

Abstract
The historical roots of Alzheimer's disease provide a sound conceptual basis for linking the behavioral and neurological symptoms of the disease with the frequently associated pathology of amyloid plaques and neurofibrillary tangles. Out of these roots has grown the “amyloid cascade hypothesis”—a vision of the etiology of Alzheimer's that has spurred the discovery of many important insights into the neurobiology of the disease. Despite these successes, the wealth of new data now available to biomedical researchers urges a full review of the origins of Alzheimer's, and such a reconsideration is offered here. It begins with the most widely accepted risk factor for developing Alzheimer's disease: age. Then, for an individual to progress from normal age-appropriate cognitive function to a condition where the full palette of clinical symptoms is expressed, three key steps are envisioned: (1) an initiating injury, (2) a chronic neuroinflammatory response, and (3) a discontinuous cellular change of state involving most, if not all, of the cell types of the brain. The amyloid cascade is integrated into this sequence, but reconfigured as an amyloid deposition cycle. In this way, the pathology of amyloid plaques is envisioned as highly correlated with, but mechanistically distinct from, the three obligatory steps leading to Alzheimer's disease. The implications of this new model are discussed with respect to our current diagnostic criteria, and suggestions are put forward for expanding our future research efforts.
A brief history of Alzheimer's disease
In 1906, Alois Alzheimer reported the first description of the dementing illness that now bears his name. He documented the progression of symptoms that beset a farmer's wife, Auguste D., as her mental status deteriorated through a complex series of behavioral and cognitive changes that left her aggressive, delusional, and unable to remember recent events. After her death, Alzheimer drew on his interest in the emerging techniques of histochemistry. He stained sections from the autopsied brain and discovered the presence of “miliar foci, which are caused by deposition of a peculiar substance in the cortex” (now recognized as neuritic or senile plaques). He also reported “very peculiar changes in the neurofibrils” (now recognized as paired helical filaments or tangles). By concerning himself with the structure of the diseased brain and the abnormal deposits that he found, he was among the early pioneers whose studies linked brain structure to function.
In considering the biology of Alzheimer's disease over 100 years later, a few aspects of this case study deserve note. Auguste D. became ill in her early 50s, meaning her symptoms emerged from a familial (i.e., genetic) form of Alzheimer's rather than the sporadic form that makes up >90% of prevalent cases (Yu et al., 2010). Second, part of what made the case noteworthy for its era was the inclusion of the neuropathological examination and the proposal that the abnormal behavior of the patient was the consequence of the abnormal deposits in her brain. As a result, from the very beginning, Alzheimer's disease research and diagnosis has been based on a tight association between the dementia we now know as Alzheimer's and the peculiar deposits we now recognize as plaques and tangles. That the presence or absence of these deposits is considered the gold standard of Alzheimer's disease (AD) diagnosis only serves to underline the broad acceptance in the field of the importance of the association.
Amyloid and the amyloid cascade hypothesis
A major advance in the study of AD came with the sequencing of the main constituent of the senile plaque—the amyloid β peptide (Aβ) (Glenner and Wong, 1984). This led in rapid sequence to four key discoveries. First, the Aβ peptide is a part of a large type I membrane protein, the amyloid precursor protein (APP), which is encoded by the APP gene on chromosome 21. Second, the APP gene is mutated in a significant fraction of the cases of familial Alzheimer's disease. Third, individuals with Down's syndrome, who have three copies of chromosome 21 and hence three copies of the APP gene, develop clinical and pathological signs of early-onset Alzheimer's. And fourth, mutations in the presenilin-1 (PSEN1) and presenilin-2 (PSEN2) genes can behave as dominant familial AD genes. Presenilin is the catalytic subunit of the γ-secretase activity that liberates the Aβ peptide from the C terminus of APP.
These findings led to the elaboration of a theory of AD known as the amyloid cascade hypothesis (Hardy and Selkoe, 2002; Citron, 2004). In familial AD (Fig. 1, top left), mutations in either APP or one of the PSEN genes lead to the brain accumulation of a 42 aa form of the amyloid peptide that has a high tendency to form β-pleated sheet structures. Amyloid aggregates form—first small oligomers and finally plaques. The amyloid cascade hypothesis proposes that these Aβ aggregates lead in turn to a series of downstream events ranging from synapse loss to plaque deposition to inflammation to the triggering of tau hyperphosphorylation to the death of susceptible neurons. The hypothesis also proposes that sporadic AD develops when the natural history of an individual accelerates a normal age-dependent process of Aβ accumulation (Fig. 1, top right). At some point, sufficient Aβ becomes deposited that the amyloid cascade is engaged. Subsequently, the sporadic disease follows the same pathway to dementia as the familial form.
