

Summary
The mTOR complex-1 (mTORC1) coordinates cell growth and metabolism, acting as a restriction point under stress conditions such as low oxygen tension (hypoxia). Hypoxia suppresses mTORC1 signaling. However, the signals by which hypoxia suppresses mTORC1 are only partially understood, and a direct link between hypoxia driven physiological stress and the regulation of mTORC1 signaling is unknown. Here we show that hypoxia results in Ataxia Telangiectasia Mutated (ATM)-dependent phosphorylation of hypoxia inducible factor 1 alpha (HIF-1α) on serine696 and mediates downregulation of mTORC1 signaling. Deregulation of these pathways in pediatric solid tumor xenografts suggest a link between mTORC1 dysregulation and solid tumor development and point to an important role for hypoxic regulation of mTORC1 activity in tumor development.
Introduction
The mammalian target of rapamycin (mTOR), is a serine/threonine kinase which in association with raptor and mLST8 forms a complex (mTORC1), regulates cap-dependent translation, transcription, cell cycle progression, and survival (Fingar and Blenis, 2004; Kim et al., 2002; Sarbassov et al., 2005). A major mechanism for regulation of mTORC1 involves growth factor signaling, which promotes mTORC1 activity, principally through the inactivation of tuberous sclerosis complex (TSC1/2) (Gao and Pan, 2001; Potter et al., 2001). In the presence of growth factors, multiple growth factor signaling pathways, including PI3K/AKT, MEK/ERK/RSK, and MAPK/MK2, inhibits TSC1/2 complex by phosphorylation which allows for the small G protein activator of mTOR, Ras homology enriched in brain (Rheb) dependent activation of mTORC1 complex. (Inoki et al., 2002; Li et al., 2003; Ma et al., 2005; Manning et al., 2002; Roux et al., 2004). Once mTORC1 complex is activated under nutrient- and energy-replete conditions, stimulates protein synthesis and cell growth (size) through phosphorylation of ribosomal S6 kinase 1 (S6K1), the eukaryotic initiation factor 4E (eIF4E)-binding protein 4E-BP1 and eukaryotic elongation factor 2 kinase (eEF2K) (Browne and Proud, 2004; Fingar et al., 2002; Hay and Sonenberg, 2004). Whereas mTORC1-mediated phosphorylation of 4E-BP1 induces its dissociation from eIF4E, the rate-limiting factor for initiation of Cap-dependent translation (Sonenberg and Gingras, 1998), phosphorylated S6K1 phosphorylates ribosomal protein S6, an essential regulator of protein translation and cell growth (Ruvinsky et al., 2005). On the other hand, under intrinsic and extrinsic stress conditions such as nutrient deprivation (amino acids, or glucose), DNA damage, low energy levels (ATP), or growth factor withdrawal, mTORC1 complex coordinates cell growth and metabolism by acting as a restriction point. Under these conditions a number of sensing pathways, in large part through activation of the TSC1/2 complex, inhibit the kinase activity of mTOR associated with mTORC1 complex (Budanov and Karin, 2008; Feng et al., 2005; Shaw et al., 2004).
Low oxygen levels (hypoxia) like the other cellular stresses discussed above, also results in the inhibition of mTORC1 signaling. In response to hypoxia, cells rapidly activate a variety of adaptive mechanisms that limit energy expenditure through inhibition of energy-intensive processes including protein translation (Liu et al., 2006; Wouters et al., 2005). A major mechanism for this effect involves the inhibition of mTORC1 activity that is observed following exposure to modest hypoxia (1% O2) (Arsham et al., 2003). Under hypoxic conditions, regulation of mTORC1 activity occurs through inhibition of the TSC1/2 complex (Brugarolas et al., 2004; Reiling and Hafen, 2004). Recent published data showed that TSC1/2-dependent regulation of mTORC1 activity in response to hypoxia occurs through dissociation of inhibitory 14–3–3 proteins from the TSC2 protein. This dissociation is mediated by the REDD1 (also know as RTP801/Dig1/ DDIT4) protein, which binds 14–3–3 and is both necessary and sufficient to induce TSC2/14–3–3 dissociation and mTORC1 inhibition (DeYoung et al., 2008).
mTORC1 signaling is critical for cells to coordinate the stimulatory signals arising from nutrients and growth factors and the inhibitory signals arising from intracellular stresses to ensure normal cell growth (size) and proliferation. Conversely, maintained mTORC1 signaling under stress, for example glucose starvation, leads to p53-mediated apoptosis (Lee et al., 2007), whereas primary fibroblasts lacking 4E-binding proteins (4E-BPs) enter p53-dependent senescence and are resistant to oncogene-induced transformation (Petroulakis et al., 2009). Inappropriate control of mTORC1 activity is, however, a hallmark of many benign and malignant human tumors, suggesting an important role in tumorigenesis (Guertin and Sabatini, 2007). Since a period of hypoxic stress is thought to occur virtually universally during human tumorigenesis (Brown and Wilson, 2004; Harris, 2002; Vaupel et al., 1998), it suggests that to maintain cellular proliferation mTORC1 must become dysregulated, and for survival p53 or downstream effectors have to be inactivated. Here, we describe that the stress sensor protein Ataxia Telangiectasia Mutated (ATM) links hypoxia-induced physiological stress to mTORC1 signaling and deregulation of mTORC1. In xenografts derived from childhood solid tumors ATM levels are low relative to leukemia xenografts, facilitating maintained mTORC1 signaling under hypoxic conditions. These data suggest a model in which partial silencing of ATM is an early step in the genesis of childhood solid tumors, followed by subsequent inactivation of either p53, or components of the extrinsic death pathway, that are found frequently in childhood cancers.
Results
ATM regulates mTORC1 signaling in response to hypoxia
To examine whether ATM contributes to mTORC1 regulation by hypoxia, the effects of hypoxia on ATM−/− mouse embryonic fibroblasts (MEFs) as well as cell lines derived from ataxia telangiectasia (AT) patients were analyzed. Western blot analyses for the level of phosphorylation at rS6(Ser235/236) as well as 4E-BP1(Ser65) were used to monitor mTORC1 activity under normoxia (21% O2) or hypoxia (0.2% or 1% O2). To overcome ATM knockout induced senescence in the MEFs, mice were intercrossed with Arf−/− mice. In the experiments that follow, ATM−/−; Arf−/− MEFs were compared ATM+/+; Arf−/− MEFs (referred to as “ATM−/−” and “ATM+/+”, respectively). In wild-type (wt) and ATM+/+ MEFs (Figure 1A,C) as well as normal human fibroblasts [NHFB (Figure 1B,D)], hypoxia inhibits mTORC1 signaling as determined by down-regulation of S6 as well as 4E-BP1 phosphorylation. In contrast, hypoxia failed to down-regulate S6 and 4E-BP1 phosphorylation in ATM−/− MEFs as well as three different human ATM (hATM−/−) deficient cell lines, indicating that mTORC1 inhibition by hypoxia requires an intact ATM protein.
Figure 1. ATM regulates mTORC1 signaling in response to hypoxia.

