the secondary amine using Pd(PPh3)4 in the presence of excess barbaturic acid to reveal the second diversity site.52 Following amine capping with a variety of sulfonyl chlorides, isocyanates, acids and aldehydes, cleavage from the Lantern was achieved by treatment with 15% HF/pyridine in THF to afford a total of 14,400 products with an average yield of 11.8 μmol.53 All library products were analyzed by UPLC and compound purity was assessed by UV detection at 210 nm. The average purity of the library was 81%, with 83% of the library being greater than 75% pure. In general all building blocks performed well during the library production with the exception of certain acids introduced at R2. When used in combination with isocyanates at R1, impurities resulting from bis-acylation were sometimes encountered.
Scheme 3.

Solid-Phase Synthesis of RCM Library
7. Analysis of structural complexity
Compounds resulting from the RCM library, as well as the SNAr and Click pathways, were analyzed to assess their structural complexity as measured by saturation and compared to other compound collections, including natural products, drugs and the MLSMR screening collection. Saturation can be calculated using various descriptors including the number of aryl, double and single bonds, or the number of aromatic rings.54 For our study we employed the method reported by Lovering et al. in which saturation is defined by the fraction sp3 (Fsp3) where Fsp3 = (number of sp3 hybridized carbons/total carbon count).1 The results of this analysis are shown in Figure 3. The mean Fsp3 value of aldol-derived DOS compounds (DOS-all) is comparable to both natural products (NPs) and drugs with a mean Fsp3 value of 0.49 (compared to 0.48 for NPs and drugs). Meanwhile the complexity of the RCM library alone is slightly higher at 0.54. In contrast, the mean Fsp3 value for the MLSMR collection is significantly lower at 0.30. Given that the MLSMR collection is largely comprised of commercial vendor libraries it is not surprising that the overall complexity of this collection would be lower.
Figure 3.

Mean Fsp3 values for various compound collections where Fsp3 = (number of sp3 hybridized carbons/total carbon count). DOS-RCM = RCM library compounds (14,400 compounds); DOS-all = All DOS compounds derived from the SNAr, Click and RCM pathways (30,099 compounds); NPs = Natural products and natural product derivatives retrieved from GVK BIO database (2,265 compounds); Drugs = Small-molecule drugs retrieved from DrugBank (4,239 compounds); MLSMR = Compounds contained in NIH's Molecular Libraries Small-Molecule Repository (307,983 compounds). Mean Fsp3 values were determined according to the method of Lovering et al.1
7. Identification of histone deacetylase inhibitors from a biochemical screen
Screening of the RCM-derived library of macrolactams in a variety of biochemical and cell-based screens led to the discovery of several low micromolar histone deacetylase (HDAC) inhibitors. HDACs remove the acetyl group from the ε-amino group of lysine side chains within the N-terminal tails of histones (and other non-histone substrates). In doing so, HDACs favor the closed, repressive state of chromatin and the further recruitment of other transcriptional co-repressors.55 There are a total of 18 HDAC enzymes in the mammalian genome.56 These enzymes can be divided into four classes, including class I, II, III and IV, based on primary sequence homology to their yeast counterparts. Class I, II and IV HDACs are the zinc-dependent hydrolases. Class I HDACs include 1, 2, 3, and 8 all of which have been well documented to exert deacetylase activity on histone substrates as well as a more limited set of non-histone substrates. Class II HDACs can be divided into class IIa members, which include HDAC 4, 5, 7 and 9, and class IIb members, which include HDAC 6 and 10. Class III HDACs (sirtuins; SIRT 1-7) are NAD(+)-dependent enzymes, which exhibit a non-overlapping sensitivity to most structural classes of inhibitors.55b
To date, most published HDAC inhibitors belong to a limited number of chemical classes: carboxylic acids (e.g. valproic acid), hydroxamic acids (SAHA; Vorinostat), thiols (FK228; Istodax) and 2-amino benzamides (e.g. MS-275; Entinostat). Additional classes include the keto epoxides (e.g. trapoxin), which were instrumental in the discovery of HDACs, ethyl ketones (apicidin), ortho-hydroxy benzamides, trifluoromethyl ketones, and α-ketoamides. Based on the structures of these inhibitors, a general model for HDAC inhibition has been put forth consisting of “cap-linker-chelator” functionalities, which is supported by structural models of HDACs and bound inhibitors and numerous structure-activity relationship studies.57 While several class specific HDAC inhibitors exist, such as the thiophene-substituted benzamides that inhibit only class I HDAC1 and HDAC2 or the recently described biphenyl hydroxamates that are selective for class IIa HDACs,58 none of these inhibitors are HDAC isoform specific limiting the precision of the functional conclusions that can be drawn about the cellular effects and therapeutic potential of HDAC inhibition. Additionally, all of the known classes of inhibitors are substrate competitive inhibitors that are presumably chelating the active site zinc as a primary component to their binding activity. Given the highly conserved nature of the HDAC active site between different class members, the discovery of HDAC inhibitors that are non-competitive in nature or that bind to HDACs with novel binding modes may provide further insight into the regulation and function of HDACs as well as attractive starting points for obtaining isoform selectivity. Beyond applications in oncology where HDAC inhibitors are the subject of both clinical and basic research, we have recently shown that HDAC2, a class I HDAC, is directly implicated in learning and memory.59
