

Abstract
Neoplastic progression is an evolutionary process driven by the generation of clonal diversity and natural selection on that diversity within a neoplasm. We hypothesized that clonal diversity is associated with the risk of progression to cancer. We obtained molecular data from a cohort of 239 participants with Barrett’s esophagus (BE), including microsatellite shifts and loss of heterozygosity, DNA content tetraploidy and aneuploidy, methylation and sequence mutations. Using these data, we tested all major diversity measurement methods, including genetic divergence and entropy based measures, to determine which measures are correlated with risk of progression to esophageal adenocarcinoma. We also tested whether the use of different sets of loci and alterations to define clones (e.g. selectively advantageous vs. evolutionarily neutral) improved the predictive value of the diversity indices. All diversity measures were strong and highly significant predictors of progression (Cox proportional hazards model, p<0.001). The type of alterations evaluated had little effect on the predictive value of most of the diversity measures. In summary, diversity measures are robust predictors of progression to cancer in this cohort.
Keywords: genetic diversity, genetic heterogeneity, clonal evolution, Barrett’s esophagus, diversity index
Introduction
Neoplastic progression is an evolutionary process (1, 2) in which genetic instability generates new variants and natural selection leads to expansion of clones containing alterations that increase survival and/or proliferation of the clones. Most biomarkers of neoplastic progression measure the presence, absence or quantity of a gene product, genetic alteration, or histopathological features (3–9). A novel class of biomarkers would measure clonal evolution, rather than any particular genetic alteration or protein. Because they measure a fundamental property of neoplastic progression, such measures of clonal evolution may be generalizable across neoplasms even when those neoplasms require different genetic alterations to cause malignancy.
Genetic heterogeneity within a neoplasm fuels the process of clonal evolution by providing the variation upon which selection can act (1, 2), but the relationship between diversity and progression has yet to be systematically evaluated. We have previously reported that clonal diversity is associated with increased risk of progression to esophageal adenocarcinoma (EA) in patients with Barrett’s esophagus (BE) (10). However, our previous study evaluated only three measures of diversity (mean pairwise genetic divergence, number of clones and Shannon index) (10) from amongst a large number of measures that have been proposed in the ecological literature (11), where they were originally developed for measuring the number of species in an ecosystem. Mean pairwise genetic divergence assesses the genetic differences that have accumulated between clones(10, 12) while many of the other indices, including the number of clones, Shannon index (also known as the Shannon-Weaver Index), and Simpson index, are based on a generalized entropy equation (13). Translation of clonal diversity measures to the clinic will require determination of which diversity measure should be used and, as new assays become available, what types of alterations should be used to define the clones.
Barrett’s esophagus provides an ideal model of human intraepithelial neoplasia in which to measure the evolutionary dynamics of neoplastic progression (14). BE is a condition in which the normal stratified squamous epithelium of the esophagus is replaced by specialized intestinal metaplasia. Approximately 0.7% of persons with BE progress to EA each year, a rate roughly 50 times higher than that of similarly-aged persons in the U.S. general population (15, 16). Because of the 8–23% mortality associated with esophagectomy reported in the Medicare database (17) and the low overall probability that a patient will progress to cancer, patients with BE undergo periodic endoscopic biopsy surveillance for early detection of cancer (18, 19). Multiple biopsies may be safely and reproducibly obtained at each endoscopy, allowing detection of spatially distinct clones and measurement of their frequencies in the Barrett’s segment (10, 20, 21).
We set out to systematically test the different diversity indices as predictors of progression to EA. In addition, we examined which loci and alterations provide the best measurements of diversity for predicting risk of progression to EA. Diversity measures were evaluated by Kaplan-Meier cancer incidence curves and Cox proportional hazard models.
Methods
Cohort Information
Biopsies were obtained from each of 263 study participants, a subset of the Seattle Barrett’s Esophagus Study cohort, according to defined protocols as previously described (22). Of the 263 participants, for our analysis we used only the 239 participants for whom we had baseline 9p LOH data, 17p LOH data, ploidy data and followed prospectively for clinical follow-up. The baseline was defined as the first endoscopic procedure occurring after 1/1/1995. The study has been approved on a continuous basis since 1983 by the Human Subjects Division of the University of Washington and/or the Institutional Review Board of the Fred Hutchinson Cancer Research Center as well as the Wistar Institute. Duration of follow-up time was based on the length of time between the baseline study visit (interview and endoscopic biopsy surveillance) and the diagnosis of esophageal adenocarcinoma, or, in participants who did not develop cancer, the last endoscopy before December 13, 2007 (average 5.2 years, range = 0.10 – 7.5 years).