A, Western blot analysis of Wild type (wt), ATM+/+ and ATM−/− MEFs exposed to hypoxia (0.2% O2) for the indicated times. Cell extracts were analyzed by western blot using antibodies as shown. B, Western blot analysis of three different human ATM deficient cell lines (hATM−/−) compared with the normal human fibroblast (NHFB). Cells were exposed to hypoxia (0.2% O2) for the indicated periods of time. At 24 hr ATM was phosphorylated (Ser1981) under hypoxia in NHFBs. C, D Experiments were repeated under mild hypoxia (1% O2)
Hypoxia-induced HIF-1α activity and REDD1 induction is attenuated in ATM−/− MEFs
The master transcriptional regulator in response to hypoxia is hypoxia inducible factor (HIF). Under hypoxia, HIF-1α becomes stabilized allowing dimerization with HIF-β and translocation of the HIF complex into the nucleus where it associates with promoters bearing hypoxia response elements (HRE) (Maxwell et al., 1999; Wang and Semenza, 1993). We next asked whether HIF-1α transcriptional activity was required for ATM-dependent inhibition of mTORC1 signaling in response to hypoxia. As shown in Figure 2A, hypoxia markedly increased the protein level of HIF-1α in ATM+/+ MEFs as well as in NHFB, whereas in ATM deficient cells, the hypoxia-induced accumulation was reduced, and translocation of HIF-1α protein into the nucleus was attenuated which is shown by using immunofluorescence and by quantitation of fluorescence intensity for HIF-1α on section images (Figure 2B–C). Consistent with the decrease in HIF-1α expression during hypoxia, transcriptional activity of HIF-1α was also significantly reduced in ATM deficient cells (Figure 2D). Moreover, we found that HIF-1α was required for the inhibition of mTORC1 signaling in response to hypoxia (Figure 2E). Taken together, these experiments strongly indicate that ATM expression is essential for HIF-1α activity and consequentially for the inhibition of mTORC1 signaling during hypoxia.
Figure 2. Hypoxia-induced HIF-1α activity is attenuated in ATM−/− MEFs.
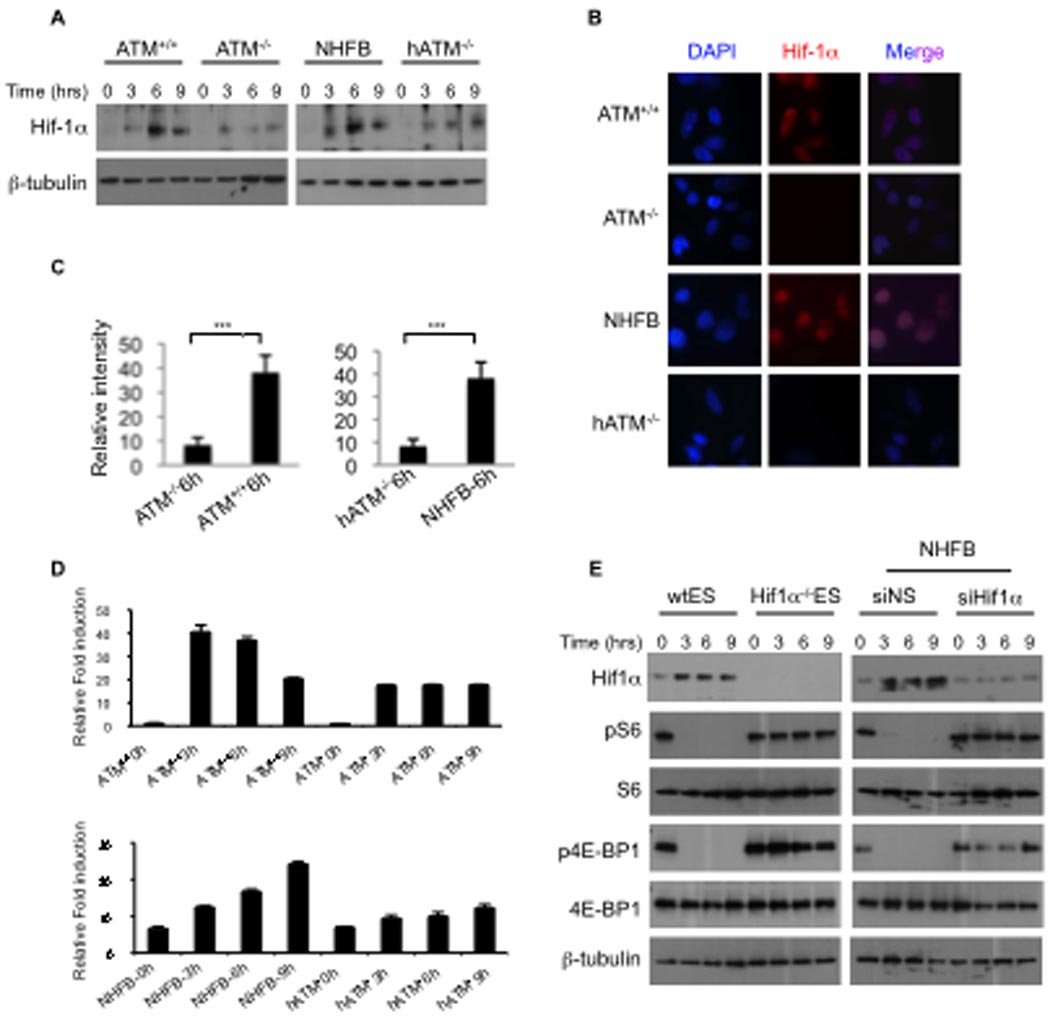
A, ATM+/+ and ATM deficient cells exposed to hypoxia (0.2% O2) for the indicated times and cell extracts were blotted for anti-Hif-1α and β-tubulin antibodies. B, Immunofluorescence staining for Hif-1α (red) of ATM+/+ and ATM deficient cells. Cells were exposed to hypoxia (0.2% O2) for 6 h and immunofluorescence was detected as described in methods. C, Quantification of fluorescence intensity for HIF-1α on section images with 150-ms exposure time (no saturation) has been shown. Relative intensity = intensity of the object (cell) – intensity of the background. For each of ATM+/+ or ATM−/−, n = 15 randomly selected nucleus were quantitated. ***P < 0.001. Mean ± s.e.m. D, Luciferase assay for ATM+/+ and ATM deficient cells transfected with HRE-Luc plasmid. After transfection, cells were incubated for 24 h under normoxic conditions (21% O2) and exposed to hypoxia (0.2% O2) for the indicated times. Cell lysates were assayed for the dual-luciferase activity. Error bars represent the mean standard deviation (s.d.), n=2. E, wtES cells, Hif-1α−/−ES and NHFB, transfected with control or siRNA against Hif-1α (Dharmacon), exposed to hypoxia (0.2% O2) for the indicated times and cell extracts were blotted for antibodies as shown.
To understand more precisely the underlying molecular mechanism of mTORC1 inhibition under hypoxic conditions, we first focused on one of the targets of HIF, Redd1 (Shoshani et al., 2002), that encodes a negative regulator of mTORC1 signaling under hypoxic conditions (Brugarolas et al., 2004; DeYoung et al., 2008; Reiling and Hafen, 2004). To determine whether hypoxia-induced Redd1 gene expression is dependent upon ATM, we compared the REDD1 protein as well as Redd1 mRNA levels of ATM−/− and ATM+/+ cells during hypoxia. As shown in Figure 3A, REDD1 protein as well as Redd1 mRNA levels (Figure S1A–B) were induced by hypoxia in wild-type cells, but not in ATM deficient cells. Like REDD1, induction of PDK1 and GLUT1 genes were also attenuated in the absence of ATM (Figure S1C–D). To determine whether in ATM−/− cells REDD1 was involved in the regulation of mTOR by hypoxia, ATM deficient cells were infected with lentiviruses expressing REDD1. ATM−/− MEFs infected with control virus, as well as human ATM−/− cells were not able to downregulate mTORC1 signaling during hypoxia. In contrast, under hypoxic conditions, overexpression of REDD1 strongly induces dephosphorylation of S6 and 4E-BP1 indicative of mTORC1 inhibition (Figure 3B). We next asked whether overexpression of ATM could also inhibit mTORC1signaling and if it required concomitant induction of hypoxia. As shown in Figure 3C, in ATM−/− MEFs, hypoxia dependent HIF-1α and REDD1 induction are strongly attenuated. Reintroducing of ATM in ATM−/− MEFs, was able to rescue hypoxia-dependent HIF-1α and REDD1 induction. Under normoxia, ATM overexpression was not sufficient to suppress mTORC1 signaling, indicating that ATM requires concomitant induction by hypoxia either in MEFs or in HEK293T cells (Figure S1E). Further, this inhibition is absolutely p53-independent (Figure 3D), consistent with data from Arf−/− MEFs (Figure 1A). Suppression of mTORC1 signaling by hypoxia has been reported to be dependent on activated AMPK phosphorylating and activating TSC1/2 before induction of REDD1 (Liu et al., 2006). Under hypoxic conditions REDD1 was induced rapidly in NHFB, and within 15 min mTORC1 signaling was inhibited, however there was only a slight transient increase in AMPK phosphorylation (Figure S1F). Downregulation of REDD1 using siRNA effectively abrogated ATM-dependent negative regulation of mTORC1 signaling by hypoxia (Figure S1G).
