

Abstract
In neurons, the endosomal system is essential for membrane receptor trafficking to dendrites and axons and thereby participates in various neuronal functions, such as neurite outgrowth and synaptic plasticity. A multitude of regulators coordinates trafficking through endosomes, but most of them have not been studied in detail in neurons. In non-neuronal cells, EHD1 (Eps15 homology-domain containing protein 1) functions in the recycling endosome and is required for endosome-to-plasma membrane transport of multiple cargos. In this study, we analyze the role of EHD1 in neurons. In particular, we investigate whether EHD1 is required for polarized trafficking of the dendritically targeted transferrin and the axonal adhesion molecule L1/NgCAM (neuron–glia cell adhesion molecule) and, if so, in what compartment it is required. We find that endosomal recycling of both L1/NgCAM and transferrin is impaired when EHD1 is downregulated. We show that EHD1 colocalizes with L1/NgCAM and transferrin mostly in EEA1 (early endosome antigen 1)-positive early endosomes and less extensively with recycling endosomes. Using live imaging, we observe that EHD1 is stably associated with endosomal membranes during their maturation into EEA1-positive compartments and often persists on them longer than EEA1. Finally, we show that downregulation of EHD1 causes a delay of L1/NgCAM in exiting EEA1-positive endosomes, resulting in impaired targeting of L1/NgCAM to the axonal membrane. We conclude that, in neurons, EHD1 functions in early endosomes rather than (or possibly in addition to) recycling endosomes. These findings point to the existence of neuronal adaptations of the endosomal system.
Introduction
Neuronal polarity requires that proteins are spatially segregated to axonal and somatodendritic domains. One of the mechanisms underlying compartmentalization of the plasma membrane is endocytosis (Lasiecka et al., 2009). Understanding the neuronal endosomal system is crucial given the multitude of processes in which endocytosis plays a role: neurotrophic signaling, degradation of proteins, axonal pathfinding, synaptic vesicle recycling, synaptic plasticity, and more.
In non-neuronal cells, the endosomal system consists of a series of heterogeneous organelles, namely early, recycling, and late endosomes, and lysosomes. The identity of endosomes is ensured by the specific localization of regulators. Live imaging demonstrated that endosomes are dynamic (Rink et al., 2005; Lakadamyali et al., 2006; Zoncu et al., 2009), so the challenge is to define compartmental boundaries and cargo flux between compartments. Endosomes in neurons are not well characterized. The most studied neuronal endosomes are those involved in synaptic vesicle recycling, retrograde transport of neurotrophins, and recycling AMPA receptors (for review, see Howe and Mobley, 2004; Kennedy and Ehlers, 2006; Schweizer and Ryan, 2006), but many questions remain even there. For all other sites and cargos, little is known.
L1 [and its chick homolog NgCAM (neuron–glia cell adhesion molecule)] is a cell adhesion molecule highly enriched on the axon. L1/NgCAM is involved in axonal pathfinding, branching, and myelination (Maness and Schachner, 2007). L1 endocytosis is required for growth cone advance on L1 substrate (Kamiguchi and Lemmon, 2000). Our laboratory has demonstrated that endosomal trafficking is also required to target L1/NgCAM properly to the axon. L1/NgCAM reaches the axon indirectly by transcytosis, involving initial somatodendritic targeting followed by endocytosis and trafficking to the axon from somatodendritic endosomes (Wisco et al., 2003; Yap et al., 2008a,b).
The EHD (Eps15 homology-domain containing protein) family, comprising EHD1–EHD4, plays crucial roles in endosomal trafficking in non-neuronal cells. Structural studies of EHD2 indicate that oligomers of EHD2 bind to lipids and deform them into tubules (Daumke et al., 2007), suggesting similarities with dynamin. EHD proteins also associate with preexisting tubules (Jović et al., 2009; Sharma et al., 2009). EHD1 can localize to recycling endosomes and also to early endosomes (Lin et al., 2001; Naslavsky et al., 2006; Blume et al., 2007; George et al., 2007). Its functions, however, have been primarily associated with the recycling endosome in nonpolarized cells based on downregulation of EHD1, expression of dominant-negative EHD1 (Grant and Caplan, 2008), and analysis of transferrin (Tfn) recycling in fibroblasts derived from EHD1-deficient mice (Rapaport et al., 2006). Dominant-negative EHD1 also delays AMPA receptor and transferrin recycling in neurons (Park et al., 2004), but the effects of downregulation of EHD1 and its site of action in neurons have not been characterized.
Recently, we found that downregulation of EHD1 impaired NgCAM targeting to the axon. It also led to increased accumulation of NgCAM in somatodendritic endosomes, potentially resulting from missorting during recycling (Yap et al., 2010). Here we analyze the identity and dynamics of EHD1-positive endosomes in neurons and determine in which compartments trafficking is impaired when EHD1 is downregulated.
Materials and Methods
Cell culture
Primary cultures of hippocampal neurons were obtained from embryonic day 18 rat hippocampi as described (Yap et al., 2008b). Cells were plated on poly-l-lysine coverslips and incubated with DMEM with 10% horse serum. After 4 h, the cells were transferred into serum-free medium supplemented with B27 (Invitrogen) and cultured for 9–12 d in vitro.
Reagents
The 8D9 anti-NgCAM hybridoma was obtained from National Institutes of Health Hybridoma Bank. Tissue culture supernatants were concentrated over a T-gel column (Thermo Fisher Scientific). Purified IgGs were coupled to Alexa Fluor-488 according to the instructions of the manufacturer (Invitrogen). Rabbit anti-Flag antibodies were purchased from Sigma, whereas mouse anti-EEA1 (early endosome antigen 1) antibody was from BD Biosciences Transduction Laboratories. FITC, cyanine 3, DyLight488, and DyLight649 rat transferrin were obtained from Jackson ImmunoResearch. Polyclonal antibody against NEEP21 (neuron-enriched endosomal protein 21 kDa) was raised using a peptide containing amino acids 7-23 of NEEP21 (Open Biosystems). Anti-NEEP21 antibody and NEEP21–green fluorescent protein (GFP) plasmid were described previously by Steiner et al. (2002). Anti-lgp120 rabbit antiserum “Mingus” was a gift from Dr. Ira Mellman (Genentech, South San Francisco, CA). Anti-myc antibodies were purchased from Santa Cruz Biotechnology. Affinity-purified anti-EHD1 antibodies were described previously (Caplan et al., 2002; Naslavsky et al., 2006, 2009; Sharma et al., 2008). Secondary antibody reagents were obtained from Invitrogen. Rat L1–myc was provided by Dr. D. Felsenfeld (Mount Sinai School of Medicine, New York, NY). GFP–EEA1 plasmid was kindly provided by Dr. Silvia Corvera (University of Massachusetts Medical School, Worchester, MA). Rab11-GFP plasmid was a gift from Dr. Ira Mellman. FLAG–EHD1 was kindly provided by Dr. Barth Grant (Rutgers University, Piscataway, NJ). mCherry–EHD1 was generated by subcloning mouse EHD1 into pCherry–C1 vector at BamHI and HindIII sites. NgCAM cDNA was described previously (Yap et al., 2008a,b).
Short hairpin RNA plasmids
To specifically knockdown the EHD1 protein expression, we used short hairpin RNA (shRNA) targeting the rat EHD1 sequences obtained from Super Array Bioscience. The shRNAs were expressed from the U1 promoter cassette of pGeneclip hMGFP vector containing GFP under the regulation of the cytomegalovirus promoter. The sequences of the two short hairpin RNA targeting the rat EHD1 were as follows: shEHD1 #1, GCC CAC CTA TGA TGA GAT CTT; shEHD1 #2, AAG GAG ATG CCC AAT GTC TTT. The scrambled sequence used as a control (shRandom) was GGA ATC TCA TTC GAT GCA TAC. For quantification and specificity of downregulation, see Yap et al. (2010).
Transfection of neuronal cultures
Neuronal cultures at 9–12 d in vitro were transfected using Lipofectamine2000 with 1 μg of DNA, 3 μl of Lipofectamine2000 for 60–90 min, washed, and incubated for 18–24 h or 4 d depending on experiment. To reduce the number of overexpressing cells, plasmids were mixed with empty plasmid containing no insert.
Immunofluorescence
Cells were fixed in 2% paraformaldehyde/3% sucrose/PBS in 50% conditioned medium at room temperature for 20 min. The fixation conditions used do not introduce holes into the overwhelming majority of cells. Coverslips were then blocked in 5% horse serum/1% BSA/PBS for 30 min. Antibodies were diluted in 1% BSA/PBS and incubated for 1–2 h. Coverslips were mounted in Vectashield (Vector Laboratories) and viewed on a Carl Zeiss Axiovert 200 with a 40× objective. Images were captured with the Orca cooled CCD camera (Hamamatsu) using Openlab software (ImproVision) and processed identically in Adobe Photoshop (Adobe Systems).