Figure 1.

The amyloid cascade hypothesis of Alzheimer's disease. This hypothesis represents the classic theory of the origins of Alzheimer's disease. Both familial forms of Alzheimer's (fAD) and later-onset forms with no known etiology (sporadic AD) lead to the production of excess Aβ1-42. Once this toxic peptide begins to aggregate, a cascade of events is triggered that produces the biological and neurological symptoms of Alzheimer's disease. The diagram is modified from that found on the AlzForum web site (http://www.alzforum.org/images/res/adh/cur/sequence2005.gif).
A new synthesis
In recent years, our knowledge base has broadened considerably, affording us the luxury of being able to revisit our hypotheses about causes of AD. Based on its prevalence, it now makes sense to begin this exercise with the more common sporadic form of the disease rather than with the rare familial forms. The precise etiology of sporadic AD is not known in detail. We do know, however, that the most critical risk factor by far is age. This makes intuitive, if not mechanistic, sense. In all species, age brings a progressive slowing of brain function in virtually every domain. Our cognition slows; our ability to form new memories is reduced; our motor functions deteriorate; even our brain's homeostatic functions become less and less robust. This functional decline is correlated with the loss of structural complexity of our brain cells. Neuronal dendrites become less complex, spine and synapse density decrease, the cell bodies of the larger neurons accumulate lipofuscin, astrocytic function declines (e.g., glutamate uptake and recycling), our immune systems become less responsive, etc. And as this list of insufficiencies grows, the brain's defenses against many diseases, Alzheimer's included, is weakened. The question is as follows: how do the signature symptoms of AD emerge from this state? The new model envisions three key events that occur sequentially when an individual develops Alzheimer's disease (Fig. 2). The first is a precipitating injury that begins the pathogenic process. This injury in turn triggers the second key event: a chronic inflammatory process that adds additional relentless stress to brain cells already weakened by age. The third event is a major shift in the cellular physiology of the brain cells. Described in more detail below, it represents a tipping point that marks the beginning of a cell's degenerative process and leads to major synaptic dysfunction and neuronal death—the final and most direct causes of Alzheimer's dementia.
Figure 2.
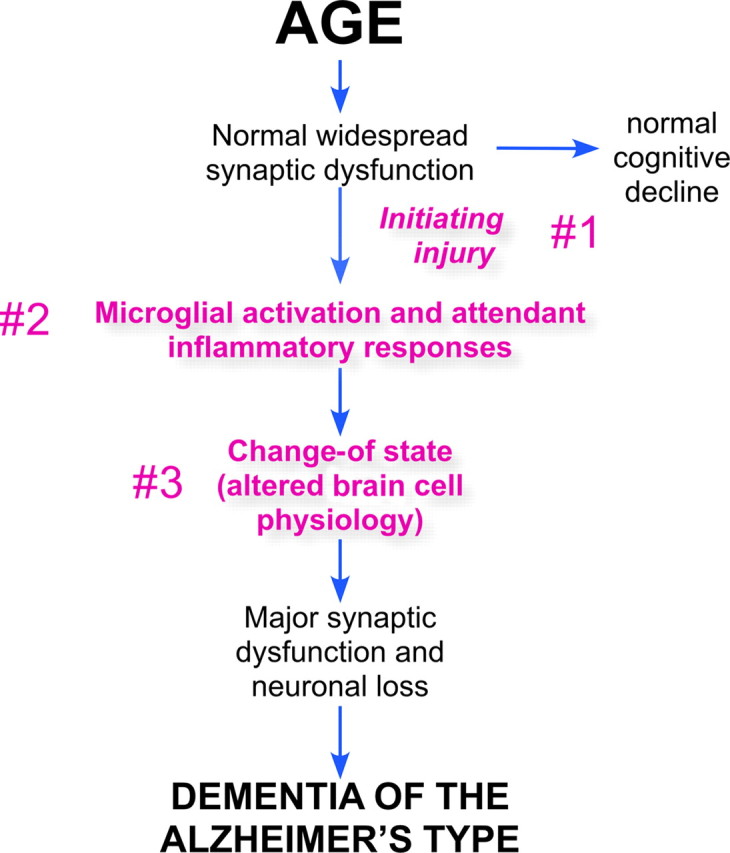
The age-dependent hypothesis of Alzheimer's disease. The new model begins with the cell biology of the brain, weakened by the normal course of aging. Under ideal circumstances, this leads only to a modest natural slowing of function. The new hypothesis postulates three events that divert the brain into the pathophysiology of Alzheimer's disease. These are numbered and highlighted in red text. The first is an initiating injury; the second is a chronic neuroinflammation; the third is a discrete change in the cell biological state of the cells of the brain that alters their normal functions and predisposes them to undergo a degenerative process.