Figure 3. ATM is necessary for hypoxia-induced expression of REDD1 protein and REDD1 is sufficient for the regulation mTORC1 signaling by hypoxia in ATM deficient cells.
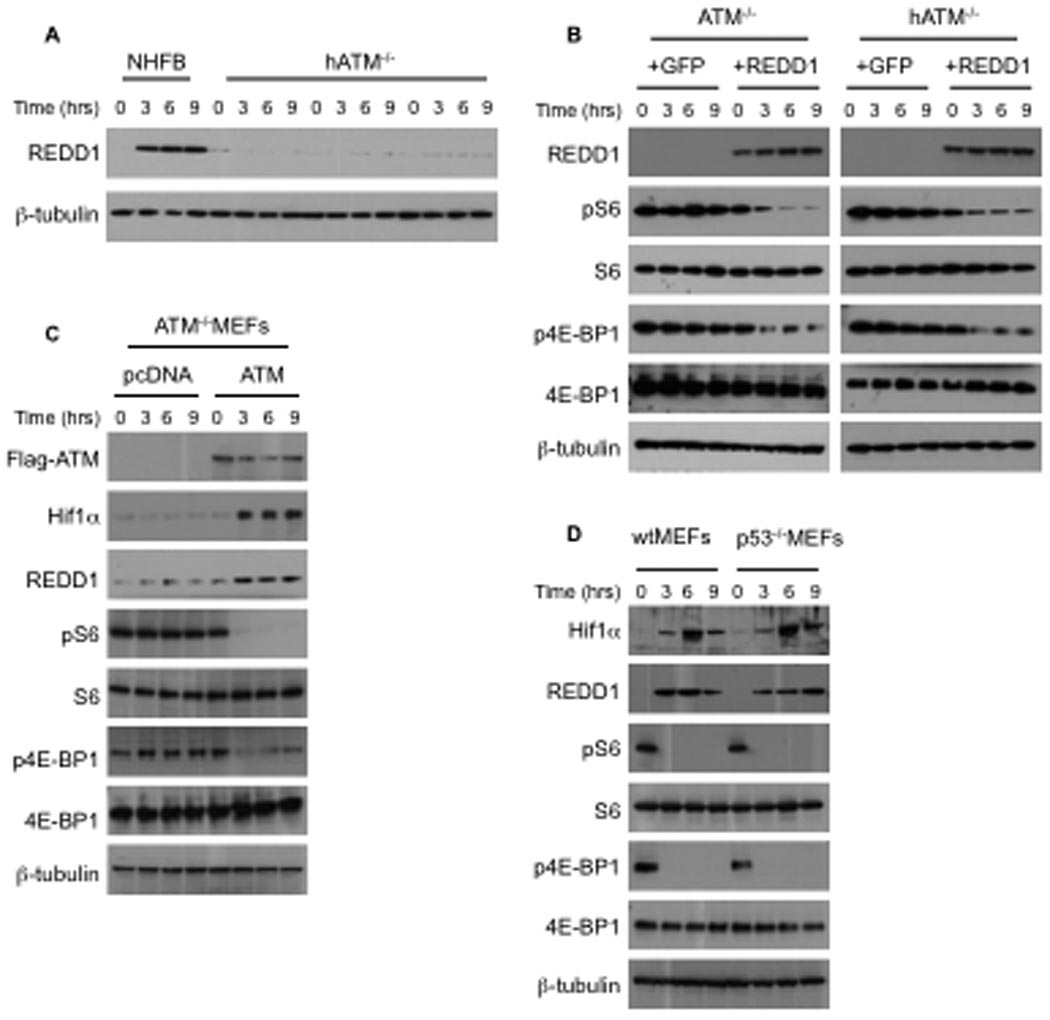
A, Western blot analysis of extracts from NHFB and a human ATM deficient cell line (GM03395) exposed to hypoxia (0.2% O2) for the indicated times and blotted for anti-REDD1 and β-tubulin antibodies. B, ATM−/− MEFs and a human ATM deficient cell line (GM03395) were transduced with either a REDD1 or a GFP expressing lentiviruses and exposed to hypoxia (0.2% O2) for the indicated times. Cell extracts were analyzed by western blot using antibodies as shown. C, ATM−/− MEFs were electrophorated by Flag-ATM or pcDNA plasmids as a control. After 30h incubation, electroporated cells were exposed to hypoxia (0.2% O2) for the indicated times. Cell extracts were analyzed by western blot using antibodies as shown. D, wtMEFs and p53−/− MEFs were treated with hypoxia for the indicated times and cell extracts were analyzed by western blot using antibodies as shown, (see also supporting data in Figure S1)
Hypoxia induces ATM kinase activity and ATM phosphorylates Hif-1α at Ser696 that results in induction of REDD1
To test whether the specific activity of ATM could be increased by hypoxia, HEK 293 cells were transfected with expression vectors encoding a Flag-tagged, wt full-length ATM protein. After 48 h, ATM was immunoprecipitated from lysed cells and used for in vitro kinase assays using as substrate a fragment of human recombinant HIF-1α (530–826) polypeptide, that includes two transactivation domains (TAD) or using p53 protein as a positive control. As shown in Figure S2A (lane 2), ATM kinase phosphorylates HIF-1α as well as its well-characterized substrate p53 (Figure S2A lane 5). To extend these data without overexpression of ATM, NHFB as well as HEK 293 cells were incubated in the presence of 0.2% O2 for 3h, and endogenous ATM was immunoprecipitated and its kinase activity measured using the human recombinant HIF-1α (530–826) polypeptide, Figure S2B. Quantitation of three independent experiments shows the results to be quite consistent (Figure S2C), and shows the specific kinase activity of ATM increased about two-fold after hypoxia treatment of NHFB cells. The increase in kinase activity was observed for both HIF-1α polypeptide and recombinant p53 protein. This level of induction of ATM activity is similar to that seen after ionizing irradiation (Canman et al., 1998) and represents an increase in specific activity because the total amounts of ATM protein did not change after hypoxia treatment (Figure S2B).
The amino acid sequence of HIF-1α contains several putative ATM substrate consensus sites TQ (Thr-Gln) and a single SQ (Ser-Gln) consensus site. To determine if ATM could phosphorylate any of these sites we performed an in-vitro kinase assay using active ATM and recombinant HIF-1α, followed by LC-MS/MS analysis to identify any sites phosphorylated on HIF-1α. As shown in the MS/MS and ESI spectra, presented in Figure S2D, we were able to detect a clear phosphorylated single SQ site (Ser696) on HIF-1α. Next, we attempted to test whether the single SQ site (Ser696) on HIF-1α is indeed phosphorylated in vivo under hypoxic conditions. As shown in MS/MS and ESI spectra (Figure S2E–G), we were able to detect the phosphorylated peptide in vivo only under hypoxic conditions. We examined the kinetics of phosphorylation using a HIF-1α (Ser696)-phospho specific antibody. Phosphorylation of HIF-1α at Ser696 was rapid, and ATM-dependent. Phosphorylation at Ser696 was detected within 15 minutes of hypoxia in NHFB cells, but was not detected in hATM −/− cells (Figure S2H).