Endocytosis assay
Neurons expressing NgCAM for 18 h or 4 d (for EHD1 downregulation experiments) were incubated with 8D9 anti-NgCAM antibodies for 20 min at 37°C and washed several times, and all antibody remaining on the surface was stripped by treatment with pH 2.0 MEM for 2 min (Fourgeaud et al., 2003), washed extensively, and returned to the incubator for various amounts of times before fixation in 2% paraformaldehyde/3%sucrose/PBS, pH 7.4. Internalized NgCAM antibody was detected by applying Alexa Fluor-568 goat anti-mouse antibody after permeabilization. Permeabilization was achieved with either 0.05% saponin or 0.2% Triton X-100 for 10 min at room temperature as in the study by Wisco et al. (2003).
Live imaging
For live imaging, cells were grown on 25-mm-diameter round coverslips (Bellco Biotechnology) or 35 mm glass-bottom microwell dishes (MatTek Corporation). For dual live imaging of endocytosed NgCAM and Cherry–EHD1, cells were transfected with NgCAM and Cherry–EHD1 plasmids. At 24 h after transfection, cells were incubated with Alexa Fluor-488-coupled anti-NgCAM antibody for 30 min, washed, and mounted in a live imaging chamber on a heated stage under 5% CO2. For dual imaging of endocytosed transferrin and Cherry–EHD1, cells were transfected with Cherry–EHD1 and, after 24 h, incubated with DyLight488 transferrin for 1 h. For dual live imaging of Cherry–EHD1 and endosomal regulators, cells were transfected with Cherry–EHD1 and either GFP–EEA1 or NEEP21–GFP or rab11–GFP and imaged 24 h later. Single and dual live imaging was performed on a Carl Zeiss Axiovert using 63× or 100× PlanApo oil lenses and Hamamatsu C9100-13 camera. For double live imaging, images from the two channels were taken sequentially at every time point as fast as the camera allowed. To avoid offset between the two channels attributable to fast motion artifacts, the shortest possible exposure times were used (usually 50–100 ms). For triple live imaging of Cherry–EHD1, NgCAM and transferrin cells were transfected with Cherry–EHD1 and NgCAM. At 24 h after transfection, cells were incubated with DyLight649 transferrin for 1 h and Alexa Fluor-488-coupled anti-NgCAM antibody for 30 min. Cells were subsequently washed, mounted on a heated stage, and imaged. Triple live imaging was performed on Leica SP5 X confocal microscope using 60× PlanApo water objective and white-light laser, with between lines sequential scanning mode. Only cells expressing low or moderate amounts of proteins were chosen for imaging to minimize overexpression artifacts. Cells in which the overexpressed protein localized abnormally, changed compartment morphology, or changed cell morphology were not used for live imaging. Expression levels of transfected proteins were compared, and, if necessary, plasmid concentration was adjusted to equalize expression.
Image quantification and analysis
For quantifying colocalization of NgCAM or L1–myc with EEA1, lgp120 and NEEP21 in 10–25 fixed cells were analyzed per condition.
L1–myc and EEA1.
Anti-myc antibody was allowed to endocytose for 20 min, unbound antibody was washed away, and internalized antibody was chased for 90 min before fixation and immunostaining. Images were taken at identical exposure settings and processed identically. Thresholds were set for each channel in Adobe Photoshop to exclude background staining, channels were merged, and overlapping endosomes were counted in each cell.
NgCAM and lgp120.
Anti-NgCAM antibody was allowed to endocytose for 20 min, internalized antibody was chased for 90 min, and surface-remaining antibodies were stripped before fixation and immunostaining. Both channels were thresholded in Adobe Photoshop, and total overlapping pixels were determined in NIH Image J and divided by the total number of NgCAM-positive pixels in the soma.
NgCAM and NEEP21.
Anti-NgCAM antibody was allowed to endocytose for 20 min, and surface-remaining antibodies were stripped before fixation and immunostaining. Both channels were thresholded in Adobe Photoshop, and endosomes containing both markers were counted in dendrites. The soma was excluded from analysis because NEEP21 is also expressed in the trans-Golgi network. Statistical tests were performed using SPSS version 13 statistics software or GraphPad Prism (GraphPad Software).
Live imaging.
For colocalization analysis of live imaging data, time-lapse sequences were saved as Volocity files, later exported, and further analyzed in NIH ImageJ. Raw regions of interest from thin dendrites were aligned vertically and bleach corrected (NIH ImageJ plugin using bleach correction optimal fitting). Kymographs were prepared using NIH ImageJ functions: “Reslice” using Z-spacing (pixels) = 1 and “Z-project” using projection-type SD. Kymographs represent a spatial position of the vesicles over time in which y-axis represents time. Therefore, vertical lines are stationary vesicles, and diagonal full or dotted lines with various angles are motile vesicles of various speeds. Speed/distance measurements were performed on kymographs. For intensity changes quantifications, kymographs were prepared using maximum intensity projection. Changes in fluorescence intensity were measured using NIH ImageJ measurement of pixel intensity along 1-pixel-wide line along the vesicle trace on the kymograph. To calculate percentage changes in fluorescence intensity during duration of the time lapse, the average of the first five frames and the last five frames were used. Intensity changes were scored as positive if the fluorescence intensity at the end of the time lapse changed by >16%. Graphs were prepared in GraphPad Prism and Microsoft Excel, and final figures were prepared in Adobe Photoshop.
Results
EHD1 colocalizes with internalized NgCAM and transferrin in neurons
Recently, a EHD1 knock-out mouse lacking all EHD1 expression was generated. This EHD1 −/− mouse shows severe fertility and eye defects (Rainey et al., 2010), but the brain was not analyzed. We showed previously that expression of short hairpin-interfering plasmids against EHD1 caused missorting of NgCAM to the somatodendritic surface and increased the accumulation of NgCAM in somatodendritic endosomes (Yap et al., 2010). Using affinity-purified antibodies specific to EHD1 (Sharma et al., 2008), we showed that endogenous EHD1 in neurons was found in presumptive endosomes and in the nucleus (Sharma et al., 2009). These antibodies were difficult to produce and showed only faint staining in neurons. However, the overall staining pattern was consistent with EHD1 distribution in non-neuronal cells, with the exception of the nuclear staining. Strikingly, we did not observe staining of tubules, which are often observed in non-neuronal cells (Yap et al., 2010). Because the endosomal staining of endogenous EHD1 is similar to the staining obtained with tagged EHD1, we used FLAG–EHD1 for all additional studies.
We first tested whether EHD1 colocalized with internalized NgCAM in the somatodendritic domain. After 20 min of anti-NgCAM antibody uptake, internalized NgCAM (shown in red) significantly overlapped with FLAG–EHD1 (cyan) staining (Fig. 1 A, arrowheads and arrows). The overlap of internalized NgCAM and FLAG–EHD1 can be visualized with fluorescence intensity scans along the dendrites. Multiple overlapping fluorescence intensity peaks of FLAG–EHD1 (cyan) and internalized NgCAM (red) could be seen (Fig. 1 A′). When colocalization was quantified, 45% of labeled endosomes costained for FLAG–EHD1 and endocytosed NgCAM (n = 496 endosomes). We note that some of the colocalizing compartments show an apparent lateral offset between NgCAM and FLAG–EHD1 (Fig. 1 A,A′, arrows). These kinds of images are reminiscent of laterally offset profiles (Miaczynska and Zerial, 2002) we noticed previously with NgCAM and transferrin (Yap et al., 2008b). To determine whether EHD1 also colocalized with the somatodendritically recycling cargo Tfn, we expressed FLAG–EHD1 and performed FITC–transferrin uptake. Internalized transferrin (red) significantly colocalized with FLAG–EHD1 (cyan) after 1 h of continuous uptake (45% colocalization, n = 571 endosomes) (Fig. 1 B, arrowheads and arrows, B′). Therefore, both somatodendritically and axonally targeted cargos endocytosed into EHD1-positive endosomal compartments in the somatodendritic domain.
Figure 1.