In the beginning—a divergence from normal aging
Although aging gradually takes its toll on our brains, the hypothesis stipulates that some event—a physical head trauma, a major illness or infection, a vascular event (possibly so small as to be clinically undetectable), the metabolic stress associated with adult-onset diabetes, or even the stress associated with a major life event such as a death in the family—is required to initiate the disease process. A genetic mutation can be such an injury, but only if it must interact with the aging process to be expressed. The injury triggers a protective response among the cells of the brain, but the age-related failure of the normal homeostatic mechanisms means that the response continues, even if the injury itself abates. The key concept is that it is the nature of the response, not the nature of the injury, that determines the outcome of Alzheimer's disease.
A useful analogy to consider is hip fracture. For a wide variety of reasons, the risk of breaking our hipbone increases dramatically with age. Bone density decreases; osteoporosis becomes more likely; balance is less sure; reaction times slow; muscles weaken; visual acuity fades; etc. Each of these is a risk factor, but the factors themselves do not cause the hipbone to break; there has to be a precipitating injury (usually a fall). Applied to AD, the analogy is meant to suggest that while any of the changes in the brain that come with advancing age may increase our risk of Alzheimer's, without an injury none can cause dementia.
The idea that AD begins with an initiating injury has both theoretical and practical relevance. Theoretically, it means that Alzheimer's is not a part of normal aging any more than breaking your hip is a part of normal aging. They are both pathological events with an underlying biology. The practical relevance is that, if research can identify the most common sources of injury, we may be able to intervene proactively and delay disease onset. Currently, it is not possible to identify a single candidate for this precipitating injury. But the frequent co-occurrence of vascular pathology with AD and the protective effects of genetic and environmental factors that improve cardiovascular health suggest that a common if not exclusive initiating injury would be a vascular event such as a head trauma or microstroke.
The role of inflammation
A cardinal feature of the neuropathology of most AD brains is the evidence for a chronic neuroinflammatory process. Many recent reviews have summarized this topic in some detail (McGeer et al., 1996; Akiyama et al., 2000; Bamberger and Landreth, 2002; Wyss-Coray and Mucke, 2002; Wyss-Coray, 2006; Glass et al., 2010). While it is true that an inflammatory response accompanies tissue damage in many brain diseases, including Parkinson's (McGeer et al., 2001; Nagatsu and Sawada, 2005), amyotrophic lateral sclerosis (Henkel et al., 2009; Ilieva et al., 2009), and others, AD is unique in the intimate association found between chronic inflammation and disease. There is also solid epidemiological evidence that inflammation serves as a cause of AD. Long-term use of high doses of certain nonsteroidal anti-inflammatory drugs (NSAIDs) lowers the lifetime risk of AD by 30–60% (McGeer et al., 1996; Stewart et al., 1997; Vlad et al., 2008), and there is biochemical evidence for elevated levels of cytokines such as Il-1, Il-6, TNFα, and S100β in human AD brain (Griffin et al., 1998; Akiyama et al., 2000). Microgliosis as well as astrocytosis are prevalent, and most plaques are surrounded by activated astrocytes and invaded by activated microglia (McGeer et al., 1987; Heneka and O'Banion, 2007; El Khoury and Luster, 2008; Rodríguez et al., 2009).
Discussions of brain inflammation tend to focus on the microglial cell; however, a variety of cell types participate in the AD inflammatory response. These cell:cell interactions have been reviewed for other diseases (Ilieva et al., 2009), and evidence for their importance appears in the AD literature as well (Griffin et al., 1998; Mucke et al., 2000; Wegiel et al., 2001; Giri et al., 2002; Griffin, 2006). Through a variety of feedforward loops, the microglial cells are assisted by the responses of astrocytes and brain vascular endothelial cells (Giri et al., 2002) in maintaining a chronic shift in the inflammation status of the brain. The result of this network-like response is a chronic stress on neurons and their function. Cell cycle proteins are activated (Wu et al., 2000), reactive oxygen species are produced (Keller et al., 1999; Fuller et al., 2010), mitochondrial function is reduced (Chen and Chan, 2009), dendritic/axonal transport is impaired (Wu et al., 2010), etc. A second tenet of the new model, therefore, is that a chronic immune response, persisting over months and years, creates the unique chemistry and cellular physiology that results in the core symptoms we recognize as dementia of the Alzheimer's type.