We next questioned whether Ser696 phosphorylation by ATM kinase is necessary for HIF-1α physiological activity under hypoxic conditions. We first generated Ser696Ala and the less conservative Ser696Arg mutant HIF-1α. Consistent with previous data (Shoshani et al., 2002), electroporation of HIF-1α-deficient ES cells by wt HIF-1α strongly induced Redd1 mRNA in response to hypoxia (Figure 4A–B). However, HIF-1α-deficient ES cells expressing mutSer696AlaHIF-1α or mutSer696ArgHIF-1 were not able to induce Redd1 mRNA in response to hypoxia (Figure 4A–B). To demonstrate directly whether Ser696 phosphorylation of HIF-1α is required for the inhibition of mTORC1 signaling under hypoxic conditions, HIF-1α-deficient ES cells were electroporated with wt HIF-1α, mutSer696AlaHIF-1α and mutSer696ArgHIF-1α, and cells were harvested during hypoxia. In contrast to previous data that used a murine hepatoma line that lacks ARNT/Hif-1β complex (Arsham et al., 2003), in HIF-1α-deficient ES expressing wt HIF-1α, hypoxia induced dephosphorylation of S6 and 4E-BP1 indicating mTORC1 inhibition (Figure 4C). Expression of either mutSer696AlaHIF-1α or mutSer696ArgHif-1α did not suppress mTORC1 function under hypoxia in ES cells (Figure 4C).
Figure 4. Ser696 phosphorylation by ATM kinase is necessary for Hif-1α physiological activity in hypoxia.
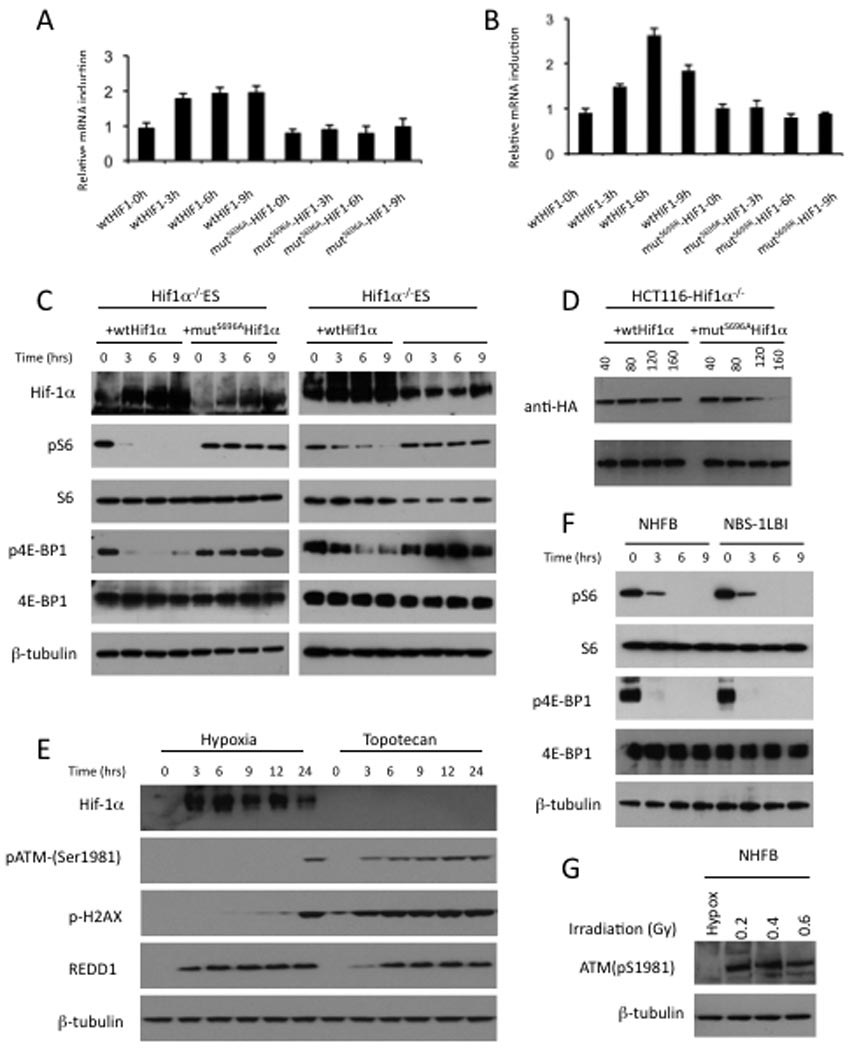
A–B, Hif-1α−/− ES cells were electroporated by wtHif-1α, mutS696AHif-1α or mutS696RHif-1α plasmids according to the manufacturer’s instructions (Amaxa Biosystems). Cells were incubated for 24h in normoxia before being exposed to hypoxia (0.2% O2) for the indicated times and finally cells were harvested for RNA analysis. Expression of Redd1 was determined by RT-PCR. Each value was normalized to GAPDH and error bars represent the mean standard deviation (s.d.). C, After Hif-1α−/− ES cells were electroporated by wtHif-1α, mutS696AHif-1α or mutS696RHif-1α plasmids, cells were incubated for 24 h in normoxia. Cells were then exposed to hypoxia (0.2% O2) for the indicated times and cell lysates were analyzed by western blotting with using antibodies as shown. D, Ser696 phosphorylation by ATM kinase effects on HIF-1α protein stability under hypoxic conditions. After HCT116-Hif-1α−/−cells were electroporated by wtHif-1α or mutS696AHif-1α plasmids, cells were first incubated for 24h in normoxia. Cells were then exposed to hypoxia (0.2% O2) for the indicated times in the presence of 100µM Cycloheximide (Sigma) and cell lysates were analyzed by low exposure western blotting with using antibodies as shown. At 120 min, mutS696AHif-1α showed 2.6 Fold decreased protein level compared to wtHif-1α protein level. Relative levels of wtHIF-1α and mutS696AHif-1α were quantitated and normalized to tubulin protein. E, NHFB were exposed to hypoxia (0.2% O2) or topotecan (4µg/ml) for the indicated times and cell lysates were analyzed by western blotting with using antibodies as shown. F, NHFB and NBS-1LBI cells were exposed to hypoxia (0.2% O2) for the indicated times and cell lysed were analyzed by western blotting with using antibodies as shown, (see also supporting data in Figure S2). G, NHFB were exposed to hypoxia (0.2% O2) or irradiated (0.2, 0.4 and 0.6 Gray). After 3 hours, cell lysates were analyzed by western blotting with using antibodies as shown.
Moreover, we analyzed the mechanism by which ATM-dependent phosphorylation of HIF-1α increased its function and found that Ser696 mutant proteins were less stable than wild type under hypoxic conditions, suggesting that Ser696 phosphorylation stabilized HIF1α (Figure 4D). Although, previously it has been shown that HIF-1α is not necessary for hypoxic regulation of the mTOR pathway in ARNT/HIF-1β null BpRc1 cells (Arsham et al., 2003), our results in HIF-1α-deficient ES (Figure 4C) clearly show that HIF-1α is required for the hypoxic regulation of the mTOR pathway.