EHD1 colocalizes with endocytosed NgCAM and transferrin and is important for transferrin recycling. A, Neurons coexpressing FLAG–EHD1 (cyan) and NgCAM (red) were allowed to endocytose anti-NgCAM antibodies for 20 min before fixation. Endocytosed NgCAM was detected with a secondary antibody, whereas FLAG–EHD1 was detected with anti-FLAG rabbit antibody. Single channels as well as overlaid channels (colocalization appears white) are shown for the soma region and part of a dendrite. Scale bar, 10 μm. A′, Representative intensity line scan of FLAG–EHD1 (cyan) and NgCAM (red) along a dendrite is shown. Endosomes correspond to the peaks on the trace. Overlapping peaks for FLAG–EHD1 and NgCAM indicate colocalization of both markers. B, Neurons expressing FLAG–EHD1 (cyan) were allowed to endocytose FITC rat transferrin (red) for 1 h before fixation. FLAG–EHD1 was detected with anti-FLAG rabbit antibody. B′, Representative intensity line scan of FLAG–EHD1 (cyan) and transferrin (red) along the dendrite is shown. Precisely coaligned peaks are marked with arrowheads. Peaks with lateral offset are marked with arrows. C, Untransfected neurons or neurons transfected with either shEHD1 #1–GFP or shEHD1 #2–GFP or shRandom–GFP were allowed to uptake cyanine 3 rat transferrin for 1 h. D, Untransfected neurons (clear rhombus) or neurons transfected with either shEHD1 #1 (blue triangle) or shEHD1 #2 (blue square) or shRandom (clear circle) were allowed to uptake cyanine 3 rat transferrin for 1 h, then unbound transferrin was washed out, and bound transferrin was chased for 0, 27, and 40 min. Percentage of transferrin retained in soma was measured and normalized to t (0). Statistics were performed using Student's t test. *p < 0.01, **p < 0.0001. Error bars indicate SEM. AU, Arbitrary units.
Downregulation of EHD1 delays transferrin recycling in neurons
EHD1 downregulation caused accumulation of NgCAM in endosomes (Yap et al., 2010). We asked whether downregulation of EHD1 also had an effect on recycling of another cargo in neurons, namely somatodendritically recycling transferrin. Neither of two shEHD1 constructs [shEHD1#1 and shEHD1#2 (for details, see Yap et al., 2010)] significantly affected steady-state transferrin loading into endosomes after 20 min (data not shown) or 60 min (Fig. 1 C) of incubation. When recycling of preloaded Tfn was measured, higher levels of transferrin remained in endosomes at 27 and 40 min of chase time (Fig. 1 D). Downregulation of EHD1, thus, caused a delay in transferrin recycling.
EHD1-positive endosomal compartments in neurons are dynamic
Because EHD1 was important for both NgCAM and transferrin recycling, we wanted to characterize the endosomal population, with which EHD1 was associated in neurons. We therefore examined the dynamic behavior of EHD1-positive endosomal compartments. Neurons were transfected with Cherry–EHD1, and live imaging was performed in dendrites. For our analysis, we chose only the low expressing cells that did not show changes in dendritic morphology (Park et al., 2004).
The Cherry–EHD1-positive compartments in dendrites resembled the compartments stained with antibodies and were mostly round (94%) rather than tubular (Fig. 2 A–C). Time-lapse imaging revealed that the vast majority of Cherry–EHD1 compartments (n = 832 endosomes) were stationary during both a 1 and 10 min imaging period. However, many smaller and dimmer compartments were motile (Table 1). The behavior of Cherry–EHD1 compartments is best appreciated by kymographs (see Materials and Methods): stationary compartments are manifest as vertical lines (Fig. 2 A–C, arrows) and moving compartments as diagonal lines (Fig. 2 A–C, arrowheads). Stationary compartments were of variable sizes with many appearing larger than ∼1.5 μm in diameter (Table 1). These large, stationary compartments might correspond to single endosomes or clusters of multiple endosomes. Moving compartments were rarely of this large size. Movements of EHD1 compartments along the dendrites were often persistent over long distances, both retrograde (toward soma) and anterograde (toward the axon tip), and appeared continuous (Fig. 2 B,C, arrowheads). Examples of individual traced movements are plotted in Figure 2 B′–C′. Motile Cherry–EHD1 compartments fell into three categories based on their speed (Table 1). Because the moving vesicles were usually fainter than the stationary endosomes, this amount of movement was likely an underestimate because fainter movements might have been missed. The motile compartments might correspond to single motile endosomes or small clusters of vesicles that move together.
Figure 2.

EHD1-positive endosomal compartments in neurons are dynamic. A–C, Neurons were transfected with Cherry–EHD1. Images were captured with either 10, 1, or 0.5 s frame rates and total duration of 10 min, 60 s, and 30 s, respectively. The first movie frame of a part of dendrite is shown with the corresponding kymographs underneath. The time dimension is shown on the y-axis, and the retrograde direction is indicated above. Arrows indicate stationary vesicles and arrowheads motile vesicles. Scale bar, 6 μm. B′, C′, Trajectories of anterograde and retrograde moving vesicles from B and C. D–F, Examples of presumptive fusion (E, F) and budding (D, F) of Cherry–EHD1 vesicles during either 30 s or 10 min time lapse. G, Examples of dynamic shape changes of stationary vesicles in the soma, which include the temporary extensions of tubular domains (arrowheads; 3 s frame rate). H, Single frames of first time lapse from G magnified 2× are shown to illustrate example of tubular extension (arrowhead). I, Examples of changes in fluorescence intensity of Cherry–EHD1 vesicles during 10 min time lapse.
Table 1.
Measurements of Cherry–EHD1 endosomes sizes, speeds, and directionality
| Total, 100% (n = 832 scored endosomes) | |||||
|---|---|---|---|---|---|
| Stationary vesicles, 71% (n = 587 scored endosomes) |
Motile vesicles, 29% (n = 245 scored endosomes) |
||||
| Average size range | Average speed range | ||||
| Small (∼0.4–0.6 μm), 37% (n = 215) | Medium (∼0.9–1.1 μm), 42% (n = 248) | Big (∼1.5–1.9 μm), 21% (n = 124) | Slow (∼0.01–0.05 μm/s), 2% (n = 4) | Medium (∼0.09–0.6 μm/s), 16% (n = 40) | Fast (∼0.8–1.6 μm/s), 82% (n = 201) |
| A. 25% (n = 1) | A. 50% (n = 20) | A. 48% (n = 97) | |||
| R. 75% (n = 3) | R. 50% (n = 20) | R. 52% (n = 104) | |||
Quantification of the appearance and behavior of Cherry–EHD1 vesicles during both 0.5–1 min and 10 min time lapse. n = 832 vesicles. A, Anterograde; R, retrograde.
In addition to continuous anterograde and retrograde movements, we also observed other behaviors. The first were events suggestive of fusion and budding of Cherry–EHD1 compartments (Fig. 2 D–F). Second, the stationary compartments in the soma occasionally underwent dynamic shape changes, which included the temporary extensions of tubular domains (Fig. 2 G,H). These tubular extensions disappeared rapidly, a result of either retraction or possibly budding of an EHD1-negative or -dim tubule. These kinds of putative tubulation events were intriguing because they are consistent with the proposed function of promoting tubule formation from liposomes observed for EHD2 (Daumke et al., 2007). Third, a subset of stationary vesicles in dendrites underwent changes in their brightness (∼15% of stationary vesicles during 10 min time lapse) (Fig. 2 I). These intensity changes could be attributable to either recruitment or dissociation of Cherry–EHD1 to/from vesicles, or fusion or budding of small Cherry–EHD1 carriers into/from stationary vesicles. Alternatively, fluorescence intensity changes of stationary vesicles could also be attributable to the movement of those vesicles out of the focal plane. The dynamic behavior of EHD1-positive endosomes thus was consistent with the notion that they were involved in sorting and trafficking of endosomal cargos (supplemental Movie 1, available at www.jneurosci.org as supplemental material).
Stationary and motile EHD1 compartments contain endocytosed cargos
Because downregulation of EHD1 affected NgCAM and transferrin recycling in somatodendritic endosomes, we asked whether internalized cargos were found in stationary EHD1 endosomes and transported in motile EHD1 endosomes. Neurons were cotransfected with Cherry–EHD1 and NgCAM for 24 h, Alexa Fluor-488 anti-NgCAM antibody was used in an endocytosis assay for 30 min, unbound antibody was washed out, and dual live imaging was performed. Endosomes were scored for movement and content of EHD1 and NgCAM (n = 1092 endosomes) (Fig. 3 E). Approximately half of the stationary Cherry–EHD1 (cyan) compartments contained endocytosed NgCAM (red) after 30 min uptake (Fig. 3 B, arrow, E). However, only few motile Cherry–EHD1 compartments contained NgCAM (Fig. 3 B, arrowheads, E). EHD1-positive, NgCAM-positive motile carriers mostly moved in the retrograde direction with slow and medium speeds (as defined in Table 1). NgCAM motile carriers devoid of detectable Cherry–EHD1 could be frequently observed, moving bidirectionally in dendrites, with mostly medium and fast speeds (Fig. 3 A,C, arrowheads). A presumptive fusion event of an EHD1-positive vesicle with an NgCAM-positive vesicle is shown in Figure 3 D. Therefore, internalized NgCAM accumulated in stationary EHD1 compartments, but was mostly transported in EHD1-negative carriers. The EHD1-motile compartments might either transport different cargos or recycle EHD1 molecules.
Figure 3.