To test for HDAC2 inhibitory activity we used a high-throughput coupled biochemical assay. The substrate for HDAC2 was a tripeptide based on histone H4 Lys12 with an acetylated lysine and a C-terminal aminomethyl coumarin (AMC) derivative (Ac)Leu-Gly-Lys-acetyl-AMC).60 Upon incubation with substrate and trypsin, HDAC2 removes the acetyl group allowing for trypsin to cleave the tripeptide substrate, liberating the AMC fluorophore whose fluorescence increases significantly. The assay was conducted in a kinetic mode using 4.6 μM substrate (the concentration of substrate that produces half-maximal velocity (Km) for HDAC2), 2.2 nM HDAC2, 150 nM trypsin, and 17 μM compound.61 A total of 22,506 DOS compounds were tested in the HDAC2 biochemical screen, including 1,604 RCM library compounds derived from scaffolds (2R,5S,6R,12R)-30a and (2R,5S,6S,12R)-30e, which vary only with respect to stereochemistry at C-6.62 Of all the DOS compounds tested, the RCM macrolactams produced the most interesting hits. The primary screening results for the RCM compounds in the HDAC2 assay are shown in Figure 4. Using a hit cut-off of 24% inhibition, 32 hits were identified (2.0% hit rate). These 32 compounds were retested in 8-point twofold dose-response (32 μM – 250 nM) and 26 had confirmed activity (81% retest rate). Interestingly, the most potent compounds with confirmed activity were the products of reductive alkylation with AL11 (piperonaldehyde) at R2. Meanwhile, the nature of the building block at R1 appeared to be less critical for HDAC inhibition. All sulfonyl chlorides (SC1-8) and formaldehyde (AL1) resulted in confirmed activity with IC50 values below 50 M. (The one urea derivative (R1 = IS7) was inactive upon retest). With respect to stereochemistry only one diastereomer was preferred in favor of the syn-aldol derived macrocycle (2R,5S,6R,12R)-30a.
Figure 4.

Primary screening data displayed as percent inhibition for the HDAC2 biochemical screen. A total of 1,604 compounds derived from two diastereomeric RCM scaffolds, (2R,5S,6R,12R)-30a and (2R,5S,6S,12R)-30e, were tested. The assay was conducted in a kinetic mode using 4.6 μM substrate, 2.2 nM HDAC2, 150 nM trypsin, and 17 μM compound. The full matrix of building blocks for R1 (y-axis) and R2 (x-axis) is shown. Piperonaldehyde (AL11) was the preferred building block at R2. Empty cells represent compounds that were not tested due to low purity or unavailability.
In light of its predicted physicochemical properties (ALogP 4.4, cLogD 2.9, PSA 83) and high solubility in aqueous buffer (>450 μM solubility in PBS pH 7), the product of reductive alkylation with formaldehyde (AL1) at R1 and piperonaldehyde (AL11) at R2 was selected for further SAR exploration (32, Figure 5). Follow-up of the screening results was expanded to include all stereoisomers of 32 tested at dose against HDAC1, 2 and 3. As highlighted in Figure 5 interesting stereo/structure-activity relationships (SSAR) for HDAC inhibition emerged.63 First, a stereoisomer with improved potency against HDAC2 was identified, (2S,5R,6R,12R)-32 (BRD-4805), which had an IC50 of 6.6 μM. This is greater than a 10-fold improvement as compared to the lead stereoisomer identified in the pilot screen, (2R,5S,6R,12R)-32, which was only weakly active against HDAC2 upon retesting (IC50 = 80 μM). To further validate the activity of BRD-4805, the compound was resynthesized in solution starting from macrolactam ent-8g using the 5-step sequence shown in Scheme 4. Preparation of the analog using solution-phase techniques obviates the need for protecting group manipulations and loading onto solid support. Follow-up testing of the resynthesized (and HPLC-purified) compound in the HDAC2 assay confirmed its activity.
Figure 5.

Stereo/structure-activity relationships for HDAC inhibition. IC50 values below 50 M are provided. A more potent stereoisomer with low micromolar activity against HDACs 1, 2 and 3 (2S,5R,6R,12R)-32 (BRD-4805) was identified. (See also Table S-1).
Scheme 4.

Solution-phase synthesis of BRD-4805
As evident by the SSAR pattern displayed in Figure 5 the stereochemistry within the macrocyclic ring of BRD-4805 appears to be most important for HDAC2 inhibition. For example, the C-2 epimer of BRD-4805, (2R,5R,6R,12R)-32, retains some activity albeit weak (IC50 = 45.5 μM). In contrast inverting only one stereocenter within the ring (C5, C6 or C12) renders the compound inactive. Notably, the enantiomer, (2R,5S,6S,12S)-32, is inactive (IC50 > 100 μM).