Hypoxia driven ATM activation is independent of DNA-damage response
We next examined whether the increase in ATM kinase activity that occurs early in the time course of hypoxic exposure is dependent on DNA double-strand breaks (DSBs). In contrast to cells treated with the DNA damaging agent topotecan, after 3h hypoxia treatment of ATM+/+ cells we did not detect either DSBs, monitored by phosphorylation of the histone variant H2AX (γH2AX) or phosphorylation of ATMSer1981. Interestingly, only 24h hypoxia treatment induced both phosphorylation of H2AX and phosphorylation of ATM kinase (Figure 4E). As shown in Figure 4E, induction of REDD1 is more rapid following hypoxia than treatment with topotecan, whereas topotecan rapidly induces detectable γH2AX, suggesting potentially different pathways are involved. To further distinguish the pathway by which hypoxia activates ATM from that induced by DNA damage, we compared NHFB to NBS1 deficient skin fibroblasts derived from a Nijmegen breakage syndrome (NBS) patient. NBS1 is an essential component of the MRE11-RAD50 complex that leads to activation and recruitment of ATM and DNA-dependent protein kinase catalytic subunit (DNA-PKCs) which phosphorylates H2AX at the site of the DNA double-strand breaks. As shown in Figure 4F, NBS1 has no impact on hypoxia-mediated downregulation of mTORC1 signaling. Further, low doses of radiation (0.2–0.6 Gy) induced ATM phosphorylation, whereas hypoxia did not (Figure 4G). Taken together, these data, supported by recently published data (Bencokova et al., 2009), indicate that the early increase of ATM kinase activity induced under hypoxic conditions is independent of DNA double-strand breaks, and suggests an alternative mode for stimulation of ATM under hypoxic conditions.
Childhood solid tumor xenografts show decreased ATM expression and elevated mTORC1 activity
In response to hypoxia, a key unanswered question is the physiologic significance of ATM regulation of mTORC1 signaling. To examine this, we screened the Affymetrix database containing expression profiles from childhood tumor xenografts established by the Pediatric Preclinical Testing Program (pptp.nchresearch.org/data) and identified regions of hypoxia in tumor xenografts following injection of EF5, a marker of hypoxic regions (Figure 5A). Interestingly, solid tumor xenografts, which have hypoxic regions, as shown by positive EF5 staining (Figure 5A), show decreased ATM expression compared to acute lymphocytic leukemia (ALL) xenografts (Figure 5B). Levels of ATM protein in these tumors were consistent with the expression levels, demonstrating significantly decreased ATM in all solid tumor models compared to ALL xenografts (Figure 5C–H). We next determined whether mTORC1 signaling was active in these pediatric tumor xenografts by immunohistochemical staining for phosphorylated S6. Phosphorylation of S6 in these xenografts is completely abolished by treatment with rapamycin (data not shown). As shown in Figure 6A, relative to control tissues (kidney, skeletal muscle, brain), kidney tumors (Wilms and rhabdoid), sarcomas, brain tumors and neuroblastomas exhibit markedly elevated levels of phospho-S6 protein indicating an elevated level mTORC1 signaling activity under physiological hypoxia. We performed parallel staining for ATM and phospho-S6 proteins (Figure 6B). Nuclear ATM was detected in each normal tissue and two acute lymphoblastic leukemia xenografts examined. In contrast, ATM was not detected in any of the solid tumor models, but there was abundant phospho-S6 detected. We next examined whether phospho-S6 could be detected in hypoxic regions denoted by EF5-positive regions (see Figure 5A). As shown in Figure 6C phospho-S6 staining was clearly detected in zones staining positive for EF5, a marker of hypoxia.
Figure 5.
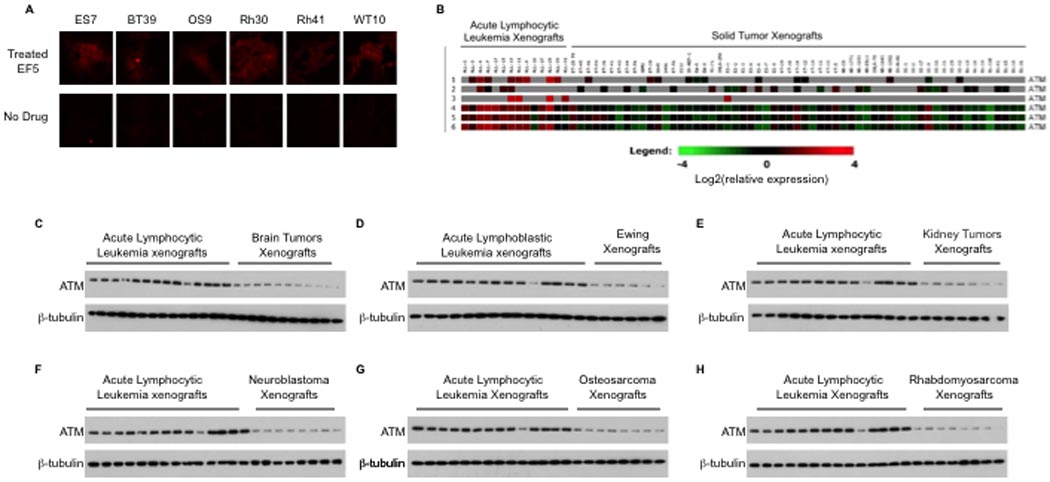
A, Tumor bearing animals were chosen randomly and treated with or without EF5 drug, given as a tail-vein injection of 10mM in 5% glucose. The injection volume (in ml) was equal to 1% of animal mass (in g). Mice were euthanized at 3hr and the tumors removed and frozen sections prepared. Sections of tumors were incubated with a Cy3-conjugated antibody to EF5 adducts (hypoxia, red). (ES: Ewing tumor, BT: brain tumor, OS: osteosarcoma, Rh: rhabdomyosarcoma, WT: Wilms’ tumor) B, ATM expression profiles of Acute Lymphocytic Leukemia (ALL) and childhood solid tumor xenografts (BT: brain tumor, ES: Ewing tumor, KT: kidney tumor, NB: neuroblastoma, OS: osteosarcoma, Rh: rhabdomyosarcoma). C-H, Immunoblot analysis of xenografts with using antibodies as shown.
Figure 6. Elevated mTORC1 signaling activity in childhood solid tumor xenografts tissues.