Stationary and motile EHD1 compartments contain endocytosed cargos. Neurons were transfected with Cherry–EHD1 and NgCAM (A–E) or Cherry–EHD1 alone (F–J), and endocytosis assays of NgCAM (A–E) or Tfn (F–J) were performed 24 h later. A–D, Dynamic behavior of Cherry–EHD1 (cyan) and endocytosed NgCAM (red) in dendrites. First frame of time-lapse of part of dendrite is shown with the respective kymograph underneath. The time dimension is shown on the y-axis, and the retrograde direction is indicated above. Arrows indicate stationary vesicles, and arrowheads indicate motile vesicles. Scale bar, 6 μm. Images were captured with 10 s frame rate (A, B) and 1 s frame rate (C, D). A, Examples of motile EHD1-positive vesicles and NgCAM-positive vesicles (arrowheads). B, Example of stationary EHD1–NgCAM-positive vesicle (arrow) and motile vesicle (arrowheads). C, Example of stationary EHD1-positive vesicle (arrow) and motile NgCAM-positive vesicle (arrowhead). D, Example of apparent fusion between NgCAM-positive vesicle and EHD1-positive vesicle. E, Quantification of colocalization and motility of all endosomes (n = 1092) in the 1 and 10 min imaging periods. F–I, Dynamic behavior of Cherry–EHD1 (cyan) and endocytosed DyLight488 transferrin (red) in dendrites. Images were captured with 1 s frame rate (F, G) and 0.5 s frame rate (H, I). F, Examples of motile EHD1-positive vesicles and transferrin-positive vesicles (arrowheads) and stationary EHD1–transferrin-positive vesicle (arrow). G, Examples of motile EHD1 vesicle, motile Tfn vesicle, and motile EHD1–transferrin-positive vesicle (arrowheads). H, Example of stationary EHD1–transferrin-positive vesicle (arrow) and motile EHD1–transferrin-positive vesicles (arrowhead). I, Example of stationary EHD1–transferrin-positive vesicle (arrow) and EHD1–transferrin-positive motile vesicle (arrowhead). J, Quantification of colocalization and motility of all endosomes (n = 556) in the 1 and 0.5 min imaging periods.
We then analyzed the extent of dynamic colocalization of EHD1 with internalized transferrin. Cells were transfected with Cherry–EHD1 for 24 h and then incubated for 1 h with DyLight488–transferrin, followed by washing out unbound transferrin and dual live imaging (Fig. 3 F–I). Endosomes were scored for movement and content of EHD1 and Tfn (n = 556) (Fig. 3 J). Approximately half of all stationary Cherry–EHD1 compartments contained internalized Tfn (red in Fig. 3 H,I, arrows, J). Interestingly, 27% of motile EHD1 carriers contained Tfn (Fig. 3 G–I, arrowheads, J). Motile carriers containing transferrin but devoid of detectable EHD1 could also be observed (Fig. 3 F,G, arrowheads). Therefore, internalized transferrin and NgCAM accumulated in stationary EHD1 compartments, but, in contrast to NgCAM, transferrin was also frequently observed in EHD1-positive motile carriers (supplemental Movies 2, 3, available at www.jneurosci.org as supplemental material).
Last, we determined whether Tfn and NgCAM can colocalize in stationary EHD1-positive endosomes by performing triple live imaging with DyLight649–Tfn, Alexa Fluor-488–anti-NgCAM antibody, and Cherry–EHD1 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We find that Tfn is much more dynamic than NgCAM and remains for a shorter time in many endosomes. This is consistent with the much shorter half-life of Tfn recycling compared with NgCAM. We are able to observe stationary endosomes (marked by EHD1 throughout the time course of the live imaging) that contain endocytosed NgCAM and endocytosed Tfn. Tfn is sometimes not present for the entire time course but colocalizes with NgCAM and EHD1 for at least part of the time (supplemental Fig. 1A,B, arrows, available at www.jneurosci.org as supplemental material).
EHD1 localizes prominently to EEA1-positive early endosomes in neurons
To determine which endosomal compartments contained EHD1 in neurons, we costained neurons with either EEA1 (a common early endosomal marker), NEEP21 (a neuronal-specific early endosomal marker), or rab11 (a recycling endosomal marker) (Fig. 4 A–C). Many antibodies against endosomal markers (rabs, EHD1 proteins) are not well suited for immunofluorescence in neurons. We therefore expressed low levels of tagged rab11 and EHD1 for colocalization studies. Again, only cells with very low expression levels of FLAG–EHD1 and rab11–GFP were used for analysis. FLAG–EHD1 (red) colocalized well with endogenous EEA1 in a large majority of EEA1 compartments (shown in cyan) (72% colocalization, n = 576 endosomes) (Fig. 4 A, arrowheads, A′). Attempts at determining the extent of colocalization of endogenous EHD1 with EEA1 were unsuccessful as a result of the faint and poor quality of the staining. The colocalization of FLAG–EHD1 with the neuronal-specific early endosome marker NEEP21 (cyan) was partial (45% colocalization, n = 377 endosomes), and only occasionally was precise overlap observed (Fig. 4 B, arrowheads, B′).
Figure 4.

EHD1 localizes prominently to EEA1-positive early endosomes in neurons. A, Neurons were transfected with FLAG–EHD1 (red) and stained for endogenous EEA1 (cyan) (colocalization appears white; arrowheads). Single channels as well as overlaid channels are shown for the somatodendritic region and a part of dendrite. A′, Representative intensity line scans of FLAG–EHD1 and EEA1 are shown for part of a dendrite. Overlapping peaks for FLAG–EHD1 (red) and EEA1 (cyan) are visible. B, Neurons were transfected with FLAG–EHD1 and stained for endogenous NEEP21. B′, Representative intensity line scans of FLAG–EHD1 and NEEP21 are shown for a piece of dendrite. C, Neurons were cotransfected with FLAG–EHD1 and rab11–GFP. C′, Representative intensity line scans of FLAG–EHD1 and rab11–GFP are shown for a piece of dendrite. AU, Arbitrary units.
Because in non-neuronal cells EHD1 regulates exit of cargo from recycling endosomes, we examined the colocalization of EHD1 with rab11. Endogenous rab11 staining was diffuse and did not resemble transferrin staining. We therefore did not trust the staining we obtained with commercially available antibodies in neurons. Instead, we performed staining of cells expressing low levels of FLAG–EHD1 and rab11–GFP. Rab11–GFP was enriched in the perinuclear area and localized to both round and tubular compartments, closely resembling the recycling compartments into which transferrin is internalized. FLAG–EHD1 (red) showed some overlap with rab11–GFP (cyan), but again the colocalization was only partial (38% colocalization, n = 985 endosomes) (Fig. 4 C, arrowheads, C′). Therefore, FLAG–EHD1 in neurons was found mostly on early endosomes, in particular EEA1-positive endosomes.
Dynamic colocalization of EHD1 with rab11- and NEEP21-positive endosomes
Our colocalization studies indicated that EHD1 only partially colocalized with rab11 and NEEP21 in neurons. However, the incomplete colocalization could reflect the dynamic state of vesicles, with rapid fusion and budding events resulting in only partial colocalization at any given time point. In fact, in non-neuronal cells, in which EHD1 showed a high level of colocalization with the rab11 effector rab11–FIP2 (Naslavsky et al., 2006), EHD1–DsRed transiently colocalized with rab11–GFP during time-lapse imaging (George et al., 2007), indicating that rab11 and EHD1 colocalization was dynamic. It was also possible that some of the motile EHD1 compartments might not be well preserved in fixed cells, but high colocalization with NEEP21 or rab11 might be detected by live imaging. Therefore, we performed live cell imaging of Cherry–EHD1 (cyan) with either rab11–GFP (red) (Fig. 5 A–D) or NEEP21–GFP (supplemental Fig. 2A–D, available at www.jneurosci.org as supplemental material). Motile EHD1-positive carriers rarely contained rab11 (10%) or NEEP21 (13%) (Fig. 5) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 5.

Dynamic colocalization of EHD1 with rab11-positive recycling endosomes. A–C, First frames of time lapse of part of a dendrite of neurons expressing Cherry–EHD1 (cyan) and rab11–GFP (red) is shown with the corresponding kymographs underneath. The time dimension is shown on the y-axis, and the retrograde direction is indicated above. Duration of time lapse is 30 s (A, B) or 60 s (C), and frame rate is 0.5 and 1 s, respectively. Scale bar, 6 μm. A, Examples of stationary EHD1-positive vesicles (arrows) and motile rab11-positive vesicles (arrowheads). B, Example of two stationary EHD1-positive vesicles that transiently colocalize with rab11. C, The example of two motile rab11-positive vesicles (arrowheads), one of them colocalizes with EHD1. D, Quantification of colocalization of EHD1 with rab11 in stationary and motile vesicles (n = 436 endosomes). There are three classes of vesicles: EHD1-alone, which are positive for only EHD1; EHD1 + rab11, which are positive for both EHD1 and rab11; and “transient overlap” vesicles, which contain EHD1 throughout time lapse but rab11 only for part of the time lapse.