While BRD-4805 did not prove to be selective for HDAC2 over HDAC 1 and 3 (with an IC50 value of 2.7 M for both HDAC 1 and 3), it was inactive against HDACs 4-8 (data not shown). This contrasts with the improved potency of the hydroxamate SAHA that also inhibits the class I HDAC8 and the class IIb HDAC6 (see Table S-2). It is interesting to note that HDAC2 inhibition was most sensitive to stereochemical changes at C2, C5 and C12. For example, in addition to retaining activity upon inversion of the C-2 methyl group, sterochemical changes within the ring at C-12 are also tolerated for HDAC 1 and 3. In fact there appears to be one stereoisomer, (2R,5R,6R,12S)-32, that is HDAC3 selective with an IC50 of 10 M. HDAC3 selective inhibitors are also of interest for applications in oncology64 and will also be useful for dissecting the contributions of HDAC3 to chromatin regulation in neurons. These results suggest that selective modulation of class I HDACs might be possible through subtle modifications to the macrocyclic ring.
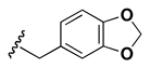
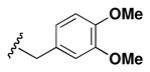
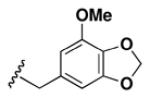
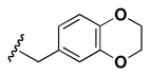
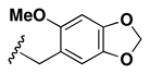
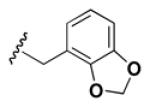
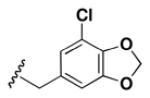
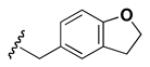
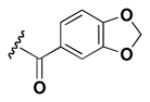
Although the nature of the building block introduced at the aniline (R1) did not seem to affect HDAC activity, modifications at the secondary amine (R2) appeared to be critical. While a variety of sulfonyl chlorides, isocyanates, acids and aldehydes were introduced at R2 as part of the original library, only the products of reductive alkylation with piperonaldehyde were consistently most potent in the HDAC assay.51 In order to further explore the importance of the benzodioxolane at R2, a series of analogs was synthesized. As evident from the data presented in Table 5, only subtle changes at this position were tolerated. HDAC activity was retained and even slightly increased (HDAC2 IC50 = 2.8 μM) with the introduction of a methoxy group at C-5 (entry 2). In contrast a 10-fold drop in potency was observed with the corresponding 6-OMe derivative (entry 3). An opposite trend was observed with corresponding chloro-substituted analogs. In this case, the 5-Cl derivative (entry 4) was 5-fold less active than BRD-4805 (HDAC2 IC50 = 37 μM), while the 6-Cl derivative (entry 5) was equally active (HDAC2 IC50 = 7.5 μM). More drastic changes such as conversion to the 3,4-bisphenol and 3,4-dimethoxy analogs (entries 6 and 7) led to a complete loss in activity. Likewise expansion of the dioxolane ring resulted in a significant drop in potency (entry 8) as did altering the position of the dioxolane ring (entry 9). Two bioisosteres, the benzopyran and benzaminothiazole, were synthesized and found to be inactive (entries 10 and 11). Lastly, the amide analog (entry 12) was also tested and displayed no activity against the three HDACs.
Table 5.
Effect of Building Block Modification on HDAC Inhibition
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | R | HDAC1 | IC50 (μM) HDAC2 |
HDAC3 | entry | R | HDAC1 | IC50 (μM) HDAC2 |
HDAC3 |
| 1 |
|
2.7 ± 0.3 | 6.6 ± 0.4 | 2.7 ± 0.2 | 7 |
|
>100 | >100 | >100 |
| 2 |
|
1.5 ± 0.7 | 2.8 ± 0.9 | 4.4 ± 2.0 | 8 |
|
59 ± 7 | 59 ± 14 | 39 ± 8 |
| 3 |
|
30 ± 7 | 65 ± 40 | 22 ± 10 | 9 |
|
>100 | >100 | >100 |
| 4 |
|
33 ± 8 | 37 ± 6 | 23 ± 15 | 10 |
|
>100 | >100 | >100 |
| 5 |

|
5.0 ± 1.1 | 7.5 ± 0.8 | 3.9 ± 0.5 | 11 |

|
>100 | >100 | >100 |
| 6 |
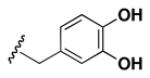
|
>100 | >100 | >100 | 12 |
|
>100 | >100 | >100 |
We have investigated the mode of inhibition by BRD-4805 and SAHA (as a control) to HDAC activity. For HDACs 1, 2 and 3, deacetylation progression in the presence of various concentrations of BRD-4805 and SAHA is linear, confirming the fast-on/fast-off nature of these inhibitors. As shown in Figure 6 the effect of BRD-4805 on the HDAC-catalyzed peptide deacetylation displayed the characteristic intersecting line pattern for mixed competitive inhibition. Similar results were obtained for HDAC1 and HDAC3 (data not shown). The macrocycle shows unequal affinity for both free HDAC and the HDAC-substrate complex. The inhibition constant (Ki) for the enzyme-substrate complex is ~5-10 fold higher than for free enzyme. The enantiomer of BRD-4805 displayed minimal inhibition against all HDACs (IC50 > 50μM). As a control, SAHA is a purely competitive inhibitor with Ki of 1nM (data not shown).