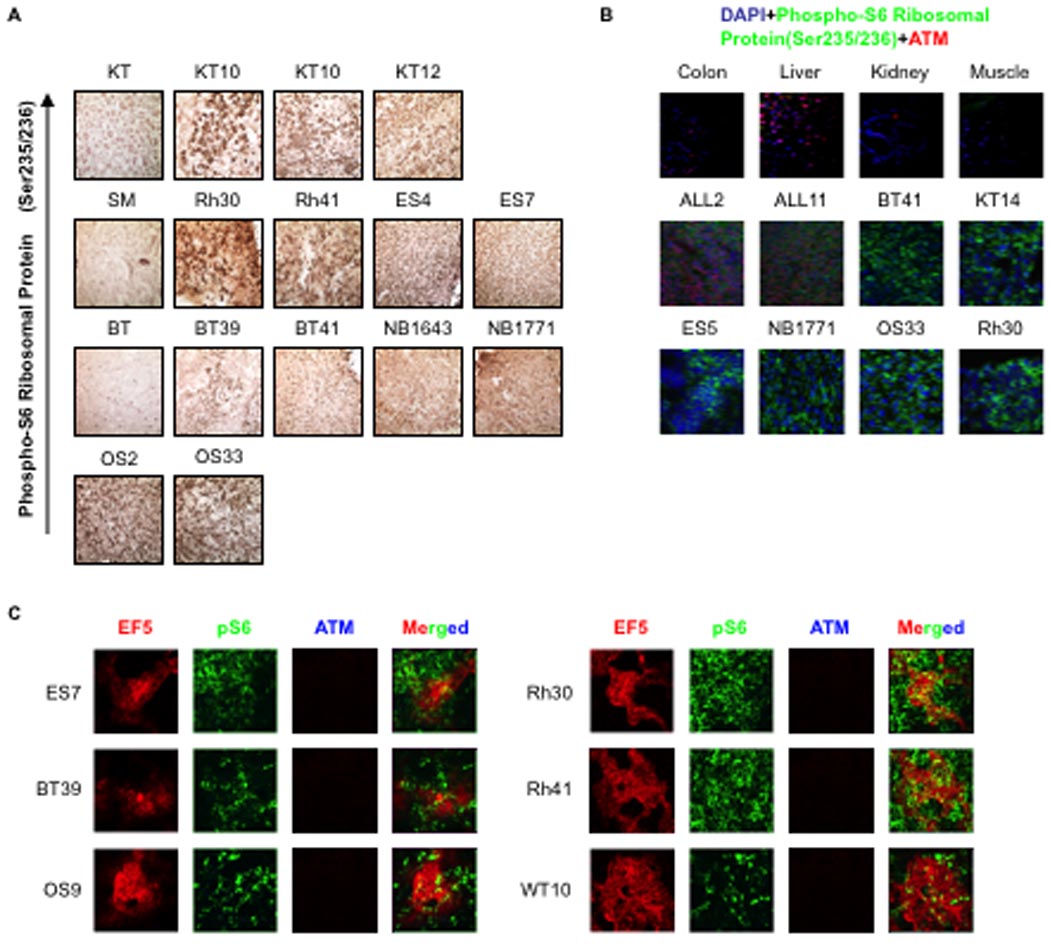
A, Immunohistochemistry (IHC) staining of solid human tumor xenografts and normal tissues were performed using phospho-S6 ribosomal protein (Ser235/236) [Cell Signaling Technology (91B2)] antibody. Paraffin embedded normal human tissue slides were obtained from ProSci Incorporated and paraffin embedded solid human tumor xenografts slides were prepared. IHC were developed according to the Cell Signaling Technology’s protocol (IHC-Paraffin). B, Sections of control tissues and tumors were parallel stained with ATM antibody (Genetex, Clon 5C2, 1:25 dilution) and phospho-S6 ribosomal protein (Ser235/236) [Cell Signaling Technology (91B2)] antibody (1:75 dilution). Slides were developed as above and covered with Vectashield Mounting Medium with DAPI (Vector Labs). Secondary antibodies used were: to visualize ATM, Alexa Fluor 546 goat anti mouse (red), to visualize phospho-S6 ribosomal protein, Alexa Fluor 488 goat anti-rabbit (green) from Invitrogen. Only merged images are shown. C, EF5 treated and Cy3-conjugated (hypoxia, red) sections (from Figure 5) were parallel stained with ATM antibody (Genetex, Clone 5C2, 1:25 dilution) and phospho-S6 ribosomal protein (Ser235/236) [Cell Signaling Technology (91B2)] antibody (1:75 dilution). Secondary antibodies used were: to visualize ATM, Alexa Fluor 350 donkey anti mouse (blue), to visualize phospho-S6 ribosomal protein, Alexa Fluor 488 goat anti-rabbit (green) from Invitrogen.
To understand more precisely the physiological role of ATM signaling under hypoxic conditions, we first addressed the contribution of ATM deficiency in cellular proliferation and survival. Under normoxic conditions, there were no significant difference in proliferation rates between wt NHFB and ATM deficient cells (Figure S3A). Surprisingly, the number of cells after four days of hypoxia was significantly lower for ATM-deficient cells compared with wt NHFB, and this difference was abrogated by the highly specific mTORC1 inhibitor, rapamycin (Figure 7A, and Figure S3B). These results suggested that maintained mTORC1 signaling in the absence of ATM contributes to the decrease in cell number (Figure 7A). This finding is consistent with data showing TCS2-deficient cells undergo apoptosis under extreme glucose starvation, an effect which is suppressed by rapamycin (Inoki et al., 2003).
Figure 7. Loss of hypoxia signaling to TORC1 complex strongly induces p53-dependent apoptosis in human ATM deficient cells.
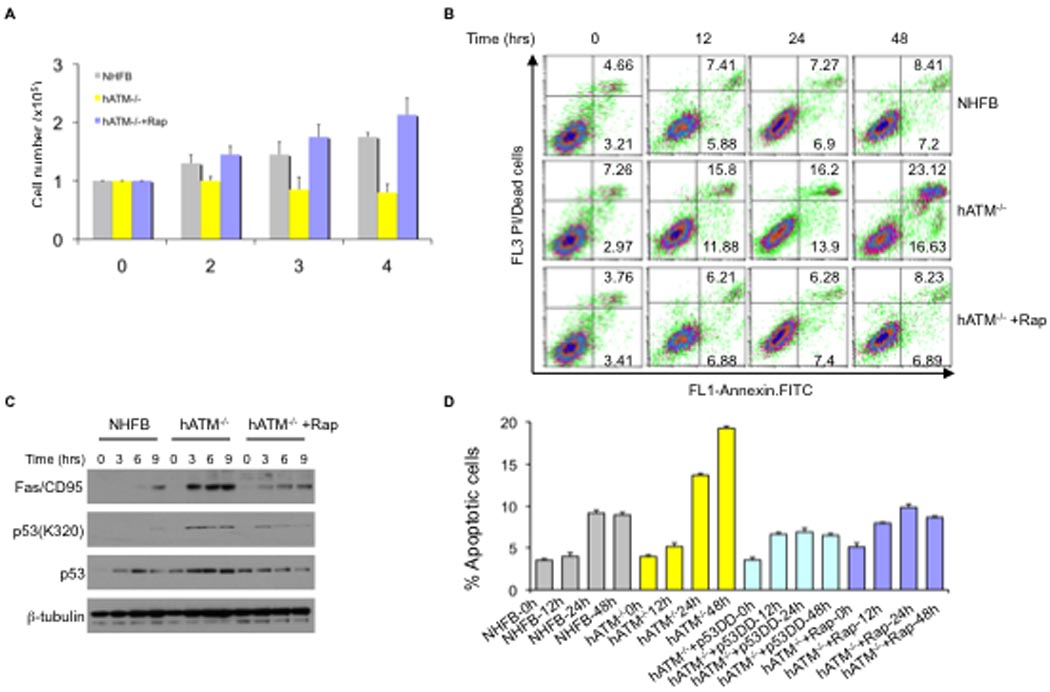
A, Equal numbers of cells were plated and exposed to hypoxia (0.2% O2) with or without rapamycin (100 nM) for the indicated time. Viable cells were counted by trypan blue exclusion assay (Coulter Vicell) and error bars show standard deviation for triplicate plates for a representative experiment. B, Annexin V-FITC staining for NHFB and a human ATM deficient cell line (GM03395). Cells were exposed to hypoxia (0.2% O2) for indicated times with or without rapamycin (100 nM). The numbers refer late or early percentage of apoptosis cells. C, NHFB and a human ATM deficient cell line (GM03395) were exposed to hypoxia (0.2% O2) for the indicated times in the presence or absence of rapamycin (100nM). Cell extracts were analyzed by western blot using antibodies as shown. D, Annexin V-FITC staining for NHFB and a human ATM deficient cell line (GM03395) transduced with or without dominant-negative p53 expressing lentiviruses and exposed to hypoxia (0.2% O2) for the indicated times in the presence or absence of rapamycin (100 nM). Error bars represent standard deviation (s.d.), n=2. (See supporting data in Figure S3)
We next asked whether, in response to hypoxia, the failure to suppress mTORC1 by ATM could sensitize the cells to hypoxia-induced apoptosis. As shown in Figure 7B, under hypoxic conditions of growth, ATM-deficient cells undergo increased apoptosis compared with wt NHFB, and apoptosis was suppressed by rapamycin. Further, pharmacological inhibition of ATM, prevented hypoxia dependent inhibition of mTORC1 signaling (Figure S3C), and enhanced hypoxia-induced apoptosis in human fibroblasts (Figure S3D), whereas expression of wt HIF-1α protected ATM-deficient MEFs (Figure S3E). Interestingly, we found that, in contrast to wt NHFB, the death receptor Fas/CD95 is upregulated in human ATM-deficient cells during hypoxia which is correlated with hypoxia-induced elevation of p53, a response also suppressed by rapamycin (Figure 7C). Inhibition of p53 function by expressing a dominant-negative allele also protected ATM−/− cells from hypoxia-induced apoptosis (Figure 7D), and resulted in increased cell survival.