The dynamic colocalization of EHD1 with rab11 differed significantly from that of EHD1 and NEEP21 in that some rab11 fluorescence overlapped dynamically with EHD1-positive endosomes only for part of the time lapse imaging (see an example in Fig. 5 B and quantification in D, “transient overlap”). Twelve percent of all EHD1-positive stationary vesicles showed dynamic, transient colocalization with rab11–GFP during a 1 min time lapse (Fig. 5 D) (n = 436 endosomes). The transient nature of the colocalization appeared to be attributable to rapidly moving rab11 vesicles stopping at or fusing with stationary EHD endosomes and then moving on or budding out. It is also possible that rab11 was recruited from a soluble pool transiently onto EHD1 compartments before dissociating again. Live imaging therefore revealed that there was a pool of rab11, which transiently colocalized with EHD1, possibly as a result of rapid budding and fusion of motile rab11-positive carriers from stationary EHD1-positive compartments, but overall the two markers behaved strikingly differently.
Dynamic colocalization of EHD1- and EEA1-positive early endosomes
Next, we analyzed the dynamic colocalization of EHD1 and EEA1. Live imaging was performed in neurons coexpressing Cherry–EHD1 (cyan) and GFP–EEA1 (red) (Fig. 6). Stationary EHD1-positive compartments highly colocalized with EEA1 (65%; n = 357 endosomes), in agreement with fixed staining (Fig. 6 A–D, arrows, F), whereas moving EHD1 vesicles contained EEA1 to a lesser extent (21%) (Fig. 6 A,B,D, arrowheads, F). On occasion, we observed apparent fusion events between EEA1-positive and EHD1-positive vesicles (Fig. 6 E). Interestingly, colocalization between EEA1 and EHD1 was even more striking when determined from the perspective of EEA1 (Fig. 6 G). However, there was a population of stationary and motile EHD1-positive compartments that did not contain EEA1, presumably either recycling endosomes or neuronal-specific early endosomal compartments (NEEP21-positive). Colocalization of Cherry–EHD1 (cyan) with GFP–EEA1 (red) was rarely observed in non-neuronal cells found in the cultures, suggesting that the high colocalization of Cherry–EHD1 and GFP–EEA1 was not the result of exogenous expression of those proteins but that the high colocalization of EHD1 with EEA1-positive compartments was specific to neuronal cells (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 6.
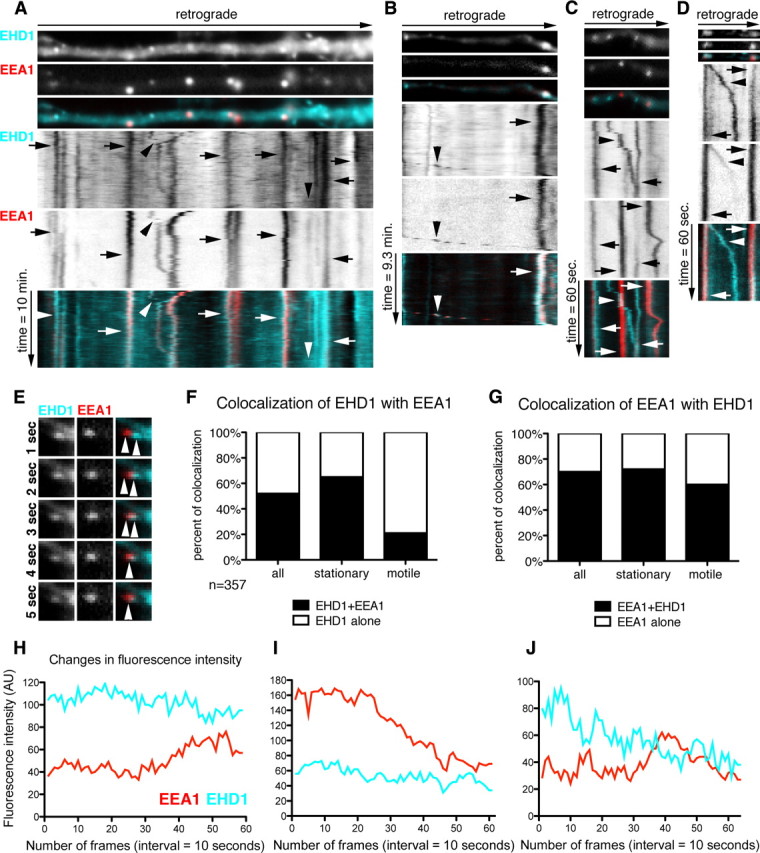
Dynamic colocalization of EHD1 with EEA1-positive early endosomes. A–D, Neurons were cotransfected with Cherry–EHD1 and GFP–EEA1. Time lapse was performed for 10 min duration (A, B) and 1 min duration (C, D). First frame of time lapse of part of dendrite and kymograph are shown. The time dimension is shown on the y-axis, and the retrograde direction is indicated above. A, General view of dynamic colocalization of Cherry–EHD1 (cyan) and GFP–EEA1 (red). Arrows indicate stationary vesicles and arrowheads motile compartments. Scale bar, 6 μm. B, Example of stationary (arrow) and motile (arrowhead) EHD1–EEA1-positive vesicles. C, Example of motile EHD1-positive vesicle (arrowhead) and stationary EHD1–EEA1-positive vesicles (arrows). D, Example of motile EHD1–EEA1-positive vesicle that is losing EEA1 during the time-lapse (arrowhead) and stationary EHD1–EEA1-positive vesicles (arrows). E, Example of presumed fusion of EEA1-positive vesicle with EHD1-positive vesicle (image is magnified 2× for comparing with images A–D). F, Quantification of colocalization of EHD1-positive stationary and motile vesicles with EEA1-positive stationary and motile vesicles (n = 357). EHD1 alone are shown in white, and EHD1 + EEA1 are shown in black. G, Quantification of colocalization of all EEA1-positive stationary and motile vesicles with EHD1-positive stationary and motile vesicles. EEA1 alone are shown in white, and EEA1 + EHD1 are shown in black. H–J, Fluorescence intensity changes in EHD1–EEA1-positive stationary vesicles. Examples of line traces of single vesicles EHD1 (cyan) and EEA1 (red) fluorescence intensity over 10 min time lapse (10 s interval between frames). H, Example of a line trace of fluorescence intensity of vesicle in which neither EHD1 nor EEA1 changed their intensity. I, Example of a vesicle in which EEA1 fluorescence intensity diminished while EHD1 remained constant. J, Example of vesicle in which EHD1 intensity diminished while EEA1 intensity was constant. AU, Arbitrary units.
EHD1 protein, similarly to EEA1 and rab proteins, is a membrane-associated molecule that can be recruited from the cytosol to the endosomal membrane (Grant and Caplan, 2008). Based on our localization data, we asked whether EHD1 might be recruited to EEA1-positive endosomes. EEA1 might then dissociate as the compartment matures to a recycling endosome still containing EHD1. To investigate this possibility, we analyzed the fluorescence intensity of stationary or slowly moving endosomes that were positive for both EEA1 and EHD1 at some point during 1 and 10 min of time-lapse imaging (n = 100 endosomes) (Table 2). The most frequent events were endosomes that did not undergo any significant changes in EHD1 and EEA1 intensity during the time lapse (36%) (Fig. 6 H). The next most frequently observed events were endosomes, which maintained constant EHD1 intensity while EEA1 intensity diminished (31%) (Fig. 6 I). Less frequently, we observed endosomes with constant EHD1 intensity but increase in EEA1 intensity (8%) or decrease in EHD1 intensity but constant EEA1 intensity (9%) (Fig. 6 J). Finally, there was a pool of vesicles that showed loss of both markers (7%). In these cases, we cannot rule out that the vesicles moved out of the focal plane during the time course of the live imaging. Overall, EEA1 fluorescence intensity changed more rapidly and more often than EHD1 intensity. EEA1 intensity changed in 53 of 100 endosomes (14 endosomes increased EEA1 and 39 decreased EEA1), whereas EHD1 intensity changed in 25 of 100 endosomes (6 increased EHD1, 19 decreased EHD1).
Table 2.
Fluorescence intensity changes of Cherry–EHD1 and GFP–EEA1 in endosomes
| EHD1 and EEA1(n = 100) | EEA1 decreasing | EEA1 constant | EEA1 increasing |
|---|---|---|---|
| EHD1 decreasing | 7 | 9 | 3 |
| EHD1 constant | 31 | 36 | 8 |
| EHD1 increasing | 1 | 2 | 3 |
Fluorescence intensity change of single vesicle was reported when fluorescence intensity of either Cherry–EHD1 or GFP–EEA1 or both markers at the end of the 1 and 10 min time lapse was equal or more than 16% of the fluorescence intensity at the beginning of the time lapse. Numbers of vesicles in which EEA1 fluorescence was either constant or decreasing or increasing are shown in columns. Numbers of vesicles in which EHD1 fluorescence was constant, decreasing, or increasing are shown in rows (n = 100).