Figure 6.

Lineweaver-Burk plot of the effect of BRD-4805 on the HDAC2-catalyzed deacetylation (Ac)Leu-Gly-Lys-acetyl-AMC peptide substrate. The experiment was performed at pH 7.0, room temperature. Inhibitor concentrations were 0, 1.56, 3.1, 6.2, 12.5, 25 and 50 μM and substrate concentrations were 2.5, 5, 10, 20, 40 and 80 μM.
Finally, we tested the ability of BRD-4805 to inhibit HDACs and induce histone acetylation in cultured mouse neuronal cells. Figure 7 shows that BRD-4805 induced robust acetylation of the N-terminal tails of histones H3 (lysine 9) and H4 (lysine 12). The 5-OMe analog, compound BRD-8172, which was more potent in vitro, was also more active in cells than BRD-4805. The inactive enantiomer (2R,5S,6S,12S)-32 (ent-BRD-4805) had no effect on histone acetylation. Thus, both RCM-derived macrocyclic compounds, BRD-4805 and the corresponding 5-OMe analog (BRD-8172), are cell-active HDAC inhibitors. Representative images of neurons stained for acetylation of histone H3 lysine 9 (in green) are shown in Figure 8.
Figure 7.

Induction of histone acetylation increase in primary mouse forebrain neurons. Neurons were treated for 24 hours with the indicated compounds (95 μM), then fixed and stained for AcH4K12 or AcH3K9. Data are shown normalized to the level of acetylation in the presence of DMSO. Error bars represent +/− the standard deviation of the measurement.
Figure 8.

Imaging of histone acetylation in primary mouse forebrain neurons. (A) Compound treated primary neuronal cultures were stained for acetylation of histone H3 lysine 9 (AcH3K9; shown in green) and MAP2B (neuronal marker; shown in red). To quantify cellular activity of compounds, the mean nuclear intensity of histone signal was measured and the percentage of cells with a value above a predefined threshold was measured. Scale bar = 50 μm. (B) Neuronal nuclei with representative levels of acetylation for each treatment (DMSO, ent-BRD-4805, BRD-4805, BRD-8172). Nuclei of cells treated with BRD-4805 and analog BRD-8172 scored as “bright green” in our analysis. Scale bar = 10 μm.
Conclusions
Expanding the diversity of small-molecule screening collections to include architecturally complex molecules with increased sp3-content requires making an upfront investment in the development of robust and practical methods for their synthesis. Moreover, it is important to select chemistry that anticipates successful discovery by developing facile synthetic routes in the initial chemical design in order to enable rapid analog synthesis. Using an aldol-based build/couple/pair strategy we synthesized a variety of library scaffolds ranging in size from 8- to 14-membered rings. Our modular design strategy enabled the generation of the complete matrix of stereoisomers for all ring systems. The value of this approach was demonstrated here, where hits from a pilot screen for HDAC2 inhibitors were immediately followed-up yielding unanticipated SSAR and increased potency. The synthetic methodology utilized to produce these compounds allowed for the expedient generation of analogs, one with increased activity against HDACs 1, 2 and 3. These compounds represent a novel class of HDAC inhibitors, which display mixed enzyme inhibition and retained biological activity in a cell-based assay.
While the objective of our study was to discover new structural classes of HDAC inhibitors whose structures are further poised for rapid synthetic optimization, which was achieved, the potency of BRD-4805 and its analogs, which lack obvious metal-chelating functional groups, is not as high as that of other known HDAC inhibitors, many of which contain zinc chelators from which the majority of the binding energy is presumably obtained. One exception is the macrocyclic tetrapeptide apicidin that contains only a ketone and lacks a metal chelator, which grossly resembles the macrocyclic structure of BRD-4805. Apicidin and related peptidic HDAC inhibitors have single digit nanomolar potency in our HDAC biochemical assay and show selectivity for class I HDACs as described in our recently published studies.60 However, the peptide bonds in this class of natural products distinguishes itself from the nonpeptidic macrocyclic structure of the RCM macrolactams. BRD-4805 is further distinguished from other classes of known HDAC inhibitors by virtue of its unique, to our knowledge, binding profile, which shows mixed inhibition kinetics that differs from the substrate competitive fast-on/fast-off kinetics of SAHA and the slow-on/slow-off kinetics of 2-amino benzamides.