Discussion
We find that the stress sensor kinase ATM transmits the hypoxic stress signal to down-regulate mTORC1 function and requires intact ATM kinase activity. Under conditions of hypoxia, ATM-deficient cells fail to negatively regulate the mTORC1 complex, resulting in continued phosphorylation of downstream substrates ribosomal S6 protein and 4E-BP1. Further, ATM deficiency prevents accumulation of HIF-1 under hypoxia. Under hypoxic conditions, down-regulation of mTORC1 signaling by ATM, depends on phosphorylation of HIF-1α at Ser696 required for transcriptional activation of REDD1 and subsequent activation of the TSC complex. Absence of ATM kinase activity under hypoxic conditions results in a marked increased cell death via deregulation mTORC1 signaling, stabilization of the tumor suppressor p53 and p53-dependent upregulation of the death receptor Fas/CD95. In pediatric solid tumor models, ATM levels are diminished, thus allowing continued mTORC1 signaling and potentially results in selection of the tumor cells that show defects of apoptotic pathways. Defects in p53 function or in the extrinsic death pathway are found in most childhood solid tumors and in their xenografts suggesting that abrogation of p53 signaling is essential for maintained mTORC1 activity and cellular proliferation (Harada et al., 2002; Kontny et al., 1998; Teitz et al., 2000). This is consistent with recent findings that cells lacking 4E-BPs became senescent if p53 remained functional (Petroulakis et al., 2009).
Hypoxia can induce unique cellular stress by driving and maintaining genetic instability (Bristow and Hill, 2008) and presents potentially the most relevant stress involved in tumor progression, as regions of hypoxia occur early and in most solid tumors. However, how hypoxia links that unique stress to mTORC1 signaling has remained unknown. In contrast to the parallel controls with very low doses (below 1GY) of ionizing radiation, hypoxia itself does not induce detectable DNA double-strand breaks (DSBs). However, it has been shown that significant levels of damage are observed in response to reoxygenation that activates ATM (Hammond and Giaccia, 2004). Moreover, although hypoxia does not induce the classic DNA damage response, it has been shown that cell cycle checkpoint protein Chk2 is phosphorylated and activated in an ATM-dependent manner (Freiberg et al., 2006; Gibson et al., 2005). Consistent with recently published data (Bencokova et al., 2009), we found that hypoxia increases ATM kinase activity and the ATM response to hypoxia is independent of the MRN complex. In this study we have shown that in early response to hypoxia (0.2% O2), ATM is activated but not phosphorylated at Ser1981 in humans (Ser1987 in mouse), a widely used biological marker to identify the active form of ATM. These results support an alternative mode for stimulation of ATM in the absence of DSBs (Soutoglou and Misteli, 2008) or indicates autophosphorylation at Ser1981 (or Ser1987) could be dispensable for human or murine ATM activation under hypoxic conditions (Pellegrini et al., 2006).
Our data demonstrate that ATM kinase phosphorylates HIF-1α at Ser696, and phosphorylation of this site on HIF-1α is required for its stability, induction of REDD1 and in turn, suppression of mTORC1 signaling under hypoxic conditions, and defines the physiological significance of this phosphorylation by ATM. ATM-deficient cells failed to induce PDK1 and GLUT1, genes also regulated by HIF1α, under hypoxia, consistent with the data of Lum et al. (Lum et al., 2007). The role of REDD1 expression in hypoxia-mediated mTORC1 regulation is somewhat controversial. Evidence has been provided (Brugarolas et al., 2004; DeYoung et al., 2008) that REDD1 is essential, whereas Lui et al. (Liu et al., 2006), demonstrated that activation of AMPK under hypoxic stress (1.5% O2 and no serum) led to phosphorylation and activation of the TSC complex prior to induction of REDD1. In our experimental conditions (0.2% O2 hypoxia, 10% serum), NHFB show a rapid induction of REDD1 within 15 min (Figure S1F) whereas there was only a slight increase in AMPK(Thr172) phosphorylation above the basal level at 15 min, but not thereafter. This suggests that rapid mTOR inhibition under hypoxia is unlikely to be caused by the AMPK hyperphosphorylation. However, overexpression of REDD1 only partially suppressed mTORC1 signaling under normoxic conditions, suggesting that it may be modified under hypoxic stress before modulating TSC complex function (DeYoung et al., 2008). This may account for the very rapid accumulation of REDD1 under hypoxic conditions.
HIF-1α has a complex role of inducing genes that permit adaptation to hypoxia (Carmeliet et al., 1998). HIF is a heterodimer consisting of the two subunits, the oxygen-sensitive HIF-1α and constitutively expressed HIF-β. The α subunit of HIF undergoes multiple posttranslational modifications. Under normal oxygen tension, the HIF-α subunits are modified by prolyl hydroxylases (PHDs), which target the hydroxylated subunit for recognition by the von Hippel-Lindau protein (VHL) and subsequent degradation by proteasomes (Huang et al., 1998; Maxwell et al., 1999; Wang et al., 1995). In hypoxic conditions, HIF-1α becomes stabilized and dimerizes with HIF-β leading to translocation to the nucleus, where the heterodimer associates with transcriptional co-activators CBP/p300, activating a complex transcriptional program. In addition to the established regulation of HIF-1α, described above, recently published data showed that hypoxia induced sumoylation of HIF-1α, which promotes its binding to a VHL protein through a proline hydroxylation-independent mechanism, leads to its ubiquitination and degradation (Cheng et al., 2007). In contrast, the sumoylation of HIF-1α by RSUME, a small RWD-containing protein, enhanced its stabilization and transcriptional activity during hypoxia (Carbia-Nagashima et al., 2007). Our finding that phosphorylation of HIF-1α by ATM is required for its stability raises the interesting question of whether this posttranslational modification has impact on its further modification such as sumoylation.
Proliferation of most cancer cells is dependent on maintained mTORC1 signaling. Our observations provide evidence that in pediatric solid tumor models, ATM levels are diminished, allowing continued mTORC1 signaling and cellular proliferation. Potentially this would enable p53 to engage the extrinsic cell death pathway in the presence of improper signaling and suggests that abrogation of p53 signaling is essential for survival when mTORC1 signaling is maintained permitting cellular proliferation in these tumors under conditions of hypoxia. Importantly, our results suggest that deregulation of this signaling pathway could strongly contribute tumor development or progression (Figure S4). Of note is that ATM is activated during the early stages of human tumorigenesis by oncogene-induced stress (Bartkova et al., 2005), which also leads to p53 activation and either senescence or apoptosis. Interestingly, inhibition of ATM suppressed the induction of senescence, and in a mouse model, the inhibition of ATM led to increased tumor size and invasiveness (Bartkova et al., 2006). These data strongly underpin our finding that childhood solid tumors have an abnormally low level ATM compared tissue of origins. As we propose in our model (Figure S4), bypassing ATM activation plus inactivation in the p53 pathway in early lesions maintains activation of mTORC1 under stress conditions such as hypoxia that could contribute to tumor progression.
In summary, we found ATM is downregulated and mTORC1 signaling is active in pediatric solid tumor xenografts relative to acute lymphoblastic leukemia xenografts that would not be anticipated to exhibit acute hypoxia. ATM was also readily detected in normal tissues, although kidney medulla and skeletal muscle may be normally hypoxic (~1% O2) (Simon and Keith, 2008). Activation of mTORC1 signaling appears to be a characteristic of many cancers (Engelman et al., 2006). This frequently occurs through activation of upstream signaling such as activating mutations of phosphatidylinositol 3’ kinase, or loss of tumor suppressors such as PTEN (Majumder et al., 2004) or PML (Bernardi et al., 2006). Our data suggests that suppression of ATM represents an important addition to the several other ways in which TORC1 activity can remain elevated in hypoxic tumors.