Two biologies—early and late stages of Alzheimer's disease
Thus far, the reenvisioning of AD has included two important tenets. The first is that Alzheimer's must be triggered by an injury. The second is that the establishment of unique type of chronic inflammation is required to set the brain's chemistry on the path to AD. To fully capture the neurobiology of the disease, however, a third tenet must be introduced: the progression to full Alzheimer's disease requires a functional discontinuity between the physiology of the brain cells in early and late stages of the disease. This dramatic change of state results in a “new normal” biology, primed toward neurodegeneration and dementia. The exact meaning of this transition in biological terms is only beginning to be understood, but some of its consequences are already becoming apparent. It is envisioned as a one-way cellular door; once a cell crosses the threshold, it can never return to its earlier state.
The best example of this change-of-state concept can be found in the paradoxical association of neuronal cell cycle events with the process of neurodegeneration. Neurons are generally considered to be permanently postmitotic cells. But when they are stressed, fully differentiated neurons can and do reinitiate the enzymatic cascades of a normal cell cycle. Curiously, the cycle stalls after DNA replication such that the neurons can neither proceed into late G2/M-phase and complete division, nor reverse and turn off the cell cycle proteins. This new neuronal “state” is of more than academic interest as it is dramatically elevated in populations of neurons that are at risk for death in several neurodegenerative diseases, the best studied of which is AD. The at-risk neurons reexpress cell cycle proteins (Vincent et al., 1996, 1997; Arendt et al., 1996; McShea et al., 1997; Busser et al., 1998; Nagy et al., 1998), accompanied by true DNA replication (Yang et al., 2001; Kingsbury et al., 2005; Yang et al., 2006; Mosch et al., 2007). These unscheduled neuronal cell cycles appear during early disease stages (Yang et al., 2003; Arendt et al., 2010), which argues that they are an integral part of the disease process. The full mechanistic implications of the appearance of cell cycle events (CCEs) in a “postmitotic” adult neuron are not yet understood, but certainly the doubling of the DNA content of any cell would seem to qualify as a profound and irreversible change of state.
Findings in the mouse models of AD enhance this view. In one carefully studied AD model, both the anatomical and temporal appearance of the CCEs follow the course of the neuropathology seen in human AD (Yang et al., 2006; Varvel et al., 2008). Further, the CCEs in this model do not appear in a slow progressive manner. Rather, in any one population of neurons, the percentage of “cycling” neurons rises rapidly from near zero to final values and then remains stable—a rapid change of state. A similar progression is likely to occur in the human AD brain (Arendt et al., 2010). The results of anti-inflammatory treatments of AD mouse models are also consistent with a change-of-state model. While 3 months of NSAID treatment can block new CCEs from appearing, even 6 months of NSAID treatment do not reverse the cell cycle protein expression pattern once it has begun (Varvel et al., 2009).
Neurons are not the only cells of the brain whose cell biology is changed during the progression of AD. Astrocytes become activated in regions of AD neuropathology, as do the microglial cells. Equally intriguing from the change-of-state perspective, the microglia can apparently adopt a phenotype found in macrophages known an “alternate activation state” (Colton et al., 2006; Cameron and Landreth, 2010). This state is accompanied by a shift from an acute proinflammatory reaction to a chronic state of activation more suited toward vascular growth and tissue repair (Allavena et al., 2008). Though not yet fully documented for microglia in AD, evidence from spinal cord injury studies suggests that this alternate activation can be achieved in CNS (Kigerl et al., 2009)
Hypothesizing that AD involves a cellular change of state from early to late disease has theoretical as well as practical importance. At the theoretical level, it encourages us to revisit the cellular events that occur after the change. The prediction is that the biology of early AD differs in qualitative ways from the biology that ultimately produces the dementia. As an analogy, if someone were to stop smoking after they developed lung cancer, they would not be likely to alter the progression of the cancer. The biology of the cells involved has changed and the process is now independent of the initiating injury and transformation. It also offers a theoretical explanation for the failure of the prospective human trials of NSAIDs: the trials were all begun after AD symptoms were manifest. With the disease already in progress, it is likely that many of the neurons in the subjects' brains had gone through their change of state. Their biology no longer required chronic inflammation to sustain their abnormal state. Coupled with the evidence cited above that the change of state may be quite abrupt in entire cohorts of neurons (Varvel et al., 2009; Arendt et al., 2010), the human NSAID trial data are consistent with the new model. At the practical level, the model predicts that there is a postamyloid, postinflammatory biology that offers important new areas for neuroprotective drug discovery.