Since the focus of our studies to date has been on the discovery of novel classes of HDAC inhibitors that are active in vitro in biochemical assays and in the context of primary mouse neurons, the pharmacokinetic properties of BRD-4805 in plasma and other tissues remain uncharacterized. Future studies will be directed to determining whether probes of this class have improved half-lives and blood-brain barrier penetration compared to other HDAC inhibitors. The synthesis and testing of additional analogs is currently underway as part of our ongoing medicinal chemistry efforts.
Supplementary Material
Acknowledgments
We thank Prof. Stuart Schreiber, Dr. Edward Holson and Dr. Florence Wagner for helpful discussions, Dr. Stephen Johnston and Anita Vrcic for analytical support, and Dr. Peter Müller for X-ray crystallography. We thank the Chemical Biology Platform Informatics group for the development of the Stereo-Structure Attribute Relationship (SSAR) visualization tool where attribute value represents activity, purity, yield or any measurement of interest. This work was funded in part by the NIGMS-sponsored Center of Excellence in Chemical Methodology and Library Development (Broad Institute CMLD; P50 GM069721). Financial support for D.M.F., S.A.R, W.-N. Z. and S.J.H. from the NIH (1R01DA028301-01) and Stanley Medical Research Institute is also gratefully acknowledged.
Footnotes
Contribution from the Chemical Biology Platform and the Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, 7 Cambridge Center, Cambridge, Massachusetts 02142, and the Center for Human Genetic Research, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02114
Supporting Information Available: Experimental procedures and spectral data for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lovering F, Bikker J, Humblet C. J. Med. Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 2.Ganesan A. Curr. Opin. Chem. Biol. 2008;12:306–317. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hübel K, Leβmann T, Waldmann H. Chem. Soc. Rev. 2008;37:1361–1374. doi: 10.1039/b704729k. [DOI] [PubMed] [Google Scholar]; (b) Koch MA, Schuffenhauer A, Scheck M, Wetzel S, Casaulta M, Odermatt A, Ertl P, Waldmann H. Proc. Natl. Acad. Sci. 2005;102:17272–17277. doi: 10.1073/pnas.0503647102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bauer RA, Wurst JM, Tan DS. Curr. Opin. Chem. Biol. 2010;14:308–314. doi: 10.1016/j.cbpa.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Marcaurelle LA, Johannes CW. Prog. Drug Res. 2008;66(187):189–216. doi: 10.1007/978-3-7643-8595-8_3. [DOI] [PubMed] [Google Scholar]; (e) Goess BC, Hannoush RN, Chan LK, Kirchhausen T, Shair M. J. Am. Chem. Soc. 2006;128:5391–5403. doi: 10.1021/ja056338g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Pelish HE, Westwood NJ, Feng Y, Kirchhausen T, Shair MD. J. Am. Chem. Soc. 2001;123:6740–6741. doi: 10.1021/ja016093h. [DOI] [PubMed] [Google Scholar]; (g) Marcaurelle LA, Johannes C, Yohannes D, Tillotson BP, Mann D. Bioorg. Med. Chem. Lett. 2009;19:2500–2503. doi: 10.1016/j.bmcl.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zhang QS, Lu HJ, Curran DP. J. Am. Chem. Soc. 2004;126:36–37. doi: 10.1021/ja038542e. [DOI] [PubMed] [Google Scholar]; (b) Curran DP, Zhang Q, Richard C, Lu H, Gudipathi V, Wilcox CS. J. Am. Chem. Soc. 2006;128:9561–9573. doi: 10.1021/ja061801q. [DOI] [PubMed] [Google Scholar]; (c) Dandapani S, Jeske M, Curran DP. J. Org. Chem. 2005;70:9447–9462. doi: 10.1021/jo051526a. [DOI] [PubMed] [Google Scholar]; (d) Wrona IE, Lowe JT, Turbyville TJ, Johnson TR, Beignet J, Beutler JA, Panek JS. J. Org. Chem. 2008;74:1897–1916. doi: 10.1021/jo802269q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Neilson TE, Schreiber SL. Angew. Chem. Int. Ed. 2007;74:48–56. [Google Scholar]; (b) Uchida T, Rodriguez M, Schreiber SL. Org. Lett. 2009;11:1559–1562. doi: 10.1021/ol900173t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schreiber SL. Nature. 2009;457:153–154. doi: 10.1038/457153a. [DOI] [PubMed] [Google Scholar]; (d) Luo T, Schreiber SL. J. Am. Chem. Soc. 2009;131:5667–5674. doi: 10.1021/ja900414s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pizzirani D, Kaya T, Clemons PA, Schreiber SL. Org. Lett. 2010;12:2822–2825. doi: 10.1021/ol100914b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Evans PA, Holmes AB. Tetrahedron. 1991;47:9131–9166. [Google Scholar]; (b) Nubbemeyer U. Topics Curr. Chem. 2001;216:126–196. [Google Scholar]; (c) Maier MC. Angew. Chem., Int. Ed. 2000;39:2073–2077. doi: 10.1002/1521-3773(20000616)39:12<2073::aid-anie2073>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Driggers EM, Hale SP, Lee J, Terrett NK. Nature Rev. Drug. Disc. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 8.Sauer WHB, Schwarz MK. J. Chem. Inf. Comput. Sci. 2003;43:987–1003. doi: 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson DM, Raber DJ. J. Am. Chem. Soc. 1989;111:4371–4378. [Google Scholar]
- 10.MOE 2007.09. Chemical Computing Group; 1010 Sherbrooke St. W, Suite 910, Montreal, Quebec, Canada H3A 2R7: http://www.chemcomp.com (accessed June 11, 2008) [Google Scholar]
- 11.Convergence was achieved by varying the maximum number of conformations generated and the failure limit that specifies number of attempts made to generate a new conformation before terminating the search.