We conclude that EHD1 is associated with the endosomal membrane for a longer duration than EEA1: EHD1 was often present on compartments before the appearance of EEA1 and persisted on endosomes after EEA1 intensity diminished. Less frequently, EHD1 was lost from EHD1–EEA1 double-positive compartments (supplemental Movie 4, available at www.jneurosci.org as supplemental material).
Downregulation of EHD1 affects L1/NgCAM trafficking through early endosomal compartments
Both the staining of fixed cells and the live imaging raised the hypothesis that the primary site of EHD1 function in neurons was the EEA1-positive early endosome rather than the recycling endosome. To establish where along the endosomal pathway EHD1 function was required, we determined in which compartments L1/NgCAM accumulated when EHD1 was downregulated.
To this end, we cotransfected neurons with L1–myc and either shEHD1 or shRandom plasmids. Then we performed anti-myc antibody uptake for 20 min, washed out unbound antibody, and chased internalized L1–myc for 90 min. We fixed cells and stained endogenous EEA1 (cyan) and internalized L1–myc (red) (Fig. 7 A). In cells expressing shRandom–GFP, colocalization of internalized L1–myc with EEA1 was low after a 90 min chase, as expected (Fig. 7 A, arrowheads, A′). In cells expressing shEHD1–GFP, colocalization of L1–myc with EEA1 was significantly increased, indicating that L1–myc was delayed in exiting EEA1-positive compartments.
Figure 7.
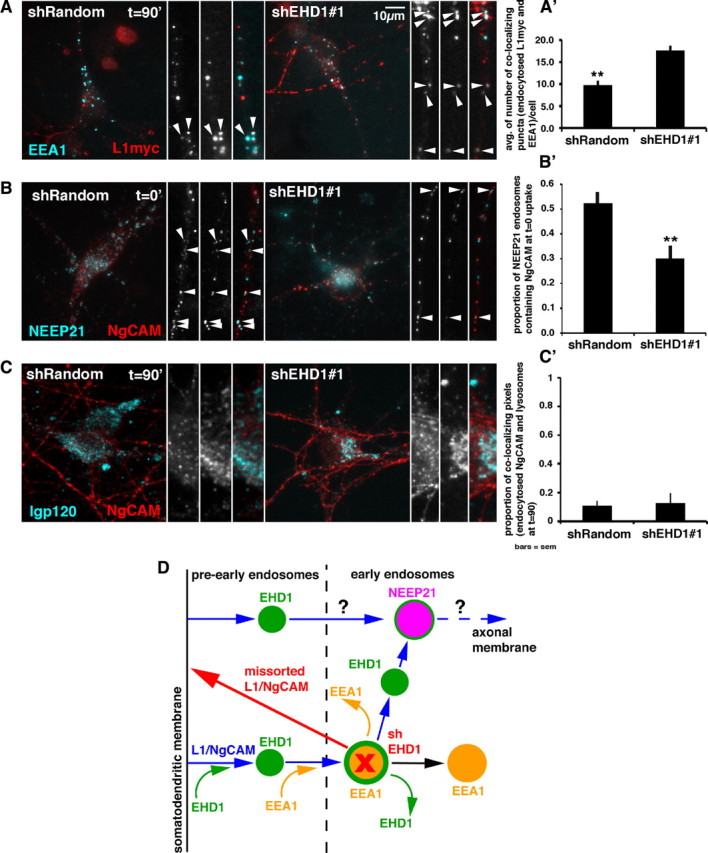
Downregulation of EHD1 affect L1/NgCAM trafficking through early endosomal compartments. A–C, Colocalization of endocytosed L1–myc/NgCAM (red) with endosomal markers (cyan) in cells expressing either shEHD1 #1–GFP or shRandom–GFP (colocalization appears white). The whole cell and piece of dendrite or cell soma are shown. A′–C′, Quantification of colocalization of endocytosed L1–myc/NgCAM with endosomal markers. Statistical significance, t test. Error bars indicate SEM; n = 10–25 cells per condition. A, Colocalization of L1–myc (20 min of anti-myc antibody uptake followed by 90 min chase) with endogenous EEA1. A′, Quantification of the extent of colocalization of endocytosed and chased L1–myc with endogenous EEA1 in cells expressing either shRandom–GFP or shEHD1 #1–GFP. B, Colocalization of NgCAM endocytosed for 20 min and fixed with endogenous NEEP21 in cells expressing either shRandom–GFP or shEHD1 #1–GFP. B′, Colocalization between endocytosed NgCAM and NEEP21 quantified as a proportion of NEEP21 endosomes containing NgCAM. C, Colocalization of NgCAM (endocytosed for 20 min and chased for 90 min) with lysosomal marker lgp120 in cell expressing either shRandom–GFP or shEHD1 #1–GFP. C′, Colocalization between endocytosed NgCAM and lysosomal marker lgp120 quantified as a proportion of colocalizing pixels of endocytosed NgCAM and lysosomes. D, Model of EHD1 localization and function in L1/NgCAM trafficking in neurons. L1/NgCAM (blue arrows) is endocytosed from the somatodendritic plasma membrane via an EHD1/EHD4-mediated pathway into EHD1-positive pre-early endosomal compartments (pre-EE) (green). These compartments then either mature or fuse into early endosomal (EE) compartments (yellow). The transition from pre-EE to EE is achieved through recruitment of EEA1 (yellow curved arrow) to EHD1 endosomal compartments. EHD1–EEA1-positive early endosomal compartments (yellow with green border) can either lose EHD1 (green curved arrow) or lose EEA1 (yellow curved arrow) and mature into EHD1-positive and/or NEEP21-positive endosomes (purple). L1/NgCAM is subsequently trafficked through NEEP21-positive neuronal early endosomes (purple). It is also possible that some L1/NgCAM enters NEEP21 compartments directly without passing through EEA1 compartments. From the NEEP21 compartment, L1/NgCAM is transported toward the axon in currently undefined endosomes (blue). Downregulation of EHD1 (red) leads to delay of L1/NgCAM exit from EEA1-positive compartments and delayed entrance into NEEP21-positive compartments, which in turn increases missorting of L1/NgCAM to the somatodendritic surface (red arrow) and therefore impairs transcytosis of L1/NgCAM to the axon.
We showed previously that internalized NgCAM highly colocalized with NEEP21 and that downregulation of NEEP21 led to missorting of L1/NgCAM to the somatodendritic domain (Yap et al., 2008b), suggesting that NgCAM traversed NEEP21-positive endosomes on its way to the axon. Therefore, we asked whether NgCAM still reached the NEEP21 compartment after EHD1 downregulation. To this end, we cotransfected neurons with NgCAM (red) and either shRandom–GFP or shEHD–GFP, performed anti-NgCAM antibody uptake for 20 min, washed out antibody, fixed cells without a chase, and stained endogenous NEEP21 (cyan). In cells expressing shRandom–GFP, colocalization of NEEP21 with internalized NgCAM was high, as expected at that time point. In cells expressing shEHD1, colocalization of NEEP21 with internalized NgCAM was significantly lower than in control cells (Fig. 7 B, arrowheads, B′), suggesting a delay of internalized NgCAM in reaching the NEEP21 compartment when EHD1 levels were decreased.
Finally, we investigated whether downregulation of EHD1 led to NgCAM missorting to lysosomes, as was the case for NEEP21 downregulation (Yap et al., 2008b). We expressed NgCAM together with either shEHD1–GFP or shRandom–GFP and then performed anti-NgCAM antibody uptake for 20 min, washed out unbound antibody, and chased internalized NgCAM for 90 min (red). We fixed cells and labeled lysosomes with the lysosomal marker lgp120 (cyan). Colocalization of internalized NgCAM with the lysosomal marker was low in cells expressing shRandom or shEHD1 (Fig. 7 C, arrowheads, C′). Thus, downregulation of EHD1 did not lead to missorting of NgCAM to lysosomes. Therefore, the increased accumulation of NgCAM in somatodendritic endosomes that we observed after downregulation of EHD1 (Yap et al., 2010) was attributable to a delay of L1/NgCAM in traversing EHD1–EEA1-positive early endosomal compartments.