The final common path—neurodegeneration and dementia
The loss of synapses and circuitry
A significant case has been made that the loss of synaptic structure and function is an integral part of the advancement of AD (Selkoe, 2002, 2008; Arendt, 2009; Palop and Mucke, 2010). The failure of these uniquely neuronal structures has been seen at the level of the single cell and also in the neural networks of the region. There is a loss of dendritic mass and a decrease in spine density in postmortem AD brain (Uylings and de Brabander, 2002). There is also a strong correlation between the sites of synaptic loss and the regions of the most dramatic neurodegenerative changes (Terry et al., 1991). The use of biochemical and immunocytochemical markers of both presynaptic and postsynaptic structures has validated this observed decrease (Hamos et al., 1989; Lippa et al., 1992). In vitro, electrophysiological studies in slice preparations have shown that when Aβ levels are elevated, they interfere with both long-term potentiation and long-term depression (Shankar et al., 2008; Li et al., 2009), and the connection has recently been made between the altered chemistry of the AD brain and the triggering of electrical activity consistent with seizure (Palop et al., 2007; Palop and Mucke, 2009).
The role of tau
Hyperphosphorylated forms of tau—modifications that weaken its affinity for microtubules—are found as the main protein constituent of the neurofibrillary tangle (NFT). This is the second of the two archetypal lesions described by Alzheimer in 1906, and the one that has proven the more reliable partner of neurodegenerative change (Gómez-Isla et al., 1997; Mitchell et al., 2002; Bennett et al., 2004). Indeed the tight correlation between the anatomical location of the NFTs and sites of greatest neuronal cell loss in AD argues for a central role of tau phosphorylation in the disease. In support of this concept, transgenic mice carrying human tau mutations develop a late-onset neurodegenerative phenotype that includes neuronal loss (Andorfer et al., 2005; Ramsden et al., 2005). The role of tau in AD is likely a complex one, however, as mutations in the tau gene itself (MAPT) have not been identified in familial forms of AD. Rather, MAPT mutations have been identified as leading to a separate late-onset form of dementia known as FTDP-17 (Hutton et al., 1998; Poorkaj et al., 1998). The simplest way of incorporating these findings into the new model is to place the role of tau in the final stages of the disease process, following the change of state. This envisions the phosphorylation of tau as a mechanistic part of the cell death program. Given the complexity of the disease, however, and the early appearance of NFTs in the progress of AD, it is highly likely that tau plays one or more additional roles in setting up the change of state in the affected neurons.
The role of autophagy and endosome dysfunction
Autophagy involves the coordination of a number of vesicle populations culminating in a process that assists the cell in degrading long-lived proteins and spent organelles. It is stimulated rapidly in response to various stresses, and results in the formation of an autophagosome, which then fuses with a lysosome and leads to the degradation of the contents. This process is of particular interest in AD research because if it is overstimulated, it can lead to cell death (Nixon, 2007). As might be expected, neurons at risk for death in AD show marked defects in their autophagic function (Cataldo et al., 1996). The presenilin mutations may also play a role here. Recent evidence has shown that one of the normal functions of presenilin is to facilitate the acidification of the cell's lysosomes, a requirement for efficient autophagy (Lee et al., 2010). As autophagy is one of the main defense mechanisms for clearing failed organelles and large protein aggregates from a cell, any compromise at this stage would only hasten the loss of cellular integrity and make the cell death process more rapid and more sure.