- 12.TIBCO Software, Inc. TIBCO Software, Inc.; Spotfire US, Somerville, MA: Available at: http://www.spotfire.com, accessed on December 1, 2009. [Google Scholar]
- 13.Pipeline Pilot, SciTegic, Inc.; 10188 Telesis Court, Suite 100, San Diego, CA 92121, USA: http://www.scitegic.com/products_services/pipeline_pilot.htm. [Google Scholar]
- 14.Bostrom J, Norrby PO, Liljefors T. J. Comput. Aided Mol. Des. 1998;12:383–396. doi: 10.1023/a:1008007507641. [DOI] [PubMed] [Google Scholar]
- 15.Paterson I. Pure & Appl. Chem. 1992;64:1821–1830. [Google Scholar]
- 16.(a) Evans DA. Aldrichimica Acta. 1982;15:23–32. references therein. [Google Scholar]; (b) Evans DA, Nelson JV, Vogel E, Taber TR. J. Am. Chem. Soc. 1981;103:3099–3111. [Google Scholar]; (c) Evans D, A.; Bartroli J, Shih TL. J. Am. Chem. Soc. 1981;103:2127–2129. [Google Scholar]; (d) Evans DA, Dart MJ, Duffy JL, Reiger DL. J. Am. Chem. Soc. 1995;117:9073–9074. [Google Scholar]; (e) Evans DA, Ratz AM, Huff BE, Sheppard GS. J. Am. Chem. Soc. 1995;117:3448–3467. [Google Scholar]; (f) Sasmal S, Geyer A, Maier ME. J. Org. Chem. 2002;67:6260–6263. doi: 10.1021/jo025889b. [DOI] [PubMed] [Google Scholar]
- 17.(a) Inoue T, Liu J-F, Buske D, Abiko A. J. Org. Chem. 2002;67:5250–5256. doi: 10.1021/jo0257896. [DOI] [PubMed] [Google Scholar]; (b) Abiko A. Org. Synth. Coll. 2004;10:343–349. [Google Scholar]; (c) Abiko A. Acc. Chem. Res. 2004;37:387–395. doi: 10.1021/ar030249w. [DOI] [PubMed] [Google Scholar]; (d) Abiko A, Liu J-F, Masamune S. J. Am. Chem. Soc. 1997;119:2586–2587. [Google Scholar]
- 18.Further details regarding the optimization of the large-scale γ-amino acid synthesis will be the subject of a separate publication: Pandya BA, Dandapani S, Duvall JR, Rowley A, Mulrooney C, Ryba T, Dombrowski M, Harton M, Young DW, Marcaurelle LA. manuscript in preparation. doi: 10.1016/j.tet.2011.06.043.
- 19.Absolute stereochemical assignment for (−)-12a was established by single crystal x-ray diffraction. Purity was >95% by HPLC and e.r. 99:1 by SFC.
- 20.The scale of the reaction was limited only by the total possible volume permitted in a jacketed 5-L round bottom flask.
- 21.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496–6511. [Google Scholar]
- 22.Chemoselective O-alkylation of D- and L-alaninol with PMBCl was achieved via the following procedure: Hu EX, Cassady JM. Synth. Comm. 1995;25:907–913. See Supporting Information for further details.
- 23.For reviews and recent applications on the use of borane reagents, see: Brown HC, Heim P. J. Org. Chem. 1973;38:912–916. Lane CF. Chem. Rev. 1976;76:773–799. Brown HC, Choi YM, Narasimhan S. J. Org. Chem. 1982;47:3153–3163. Amedia JC, Jr., Bernard PJ, Fountain M, Van Wagenen G., Jr. Syn. Commun. 1999;29:2377–2391. Ostresh JM, Schroner CC, Hamashin VT, Nefzi A, Meyer J-P, Houghten RA. J. Org. Chem. 1998;63:8622–8623.
- 24.This coupling protocol was routinely performed in >100-gram batches with consistent results for all stereochemical permutations. Purity was determined by LCMS and SFC prior to further reaction.
- 25.Initially we investigated the conversion of 13 to 3 using an excess (6 equiv) of borane-tetrahydrofuran (BH3-THF). Unfortunately <50% of the desired product was observed under these conditions along with decomposition of the amide.