Experimental procedures
Cells and culture conditions
Wild-type, Arf−/−, Arf−/−ATM−/− MEFs, NBS1-1LB, HCT116-HIF-1α−/− cells as well as HEK-293 cells were cultured in DMEM (Cellgro) supplemented with 4.5 g/L glucose, and 10% heat-inactivated FBS (Hyclone). Human ATM deficient fibroblast cell lines (GM02052, GM03395 and GM03396 were obtained from Coriell Institute) as well as normal human fibroblasts (PromoCell) were cultured in Eagle’s MEM (GIBCO) containing Earle’s salts, nonessential amino acids, and supplemented with 10% heat-inactivated FBS (Hyclone). Hif-1α−/− ES cells were maintained as described (Ateghang et al., 2006). For hypoxia experiments, cells were grown in 10% serum and when nearly 60% confluent, the cells were cultured in an hypoxia chamber (Invivo2 300; Ruskinn) with conditions 0.2% oxygen and 10% carbon dioxide at 37°C.
In vitro kinase assays and mass spectrometry analysis
In vitro kinase assay for ATM were performed as described in Yang D. et, al.(Yang and Kastan, 2000). For kinase reactions with endogenous ATM, endogenous ATM was immunoprecipitated with monoclonal antibody D16.11. For Mass Spectrometry analysis, the Hif-1α protein band was excised from a 1D gel followed by in-gel tryptic digestion as described (Galea et al., 2006). LC-MS/MS analysis was carried out using a ThermoFinnigan LTQ linear ion trap mass spectrometer in line with a nanoAcquity ultra performance LC system (Waters Corporation, Milford, MA). Tryptic peptides were loaded onto a “precolumn” (Symmetry C18, 180um i.d X 20mm, 5um particle) (Waters Corporation, Milford, MA) connected to the analytical column (BEH C18, 75um i.d X 100mm, 1.7um particle) (Waters Corporation, Milford, MA). Tryptic peptides were eluted over a gradient (0–70% B in 60 minutes, 70–100% B in 10 minutes, where B = 70%Acetonitrile, 0.2% formic acid) at a flow rate of 250nL/min and introduced online into a linear ion trap mass spectrometer (ThermoFisher Corporation, San Jose, CA). Data dependent scanning was then performed. For database searching, product ions generated by fragmentation along the peptide backbone by collision activated dissociation (CAD) (b/y-type ions) were used in an automated database search against the SwissProt human subset database and a user defined protein database that contained the relevant sequence of the Hif-1α protein using the Mascot search routine with following residue modifications being allowed: carbamidomethylation on cysteine, oxidation on methionine, and phosphorylation on serine, threonine, and tyrosine. The identifications from the automated search were further validated through manual inspection of the raw data.
Solid tumor xenografts studies
CB17SC-M scid−/− female mice (Taconic Farms, Germantown, NY) were used to propagate subcutaneously implanted tumors (Houghton et al., 2007). Tumor sections were processed for immunohistochemistry using standard procedures. For studies marking hypoxic regions in tumors, EF5 was injected 3 h before tumor excision. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee.
Standard immunoblot and immunofluorescence staining, plasmids and generation of lentiviruses, RNA isolation and apoptosis assays are described under Supplemental Experimental Procedures.
Highlights
Negative regulation of mTORC1 signaling by hypoxia is ATM-dependent.
ATM-dependent phosphorylation of HIF1α(Ser696) is required for mTORC1 regulation
Maintained mTORC1 signaling in ATM−/− cells under hypoxia leads to apoptosis
Childhood solid tumors show 10- to 20-fold lower ATM levels than leukemias
Supplementary Material
Acknowledgements
We thank M.B Kastan, P.J. McKinnon, A. R. Tee, W.G Kaelin, L.W. Ellisen, R.L Giles, M. Gassmann, M.Z. Zidzienicka, L.H. Dang. C.J. Koch and T.M Gilmer for reagents described in Methods. We are grateful to Dr. Peter J. McKinnon and Dr. Robert G. Bristow for helpful comments. We also thank Chris Morton, Kelli Boyd for histology slides, St. Jude flow cytometry core facility for performing flow cytometric analysis, and the St. Jude proteomics core facility in the Hartwell Center for performing mass spectrometry analysis of Hif-1α. This work was supported USPHS Grants CA77776 and CA21675 (Cancer Center Support Grant), NO1-CM42216, and NO1-CM91001-03 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors are unaware of any actual or perceived conflict of interests.
References
- Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- Ateghang B, Wartenberg M, Gassmann M, Sauer H. Regulation of cardiotrophin-1 expression in mouse embryonic stem cells by HIF-1alpha and intracellular reactive oxygen species. J Cell Sci. 2006;119:1043–1052. doi: 10.1242/jcs.02798. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bencokova Z, Kaufmann MR, Pires IM, Lecane PS, Giaccia AJ, Hammond EM. ATM activation and signaling under hypoxic conditions. Mol Cell Biol. 2009;29:526–537. doi: 10.1128/MCB.01301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, Holsboer F, Arzt E. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131:309–323. doi: 10.1016/j.cell.2007.07.044. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiberg RA, Hammond EM, Dorie MJ, Welford SM, Giaccia AJ. DNA damage during reoxygenation elicits a Chk2-dependent checkpoint response. Mol Cell Biol. 2006;26:1598–1609. doi: 10.1128/MCB.26.5.1598-1609.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea CA, Pagala VR, Obenauer JC, Park CG, Slaughter CA, Kriwacki RW. Proteomic studies of the intrinsically unstructured mammalian proteome. J Proteome Res. 2006;5:2839–2848. doi: 10.1021/pr060328c. [DOI] [PubMed] [Google Scholar]
- Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001;15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson SL, Bindra RS, Glazer PM. Hypoxia-induced phosphorylation of Chk2 in an ataxia telangiectasia mutated-dependent manner. Cancer Res. 2005;65:10734–10741. doi: 10.1158/0008-5472.CAN-05-1160. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hammond EM, Giaccia AJ. The role of ATM and ATR in the cellular response to hypoxia and re-oxygenation. DNA Repair (Amst) 2004;3:1117–1122. doi: 10.1016/j.dnarep.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, et al. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002;62:5897–5901. [PubMed] [Google Scholar]
- Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–940. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kontny HU, Lehrnbecher TM, Chanock SJ, Mackall CL. Simultaneous expression of Fas and nonfunctional Fas ligand in Ewing's sarcoma. Cancer Res. 1998;58:5842–5849. [PubMed] [Google Scholar]
- Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, Guan KL. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007;26:4812–4823. doi: 10.1038/sj.emboj.7601900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Inoki K, Vacratsis P, Guan KL. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J Biol Chem. 2003;278:13663–13671. doi: 10.1074/jbc.M300862200. [DOI] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–225. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- Petroulakis E, Parsyan A, Dowling RJ, LeBacquer O, Martineau Y, Bidinosti M, Larsson O, Alain T, Rong L, Mamane Y, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–446. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–2892. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Gingras AC. The mRNA 5' cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol. 1998;10:268–275. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008;320:1507–1510. doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Thews O, Kelleher DK, Hoeckel M. Current status of knowledge and critical issues in tumor oxygenation. Results from 25 years research in tumor pathophysiology. Adv Exp Med Biol. 1998;454:591–602. doi: 10.1007/978-1-4615-4863-8_70. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters BG, van den, Beucken T, Magagnin MG, Koritzinsky M, Fels D, Koumenis C. Control of the hypoxic response through regulation of mRNA translation. Semin Cell Dev Biol. 2005;16:487–501. doi: 10.1016/j.semcdb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Yang DQ, Kastan MB. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol. 2000;2:893–898. doi: 10.1038/35046542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.