Amyloid and the amyloid cascade hypothesis revisited
It is gratifying that the new model comfortably incorporates the amyloid cascade as a contributing cause of AD pathogenesis. Extracellular Aβ naturally accumulates with age, and with time, the non-neuronal cells of the brain would be expected to sense its presence and react (Matsuoka et al., 2001). The associated cytokines then enhance the production of the Aβ peptide. This creates a feedforward reaction described previously in the writings of Griffin (Griffin et al., 1998; Griffin, 2006). Other factors, such as excessive synaptic activity of the type found during epilepsy or excitotoxic injury, can also enhance Aβ production. Thus, driven by one or more of these means, Aβ aggregates stimulate the immune response, and the immune response stimulates more Aβ production. In this way a cycle is created—the amyloid deposition cycle (Fig. 3). While this description requires no change in the well described chemistry of APP metabolism, recasting the amyloid cascade as an amyloid deposition cycle presents a new view of the linkage between this chemistry and the biology of AD. In this view, the deposition of amyloid is tightly linked to, but mechanistically distinct from, the forces that drive the development of dementia. Thus, the model predicts that while Aβ deposition and chronic inflammation each renders the other more likely, neither one has an absolute requirement for the other to be present in the brain.
Figure 3.

The amyloid deposition cycle. In this diagram, the amyloid cascade of Figure 1 is reenvisioned as a feedforward cycle of amyloid deposition and inflammatory responses. The inflammation step of the cycle connects it directly to the age-dependent hypothesis (grayed text on the right side of the diagram), integrating the two models. As the processes are mechanistically distinct, however, the presence of amyloid is not required for the inflammation, and the chronic inflammation is not required for the deposition of amyloid.
Note that the proposed close linkage between chronic inflammation and the Aβ deposition cycle offers a pathway by which familial AD genes can lead to disease. By accelerating the deposition of Aβ, these mutations enhance inflammation and thus drive the amyloid deposition cycle earlier and harder than normal, strongly favoring the development of a chronic inflammation. The ApoE protein is likely to play an important role in this process at this stage. ApoE is closely involved in mediating the clearance of Aβ from brain (Shibata et al., 2000; Deane et al., 2008). Thus any change that reduced ApoE-dependent clearance of Aβ, such as the E4 variant of the gene, would enhance deposition, encouraging the establishment of a chronic inflammation and driving the amyloid deposition cycle. ApoE genotype is also recognized for its impact on the cardiovascular system, and since the first step of the new model is postulated to involve a vascular injury, ApoE genotype might independently act to alter the probability of the occurrence of an initiating injury.
This recasting of the role of Aβ in AD has both theoretical and practical significance. At a theoretical level, the prediction that the amyloid deposition cycle can run independently offers a fresh way of looking at the 30% of elderly individuals who are cognitively normal but are found to have significant plaque deposits in their brains. Rather than characterizing them as preclinical Alzheimer's, the new hypothesis suggests that it would be more accurate to say that they are cognitively normal, but their high plaque burden places them at enhanced risk for developing AD in the future. At a practical level, the model makes another important statement: the presence of plaques in the brain, while highly correlated with AD, should not be an essential part of an Alzheimer's diagnosis. This is a major departure from current thinking and thus deserves careful consideration, as it will impact both our premortem and postmortem diagnostic criteria in substantial ways. One attractive feature of adopting this idea is that it removes a troubling piece of circular logic from our current models. Presently, we are comfortable identifying individuals as having plaques without Alzheimer's, but we have stipulated that it is impossible to have Alzheimer's without plaques. There is no inherent biological reason for this; we have simply defined away this category. If an individual presents with a classical behavioral and neurological course of AD symptoms, but their brain does not contain the requisite plaque burden and tangle density at autopsy, the dementia that they had was not Alzheimer's disease—by definition. Note that the new model predicts that this total discordance between plaques and AD will be rare in practice since the amyloid deposition cycle is stimulated by the same immune response that drives the neurodegeneration. This encourages us to continue to use Aβ and plaque-based outcome measures in our diagnoses. But just as there can be Parkinson's without Lewy bodies (Gomez and Ferrer, 2010), it may be wise for us to consider the possibility that there can be Alzheimer's without plaques.
Conclusions
Viewed in its entirety (Fig. 4), the new model is a complex web of interactions. This complexity makes viewing the diagram difficult, but it probably represents an accurate reflection of the biology underlying Alzheimer's disease, arguably one of the most complex diseases of the human nervous system. A key strength of the new framework is that it begins not with pathology, but with age, a well correlated risk factor. In doing so, it encourages us to draw on the rapid advances that have been made in the biology of aging and incorporate these insights into the design of new therapeutic approaches to Alzheimer's. The three tenets of the new model (Figs. 2, 4) can each be seen as enlarging the number of targets for future intervention. The requirement for an initiating injury may not offer specific pharmaceutical targets, but it suggests that adjunct therapies and treatments may well be used to improve the action of more targeted drugs.