- 26.Holsworth DD, Powell NA, Downing DM, Cai C, Cody WL, Ryan JM, Ostroski R, Jalaie M, Bryant JW, Edmunds JJ. Bioorg. Med. Chem. 2005;13:2657–2664. doi: 10.1016/j.bmc.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 27.The use of commonly employed protolytic conditions for dissociating amine-borane complexes, such as TFA or HCl, was incompatible with our substrate due to the presence of acid-sensitive protecting groups (Boc, PMB and TBS) required for downstream chemistry.
- 28.Comer E, Rohan E, Deng L, Porco JA. Org. Lett. 2007;9:2123–2126. doi: 10.1021/ol070606t. [DOI] [PubMed] [Google Scholar]
- 29.For reviews on the synthesis of biaryl ethers via intramolecular SNAr, see: Rao AVR, Gurjar MK, Reddy L, Rao AS. Chem. Rev. 1995;95:2135–2167. Burgess K, Lim D, Martinez CI. Angew. Chem., Int. Ed. 1996;35:1077–1078. Zhu J. Synlett. 1997:133–144. Nicolaou KC, Boddy CNC, Bräse S, Winssinger N. Angew. Chem., Int. Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. Sawyer JS. Tetrahedron. 2000;56:5045–5065.
- 30.For the synthesis of aryl—alkyl ethers via intramolecular SNAr, see: Goldberg M, Smith L, II, Tamayo N, Kiselyov AS. Tetrahedron. 1999;55:13887–13898. Jefferson EA, Swayze EE. Tetrahedron Lett. 1999;40:7757–7760. Temal-Laieb T, Chastanet J, Zhu J. J. Am. Chem. Soc. 2002;124:583–590. doi: 10.1021/ja0170807. Abrous L, Jokiel PA, Friedrich SR, Hynes J, Jr., Smith AB, III, Hirschmann R. J. Org. Chem. 2004;69:280–302. doi: 10.1021/jo0352068. Tempest P, Ma V, Kelly MG, Jones W, Hulme C. Tetrahedron Lett. 2001;42:4963–4968. Rolfe A, Samarakoon TB, Hanson PR. Org. Lett. 2010;12:1216–1219. doi: 10.1021/ol100035e.
- 31.The acid chloride was prepared via treatment of the commercially available acid with thionyl chloride. See Supporting Information.
- 32.Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7–17. [Google Scholar]; (b) Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (c) Meldal M, Tornoe WC. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 33.(a) Zhang L, Chen X, Xue P, Sun HHY, Williams ID, Sharpless KB, Fokin VV, Jia G. J. Am. Chem. Soc. 2005;127:15998–15999. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]; (b) Rasmussen LK, Boren BC, Fokin VV. Org. Lett. 2007;9:5337–5339. doi: 10.1021/ol701912s. [DOI] [PubMed] [Google Scholar]; (c) Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J. Am. Chem. Soc. 2008;130:8923–8930. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 34.Kelly AR, Wei J, Kesavan S, Marie J-C, Windmon N, Young DW, Marcaurelle LA. Org. Lett. 2009;11:2257–2260. doi: 10.1021/ol900562u. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
35.Structure of oxazolidine by-product formed under basic alkylation conditions:

- 36.Ghorai S, Mukhodopadhyay R, Kundu PA, Bhattacharjya A. Tetrahedron. 2005;61:2999–3012. [Google Scholar]
- 37.[Cp*Ru(PPh3)2Cl], [Cp*Ru(COD)Cl] and [Cp*RuCl]4 were screened and all three catalysts effected the regioselective macrocyclization with Cp*RuCl4 providing the highest yields for all diastereomers.
- 38.Gonthier E, Breinbauer R. Molecular Diversity. 2005;9:51–62. doi: 10.1007/s11030-005-1308-8. [DOI] [PubMed] [Google Scholar]
- 39.(a) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem., Int. Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]; (b) Deiters A, Martin SF. Chem. Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; (c) Piscopio AD, Robinson JE. Curr. Opin. Chem. Biol. 2004;8:245–254. doi: 10.1016/j.cbpa.2004.04.001. [DOI] [PubMed] [Google Scholar]; (d) Gradillas A, Perez-Castells J. Angew. Chem., Int. Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]; (e) Micalizio GC, Schreiber SL. Angew. Chem., Int. Ed. 2002;41:152–154. doi: 10.1002/1521-3773(20020104)41:1<152::aid-anie152>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; (f) Kim YK, Arai T, Lamenzo JO, Dean EF, Patterson N, Clemons PA, Schreiber SL. J. Am. Chem. Soc. 2004;126:14740–14745. doi: 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]; (g) Speigel DA, Schroeder FC, Duvall JR, Schreiber SL. J. Am. Chem. Soc. 2006;128:14766–14767. doi: 10.1021/ja065724a. [DOI] [PubMed] [Google Scholar]; (h) Morton D, Leach S, Cordier C, Warriner S, Nelson A. Angew. Chem., Int. Ed. 2009;48:104–109. doi: 10.1002/anie.200804486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Nicola T, Brenner M, Donsbach K, Kreye P. Org. Process Res. Dev. 2005;9:513–515. [Google Scholar]; (b) Farina V, Shu C, Zeng X, Wei X, Han Z, Yee NK, Senanayake CH. Org. Process Res. Dev. 2009;13:250–254. [Google Scholar]; (c) Wang H, Goodman SN, Dai Q, Stockdale GW, Clark WM., Jr. Org. Process Res. Dev. 2008;12:226–234. [Google Scholar]
- 41.See Supporting Information for the preparation of 20a and 20b.