Discussion
Regulating which membrane proteins are present at what levels in which locations is a major task for neurons and necessary for modulating neuronal morphology and physiology. One of the major mechanisms of adjusting receptor levels and localization is by membrane trafficking through endosomes. Consequently, it is not surprising that a large number of neurological pathologies result from disturbances of membrane traffic (Aridor and Hannan, 2000). The endosomal system in non-neuronal cells is relatively well understood; after endocytosis into early endosomes, receptors are either trafficked to the late endosome and then to the lysosome for degradation, or sorted to recycling endosomes from which they can return to the plasma membrane (Jović et al., 2010). Recycling can also take place from the early endosome. Endosomes in neurons, conversely, are not well understood, nor are the functions of the proteins known to regulate endosomal transport in non-neuronal cells. We report here that in neurons, EHD1, a putative dynamin-like endosomal regulator, is important for proper trafficking of both axonally targeted L1/NgCAM and dendritically targeted transferrin. EHD1 is localized mainly to EEA1-positive early endosomes in neurons. We show that internalized L1/NgCAM exits EEA1 compartments more slowly and reaches NEEP21 compartments with a delay when EHD1 levels are decreased, suggesting that EHD1 functions in early endosomes in neurons, unlike in fibroblasts in which its functions have been localized to the recycling endosome. Interestingly, interference with EHD1 function by expressing an EHD1 dominant-negative construct also inhibits the recycling of synaptic AMPA receptors (Park et al., 2004). In neurons [as in other cells (Gudmundsson et al., 2010)], EHD1 is therefore important in trafficking of many different cargos and is likely to be involved in vital neuronal functions, such as axon outgrowth and pathfinding, as well as synaptic plasticity.
EHD1 endosomes are dynamic in neurons
Endosomes are highly dynamic (Rink et al., 2005; Lakadamyali et al., 2006; Zoncu et al., 2009), and live imaging is necessary to understand their composition and function. Most of the live imaging studies of endosomal systems so far have been done in non-neuronal cells, and not much is known about the dynamic behavior of the neuronal endosomal system (Prekeris et al., 1999; Hoogenraad et al., 2010). Even in non-neuronal cells, our understanding of the dynamic nature and regulation of the endosomal system is still emerging. The unique morphology of neurons enabled us to visualize and analyze trafficking processes that are difficult to look at even in non-neuronal cells: trafficking events occur along fairly linear tracks in dendrites and axons and can be easily quantified by kymographs.
Given the importance of EHD1 in neuronal function, we embarked on understanding its localization and behavior in neuronal cells. Live-cell imaging of GFP–EHD1 in non-neuronal cells showed GFP–EHD1 tubules with limited motility and smaller tubular and vesicular structures with more motility (Caplan et al., 2002). Furthermore, fluorescence recovery after photobleaching experiments suggested that GFP–EHD1 could be recruited to the tubule from the cytosol and/or is delivered there by vesicular transport (Caplan et al., 2002). Using live imaging, we observed that EHD1-positive endosomes in neurons are either stationary or motile and are rarely tubular. We also noticed EHD1-positive compartments producing transient tubules, as well as fusing and budding with each other. Presumably, motile carriers traffic between stationary compartments, either carrying cargo or endosomal regulators over long distances along dendrites in both retrograde and anterograde directions.
EHD1 in neurons is localized to multiple endosomal populations
In non-neuronal cells, EHD1 is localized to and acts at recycling endosomes. In some cell types, EHD1 colocalizes with both perinuclear, tubular, rab11-positive recycling endosomes and dispersed vesicular compartments, presumably early endosomes (Lin et al., 2001; George et al., 2007). In other cells, colocalization with EEA1-positive early endosomes is not observed; rather EHD1 is found in the recycling endosome in which it colocalizes well with rab11 (Blume et al., 2007) or its effector rab11–FIP2 (Naslavsky et al., 2006). However, in neurons, EHD1 highly localized to EEA1-positive early endosomes. One needs to be careful, therefore, in assuming that the endosomal system of neurons is identical to that of non-neuronal cells. Live imaging revealed that almost all EEA1-positive compartments, both stationary and motile, also contained EHD1. However, there was a pool of EHD1, especially in motile vesicles that did not contain EEA1. Some of these compartments contained the recycling endosomal marker rab11 or neuronal-specific early endosomal marker NEEP21. Interestingly, transferrin was frequently found in motile EHD1 carriers, whereas NgCAM only rarely was. This is consistent with previous studies of transferrin in non-neuronal cells (Sharma et al., 2009).
EHD1 and endosomal maturation
Part of the challenge for understanding the endosomal system is to define compartmental boundaries and cargo flux between compartments. Compartment identity is ensured by the specific localization of specific endosomal regulators. For instance, the early endosomal regulator EEA1, a rab5 effector, is localized to the cytoplasmic side of early endosomal compartments and is thought to be essential for fusion of early endosomes in non-neuronal cells (Simonsen et al., 1998; Christoforidis et al., 1999). One issue of long-standing controversy is the question of how constant or dynamic endosomal compartments are. Much evidence has accumulated to support a “maturation” model, in which earlier compartments in the pathway recruit successively new regulators, which change the compartment identity over time. Recently, live-cell imaging demonstrated that, in non-neuronal cells, the pre-early endosomal compartments mature into early endosomal compartments through the exchange of the pre-early endosomal regulator APPL1, to the early endosomal regulator EEA1 (Zoncu et al., 2009). Similarly, an exchange from rab5 to rab7 occurs when early endosomes mature into late endosomes (Zerial and McBride, 2001; Rink et al., 2005).
Our live imaging established that, in dendrites, many of the compartments containing EEA1 and/or EHD1 are stable in space and time on the order of many minutes (>10 min). Rab11 compartments, in contrast, are mostly highly mobile and do not persist in time at the same location. Whether rab11 vesicles fuse with and bud from stationary EHD1 compartments requires additional experimentation. In addition to stable compartments, we also observed events suggestive of endosomal maturation. Intensity levels of EEA1 and EHD1 on stationary endosomes frequently changed in the course of minutes, suggesting recruitment and dissociation (or budding and fusion) of EHD1 and EEA1. Our observations suggest the model depicted in Figure 7 D: EHD1 precedes EEA1 on early endosomes and persists on endosomes after EEA1 dissociates. We propose that EHD1–EEA1 double-positive compartments mature into EEA1-negative, EHD1-positive endosomes. Other regulators might subsequently be recruited to affect change of endosomal fate. Significantly, EEA1 has a phosphatidyl-insotol-3-phosphate-binding FYVE domain (Gaullier et al., 1998), and the EH domain of EHD1 itself is capable of directly binding phosphoinositides (Naslavsky et al., 2007), aiding recruitment to membranes. How the transient association of EHD1 with specific endosomal membranes is regulated is not understood.
EHD1 is important for trafficking of L1/NgCAM through EEA1-positive early endosomes
Finally, we showed that missorting of NgCAM to the somatodendritic membrane after downregulation of EHD1 occurs through the delay of L1/NgCAM exiting from EEA1-positive endosomes, suggesting that EHD1 acts at the EEA1-positive early endosomes in neurons (see model in Fig. 7 D). This observation differs from the observations made in non-neuronal cells in which EHD1 is primarily important for trafficking of cargo from recycling endosomes (Grant and Caplan, 2008). Consistent with our findings in neurons, EHD1 might at least be transiently found in early endosomes in non-neuronal cells because depletion of the EHD1-binding paralog EHD4 leads to EHD1 retention in EEA1-containing early endosomes (Sharma et al., 2008).
Interestingly, we also observed that, after downregulation of EHD1, there is a delay in the arrival of NgCAM in NEEP21 compartments. This observation suggested that NEEP21 compartments might be positioned later along the endosomal pathway than EEA1–EHD1-positive compartments (Steiner et al., 2002, 2005). These results suggested that delay of L1/NgCAM exiting EEA1-positive early compartments and delay in entering NEEP21-positive compartments led to local L1/NgCAM recycling to the somatodendritic membrane rather than transcytosis to the axon when EHD1 was downregulated. Clearly, precise trafficking through various stations of the endosomal system is tightly regulated. Therefore, it is not only important whether or not receptors are internalized but along which specific endosomal pathway they travel.
Multiple places of action for EHD1 in neurons
EHD1 plays additional neuronal- and cargo-specific roles in endocytosis together with its close family member EHD4 (Yap et al., 2010). We found previously that overexpression of EHD1 or EHD4 leads to decreased endocytosis of NgCAM but not of Tfn. Because EHD1 overexpression did not impair Tfn endocytosis or recycling rates, we suggested that EHD1 homodimers might be responsible for Tfn recycling. Consistent with this idea, downregulation of EHD1 slows the recycling rate of Tfn. NgCAM recycling is also affected when EHD1 is downregulated. Because NgCAM endocytosed poorly when EHD1 was overexpressed, we could not determine whether NgCAM recycling was impaired by EHD1 overexpression (suggestive of EHD1-containing heterodimers) or was unimpaired (suggestive of EHD1 homodimers). NgCAM and Tfn behave similarly when EHD1 is downregulated, whereas they behaved strikingly different during overexpression. EHD dimers are therefore differentially involved in regulating NgCAM and Tfn endocytosis. The oligomeric partner for EHD1 in neuronal early endosomes is not known. EHD1 therefore has multiple sites of action in neurons, including the plasma membrane (in concert with EHD4), and the early endosome.