Figure 4.

The expanded age-dependent hypothesis of Alzheimer's disease. The three tenets of the new hypothesis are displayed in bold type on blue ovals and numbered for clarity. The interconnected amyloid deposition cycle is displayed in a similar fashion. Added to the diagram are a few of the cell biological and biochemical processes that play significant roles in Alzheimer's disease. Blue arrows (single- and double-headed) provide a small bit of the interactive nature of the various events. For clarity, some events are illustrated twice (e.g., “autophagy defects”). The light blue text and arrows at the top of the diagram are meant to symbolize the possibility discussed in the text that other late-onset neurodegenerative diseases may begin from the same origin as Alzheimer's disease, but differ based on their initiating injury and downstream response.
The requirement for chronic inflammation opens a wide array of well studied targets for potential intervention. Many laboratories are already exploring this pathway, but there is much to learn. A critical unanswered question, indeed a key challenge for future research, is how does a process as nonspecific and prevalent as brain inflammation reproducibly create the unique set of symptoms we recognize as Alzheimer's disease? While part of the answer is likely to lie in the persistent nature of the response, the bulk of the answer is most probably found in an Alzheimer-specific quality to the “marinade” of cytokines and chemokines that are produced by the aging astrocytes, microglia, endothelial cells, and others. A useful context in which to view this challenge can be found in the observation that experimental allergic encephalomyelitis (EAE) and multiple sclerosis (MS) are both demyelinating conditions caused by brain inflammation. Yet, EAE is now recognized as an imperfect model of MS with treatments that function as cures in the model proving ineffective or worse in the human disease (Sriram and Steiner, 2005; Baxter, 2007). This emphasizes the diversity of possible neuroinflammatory responses in different conditions and urges us to learn their details in the context of the AD brain.
The proposal for a cellular change of state is a call to explore the cell biological changes that move the degenerative process after the change of state has occurred. The model proposes that cell cycle events, defects in autophagy, and alterations of tau are part of this process, but because they exert their actions after the change of state, they must now be reexamined with the assumption that the processes with which they are involved can be independent of both amyloid and inflammation.
Finally, beyond Alzheimer's, the new model contains the seeds of a reexamination of other diseases. The proposition of an initiating injury that is required to begin the process of Alzheimer's disease comes with the strong implication that different injuries to the same age-weakened brain will lead to different responses of the cells of the brain and in so doing begin different disease processes (Fig. 4, light blue arrows). The most clear-cut example of this would be Parkinson's disease. In this case, the ability of paraquat and MPTP to mimic the symptoms of the disease hints that Parkinson's may represent the brain's reaction to oxidative damage and mitochondrial malfunctions rather than inflammatory changes. What makes this last notion particularly appealing is the prediction that if different late-onset neurodegenerative diseases can evolve from a common origin but take different pathways to neuronal loss, then the late-life dementias should often appear as mixed dementias. This follows because it is unlikely that one type of injury precludes a second. The occurrence of these mixed dementias vexed researchers performing the early studies of Alzheimer's disease, but can be viewed in the context of the new model as a predictable consequence of how the diseases begin.
Footnotes
Editor's Note: Disease Focus articles provide brief overviews of a neural disease or syndrome, emphasizing potential links to basic neural mechanisms. They are presented in the hope of helping researchers identify clinical implications of their research. For more information, see http://www.jneurosci.org/misc/ifa_minireviews.dtl.