- 42.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Jr., Hoveyda AH. J. Am. Chem. Soc. 1999;121:791–799. [Google Scholar]
- 43.All RCM products (8) were isolated as mixtures of geometric isomers of the double bond. No attempt was made to separate the E/Z product mixture as our plan called for reduction of the olefin in subsequent steps.
- 44.A by-product with the same MW as the starting olefin was observed which did not react with the catalyst under the reaction conditions. NMR analysis of this by-product seemed to indicate that both the terminal olefins had isomerized.
- 45.Hong SH, Sanders DP, Lee CW, Grubbs RH. J. Am. Chem. Soc. 2005;127:17160–17161. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- 46.The acidic conditions required for Boc removal and nitro reduction are not compatible with the use of the alkylsilyl linker to be used for downstream solid-phase synthesis.
- 47.Ryba TD, Depew KM, Marcaurelle LA. J. Comb. Chem. 2009;11:110–116. doi: 10.1021/cc8000986. [DOI] [PubMed] [Google Scholar]
- 48.(a) Sakaitani M, Ohfune Y. J. Org. Chem. 1990;55:870–876. [Google Scholar]; (b) Chen J, Forsythe CJ. Angew. Chem., Int. Ed. 2004;43:2148–2152. doi: 10.1002/anie.200453663. [DOI] [PubMed] [Google Scholar]
- 49. www.mimotopes.com, accessed on Nov 28, 2009.
- 50.Characterization details for all 16 stereoisomers of the RCM scaffold (30a-h and ent-30a-h) are provided in the Supporting Information.
- 51.See Supporting Information for a full list of building blocks used for the library production.
- 52.The use of phenylsilane as the π-allyl scavenger was also explored, however the formation of minor amounts of N-allyl side product were observed.
- 53.See Supporting Information for a detailed account of purity and yield for all library members according to scaffold/building block.
- 54.Badertscher M, Bischofberger K, Munk ME, Pretsch EA. J. Chem. Inf. Comp. Sci. 2001;41:889–893. doi: 10.1021/ci000135o. [DOI] [PubMed] [Google Scholar]
- 55.(a) Grozinger CM, Schreiber SL. Chem. Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]; (b) Ruthenburg AJ, Li H, Patel DJ, Allis CD. Nat. Rev. Mol. Cell. Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.(a) de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Biochem. J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith BC, Hallows WC, Denu JM. Chem. Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.(a) Bieliauskas AV, Pflum MK. Chem. Soc. Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]; (c) Yoshida M, Matsuyama A, Komatsu Y, Nishino N. Curr. Med. Chem. 2003;10:2351–2358. doi: 10.2174/0929867033456602. [DOI] [PubMed] [Google Scholar]
- 58.Tessier P, Smil DV, Wahhab A, Leit S, Rahil J, Li Z, Déziel R, Besterman JM. Bioorg. Med. Chem. Lett. 2009;19:5684–5688. doi: 10.1016/j.bmcl.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 59.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Matzischek R. Nature Chem. Bio. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.This concentration of compound was chosen for the primary screens based upon the fact that robotic pin transfer used for compound handling led to the transfer of 100 nL of compound at 5 mM into a reaction volume of 30 μL. See details provided in Suppporting Information describing compound handling.
- 62.At the time the HDAC2 assay was run only a subset of the RCM library was available for screening. Therefore, compounds derived from only 2 of the 16 stereoisomeric RMC scaffolds were tested during the primary screen. The remainder of the compounds screened were derived from the SNAr and Click libraries.
- 63.All analogs were resynthesized and subjected to HPLC purification prior to testing.
- 64.(a) Godman CA, Joshi R, Tierney BR, Greenspan E, Rasmussen TP, Wang HW, Shin D.G.; Rosenberg DW, Giardina C. Cancer Biol. Ther. 2008;7:1570–80. doi: 10.4161/cbt.7.10.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Narita N, Fujieda S, Kimura Y, Ito Y, Imoto Y, Ogi K, Takahashi N, Tanaka T, Tsuzuki H, Yamada T, Matsumoto H. 2010;396:310–316. doi: 10.1016/j.bbrc.2010.04.089. [DOI] [PubMed] [Google Scholar]; (c) Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, Rice VM. Cancer Biol. Ther. 2007;6:795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.