Footnotes
This work was supported by National Institutes of Health Grant GM086913 (B.W.) and National Center for Research Resources Grants GM074876 and P20 RR018759 (both to S.C). We thank Dr. Heike Folsch (Northwestern University, Chicago, IL) for critical reading of this manuscript. We are grateful to Drs. Barth Grant, Dan Felsenfeld, Ira Mellman, and Sylvia Corvera for generously supplying reagents. Last, we thank the members of the Winckler Laboratory for helpful advice. We acknowledge the Keck Center for Cellular Imaging for help with the Leica 5P5X confocal microscopy system (NIH-NCRR-RR025616).
References
- Aridor M, Hannan LA. Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic. 2000;1:836–851. doi: 10.1034/j.1600-0854.2000.011104.x. [DOI] [PubMed] [Google Scholar]
- Blume JJ, Halbach A, Behrendt D, Paulsson M, Plomann M. EHD proteins are associated with tubular and vesicular compartments and interact with specific phospholipids. Exp Cell Res. 2007;313:219–231. doi: 10.1016/j.yexcr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, Donaldson JG, Bonifacino JS. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J. 2002;21:2557–2567. doi: 10.1093/emboj/21.11.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Bessis AS, Rossignol F, Pin JP, Olivo-Marin JC, Hémar A. The metabotropic glutamate receptor mGluR5 is endocytosed by a clathrin-independent pathway. J Biol Chem. 2003;278:12222–12230. doi: 10.1074/jbc.M205663200. [DOI] [PubMed] [Google Scholar]
- Gaullier JM, Simonsen A, D'Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- George M, Ying G, Rainey MA, Solomon A, Parikh PT, Gao Q, Band V, Band H. Shared as well as distinct roles of EHD proteins revealed by biochemical and functional comparisons in mammalian cells and C. elegans . BMC Cell Biol. 2007;8:3. doi: 10.1186/1471-2121-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Caplan S. Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic. 2008;9:2043–2052. doi: 10.1111/j.1600-0854.2008.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, Koval OM, Cunha SR, George M, Rainey MA, Kashef FE, Dun W, Boyden PA, Anderson ME, Band H, Mohler PJ. EH domain proteins regulate cardiac membrane protein targeting. Circ Res. 2010;107:84–95. doi: 10.1161/CIRCRESAHA.110.216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Popa I, Futai K, Martinez-Sanchez E, Sanchez-Martinez E, Wulf PS, van Vlijmen T, Dortland BR, Oorschot V, Govers R, Monti M, Heck AJ, Sheng M, Klumperman J, Rehmann H, Jaarsma D, Kapitein LC, van der Sluijs P. Neuron specific Rab4 effector GRASP-1 coordinates membrane specialization and maturation of recycling endosomes. PLoS Biol. 2010;8:e1000283. doi: 10.1371/journal.pbio.1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, Mobley WC. Signaling endosome hypothesis: a cellular mechanism for long distance communication. J Neurobiol. 2004;58:207–216. doi: 10.1002/neu.10323. [DOI] [PubMed] [Google Scholar]
- Jović M, Kieken F, Naslavsky N, Sorgen PL, Caplan S. Eps15 homology domain 1-associated tubules contain phosphatidylinositol-4-phosphate and phosphatidylinositol-(4,5)-bisphosphate and are required for efficient recycling. Mol Biol Cell. 2009;20:2731–2743. doi: 10.1091/mbc.E08-11-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jović M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol. 2010;25:99–112. doi: 10.14670/hh-25.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. Recycling of the cell adhesion molecule L1 in axonal growth cones. J Neurosci. 2000;20:3676–3686. doi: 10.1523/JNEUROSCI.20-10-03676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasiecka ZM, Yap CC, Vakulenko M, Winckler B. Compartmentalizing the neuronal plasma membrane from axon initial segments to synapses. Int Rev Cell Mol Biol. 2009;272:303–389. doi: 10.1016/S1937-6448(08)01607-9. [DOI] [PubMed] [Google Scholar]
- Lin SX, Grant B, Hirsh D, Maxfield FR. Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nat Cell Biol. 2001;3:567–572. doi: 10.1038/35078543. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Zerial M. Mosaic organization of the endocytic pathway. Exp Cell Res. 2002;272:8–14. doi: 10.1006/excr.2001.5401. [DOI] [PubMed] [Google Scholar]
- Naslavsky N, Rahajeng J, Sharma M, Jovic M, Caplan S. Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol Biol Cell. 2006;17:163–177. doi: 10.1091/mbc.E05-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Rahajeng J, Chenavas S, Sorgen PL, Caplan S. EHD1 and Eps15 interact with phosphatidylinositols via their Eps15 homology domains. J Biol Chem. 2007;282:16612–16622. doi: 10.1074/jbc.M609493200. [DOI] [PubMed] [Google Scholar]
- Naslavsky N, McKenzie J, Altan-Bonnet N, Sheff D, Caplan S. EHD3 regulates early-endosome-to-Golgi transport and preserves Golgi morphology. J Cell Sci. 2009;122:389–400. doi: 10.1242/jcs.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Prekeris R, Foletti DL, Scheller RH. Dynamics of tubulovesicular recycling endosomes in hippocampal neurons. J Neurosci. 1999;19:10324–10337. doi: 10.1523/JNEUROSCI.19-23-10324.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey MA, George M, Ying G, Akakura R, Burgess DJ, Siefker E, Bargar T, Doglio L, Crawford SE, Todd GL, Govindarajan V, Hess RA, Band V, Naramura M, Band H. The endocytic recycling regulator EHD1 is essential for spermatogenesis and male fertility in mice. BMC Dev Biol. 2010;10:37. doi: 10.1186/1471-213X-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport D, Auerbach W, Naslavsky N, Pasmanik-Chor M, Galperin E, Fein A, Caplan S, Joyner AL, Horowitz M. Recycling to the plasma membrane is delayed in EHD1 knockout mice. Traffic. 2006;7:52–60. doi: 10.1111/j.1600-0854.2005.00359.x. [DOI] [PubMed] [Google Scholar]
- Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Schweizer FE, Ryan TA. The synaptic vesicle: cycle of exocytosis and endocytosis. Curr Opin Neurobiol. 2006;16:298–304. doi: 10.1016/j.conb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Sharma M, Naslavsky N, Caplan S. A role for EHD4 in the regulation of early endosomal transport. Traffic. 2008;9:995–1018. doi: 10.1111/j.1600-0854.2008.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Giridharan SS, Rahajeng J, Naslavsky N, Caplan S. MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol Biol Cell. 2009;20:5181–5194. doi: 10.1091/mbc.E09-06-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Lippé R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- Steiner P, Sarria JC, Glauser L, Magnin S, Catsicas S, Hirling H. Modulation of receptor cycling by neuron-enriched endosomal protein of 21 kD. J Cell Biol. 2002;157:1197–1209. doi: 10.1083/jcb.200202022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Alberi S, Kulangara K, Yersin A, Sarria JC, Regulier E, Kasas S, Dietler G, Muller D, Catsicas S, Hirling H. Interactions between NEEP21, GRIP1 and GluR2 regulate sorting and recycling of the glutamate receptor subunit GluR2. EMBO J. 2005;24:2873–2884. doi: 10.1038/sj.emboj.7600755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisco D, Anderson ED, Chang MC, Norden C, Boiko T, Fölsch H, Winckler B. Uncovering multiple axonal targeting pathways in hippocampal neurons. J Cell Biol. 2003;162:1317–1328. doi: 10.1083/jcb.200307069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CC, Nokes RL, Wisco D, Anderson E, Fölsch H, Winckler B. Pathway selection to the axon depends on multiple targeting signals in NgCAM. J Cell Sci. 2008a;121:1514–1525. doi: 10.1242/jcs.022442. [DOI] [PubMed] [Google Scholar]
- Yap CC, Wisco D, Kujala P, Lasiecka ZM, Cannon JT, Chang MC, Hirling H, Klumperman J, Winckler B. The somatodendritic endosomal regulator NEEP21 facilitates axonal targeting of L1/NgCAM. J Cell Biol. 2008b;180:827–842. doi: 10.1083/jcb.200707143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CC, Lasiecka ZM, Caplan S, Winckler B. Alterations of EHD1/EHD4 protein levels interfere with L1/NgCAM endocytosis in neurons and disrupt axonal targeting. J Neurosci. 2010;30:6646–6657. doi: 10.1523/JNEUROSCI.5428-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell. 2009;136:1110–1121. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]