Financial support from Rutgers University, the Alzheimer's Association, and the National Institutes of Health (AG029449; NS20591) is gratefully acknowledged. I thank my friends and colleagues, including Jianmin Chen, Jiali Li Jie Zhang, and Haley Fitzpatrick, for their helpful comments during the preparation of this manuscript. Special thanks go to the contributions of Gary Landreth for insightful and critical comments; to Maria Carrillo for helpful discussions and suggestions; and to Gina Kolata, whose beautifully written series of articles in the New York Times triggered the urge to present this model in a formal way.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–161. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- Arendt T, Rödel L, Gärtner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. Neuroreport. 1996;7:3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- Arendt T, Brückner MK, Mosch B, Lösche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010;177:15–20. doi: 10.2353/ajpath.2010.090955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer's disease. Neuroscientist. 2002;8:276–283. doi: 10.1177/1073858402008003013. [DOI] [PubMed] [Google Scholar]
- Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol. 2007;7:904–912. doi: 10.1038/nri2190. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–384. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci. 2004;5:677–685. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer's disease: therapeutic implications. Trends Pharmacol Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Fuller S, Steele M, Münch G. Activated astroglia during chronic inflammation in Alzheimer's disease—do they neglect their neurosupportive roles? Mutat Res. 2010;690:40–49. doi: 10.1016/j.mrfmmm.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Giri R, Selvaraj S, Miller CA, Hofman F, Yan SD, Stern D, Zlokovic BV, Kalra VK. Effect of endothelial cell polarity on beta-amyloid-induced migration of monocytes across normal and AD endothelium. Am J Physiol Cell Physiol. 2002;283:C895–904. doi: 10.1152/ajpcell.00293.2001. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gomez A, Ferrer I. Involvement of the cerebral cortex in Parkinson disease linked with G2019S LRRK2 mutation without cognitive impairment. Acta Neuropathol. 2010;120:155–167. doi: 10.1007/s00401-010-0669-y. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Griffin WS. Inflammation and neurodegenerative diseases. Am J Clin Nutr. 2006;83:470S–474S. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamos JE, DeGennaro LJ, Drachman DA. Synaptic loss in Alzheimer's disease and other dementias. Neurology. 1989;39:355–361. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol. 2009;4:389–398. doi: 10.1007/s11481-009-9171-5. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Gabbita SP, Friebe V, Mattson MP, Kindy MS. Oxidized lipoproteins increase reactive oxygen species formation in microglia and astrocyte cell lines. Brain Res. 1999;830:10–15. doi: 10.1016/s0006-8993(99)01272-x. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury MA, Friedman B, McConnell MJ, Rehen SK, Yang AH, Kaushal D, Chun J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc Natl Acad Sci U S A. 2005;102:6143–6147. doi: 10.1073/pnas.0408171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa CF, Hamos JE, Pulaski-Salo D, DeGennaro LJ, Drachman DA. Alzheimer's disease and aging: effects on perforant pathway perikarya and synapses. Neurobiol Aging. 1992;13:405–411. doi: 10.1016/0197-4580(92)90115-e. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemere CA, Duff K. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 2001;158:1345–1354. doi: 10.1016/S0002-9440(10)64085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies [see comments] Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson's disease. Adv Neurol. 2001;86:83–89. [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- Mitchell TW, Mufson EJ, Schneider JA, Cochran EJ, Nissanov J, Han LY, Bienias JL, Lee VM, Trojanowski JQ, Bennett DA, Arnold SE. Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Ann Neurol. 2002;51:182–189. doi: 10.1002/ana.10086. [DOI] [PubMed] [Google Scholar]
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci. 2007;27:6859–6867. doi: 10.1523/JNEUROSCI.0379-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Yu GQ, McConlogue L, Rockenstein EM, Abraham CR, Masliah E. Astroglial expression of human alpha(1)-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic mice. Am J Pathol. 2000;157:2003–2010. doi: 10.1016/s0002-9440(10)64839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Inflammatory process in Parkinson's disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Smith AD. The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience. 1998;87:731–739. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer's disease. Cell Death Differ. 2009;16:378–385. doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram S, Steiner I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol. 2005;58:939–945. doi: 10.1002/ana.20743. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Uylings HB, de Brabander JM. Neuronal changes in normal human aging and Alzheimer's disease. Brain Cogn. 2002;49:268–276. doi: 10.1006/brcg.2001.1500. [DOI] [PubMed] [Google Scholar]
- Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT. Abeta oligomers induce neuronal cell cycle events in Alzheimer's disease. J Neurosci. 2008;28:10786–10793. doi: 10.1523/JNEUROSCI.2441-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Bhaskar K, Kounnas MZ, Wagner SL, Yang Y, Lamb BT, Herrup K. NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Invest. 2009;119:3692–3702. doi: 10.1172/JCI39716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging. 2000;21:797–806. doi: 10.1016/s0197-4580(00)00219-0. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer's disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–784. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Marchani E, Nikisch G, Müller U, Nolte D, Hertel A, Wijsman EM, Bird TD. The N141I mutation in PSEN2: implications for the quintessential case of Alzheimer disease. Arch Neurol. 2010;67:631–633. doi: 10.1001/archneurol.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]