Molecular assays
One fresh, frozen biopsy obtained at every 2cm interval along the entire length of each participant’s BE segment at their baseline endoscopy was purified from underlying non-proliferating stroma into proliferating diploid (2N, G1), 4N (G2/tetraploid) and/or aneuploid fractions (10). DNA samples are defined as any DNA extracted from flow-purified nuclei after ki67/DNA content multiparameter flow-cytometry. Sampling density was normalized to a maximum of two samples per level every two centimeters (cm) spanning the ora serrata to the lower esophageal sphincter. Tetraploidy (4N) was defined as fractions with >6% of cells from the biopsy with DNA content between 3.85 and 4.10N, and aneuploidy as >2.5% of cells with DNA content at least 0.2N different from 2N and 4N fractions, as previously described (22, 23). The sorted fractions were assayed for 1) loss of heterozygosity and changes in microsatellite lengths (shifts) at 18 microsatellite loci spanning chromosome 17 (ten loci) and chromosome 9 (eight loci), 2) mutations in the TP53 and CDKN2A genes determined by sequencing of exons 5–9, and 2, respectively, and 3) methylation status of the CDKN2A promoter determined by methylation specific PCR performed in a subset of the cohort using bisulfite treated DNA, as previously described (22).
Genotyping
DNA was extracted from flow sorted fractions and subject to primer extension whole genome amplification as previously described (22). Genotyping was performed using 18 short tandem repeat (STR) microsatellite polymorphisms on chromosome arms 17p (D17S1298, D17S1537, TP53VNTR, D17S786, D17S974, D17S1303, D17S1294, D17S1301), 17q (D17S1290, 17S1293), 9p (D9S935, D9GATA62F03, D9S925, D9S932, D9S1121, D9S1118), and 9q (D9S301, D9S930). For each locus, gastric control samples for each participant were genotyped and the ratio of the peak signals of two alleles was calculated. Our data showed that the distribution of allele ratios (log-transformed) of each locus (from different human research participants) satisfied normal distribution well. Therefore, for a given locus (i) it had a locus-specific normal distribution of allele peak ratio values with mean (mi) and standard deviation (si) which was measured from different normal samples. To evaluate the LOH of somatic tissue at a specific locus i for a given patient, the ratio of two signal peak values was calculated and log transformed (ri). The p-value of the normal sample is estimated as , where Φ is the cumulative distribution function of normal (24).The p-value for LOH is estimated by 1 − pn. The p-values were adjusted (25) for multiple comparison with a false discovery rate of 1% since multiple loci were evaluated. To determine informativity for each participant/locus, the number and basepair difference between all normal controls was determined. For any sample to be evaluated for LOH at a specific locus we required that all normal controls were in agreement or that locus was not further evaluated for that participant. Only 1 biopsy or up to 2 DNAs per every 2cm level was used. Allele peaks were called automatically using Genescan/Genotyper software (Applied Biosystems). For informative loci, the lower limit of peak detection was set to 125 fluorescent units for LOH results with only one detectable peak in order to limit false positive LOH results.
Diversity indices
Mean pairwise divergence was determined by the number of non-contiguous molecular differences between two samples divided by the number of informative assays, averaged over all pairs of samples, as previously described (10). Clones were distinguished by differences in 1) DNA content, LOH, microsatellite shifts and sequence mutations in CDKN2A and TP53, 2) LOH and DNA content only, 3) selected loci (only LOH and mutation of TP53, and LOH, mutation, and methylation of CDKN2A were considered), and 4) neutral loci. We have previously shown that LOH on the q arms of chromosomes 9 and 17, DNA content abnormalities, as well as shifts in the lengths of all 19 microsatellites are evolutionarily neutral and are not associated with clonal expansions in BE (though ploidy is predictive of progression) (20). For conditions 1, 2 and 4, a DNA content difference of >0.2N was sufficient to distinguish a clone (26). The frequency of each clone was calculated from the proportion of the clone in each of the flow-sorted fractions as previously described (10).
Diversity indices (H) of order (Hill number) q were calculated by the generalized expression (Equation 1):
| (1) |
where S is the total number of clones and pi is the frequency of clone i (13). Though Hill cautions against using non-integer values of q (27) to avoid unnecessary complication, we have tested a complete range from 0 to 3 in increments of 0.2 to examine how the performance of the diversity indices change across that range.
Data analysis methods
A Cox proportional hazards regression model was used to assess the significance of each diversity index in predicting progression to cancer. Kaplan-Meier (KM) curves were used to estimate survival probabilities of the strata stratified by the different diversity indices. Differences between KM curves were evaluated with by log-rank test (28). Analyses were performed using the R statistical package.
Results
Cohort characteristics are shown in Table 1. The large relative proportion of males to females in the cohort is characteristic of patients with BE. Average participant age is 63 (median=65), and average segment size is 4.6 cm (median=3 cm). Of the 239 participants, 33 developed esophageal adenocarcinoma in follow-up. We tested the association between clonal diversity and risk of progression to cancer in univariate analyses using two qualitatively different types of diversity measures. The first is a measure of the number of genetic differences between clones in the same neoplasm (sometimes called taxonomic distinctness (12)). To measure those differences, we used mean pairwise genetic divergence (10), which is simply the number of loci that differ between two samples, divided by the number of informative loci (heterozygous loci in the normal tissue). LOH alterations that span contiguous loci are considered single events for calculating divergence. This value is averaged over every pair of samples from a Barrett’s segment.
Table 1.
Cohort Characteristics
| Characteristic | Group | Number | Percentage |
|---|---|---|---|
| Sex* | Male | 185 | 77 |
| Female | 52 | 22 | |
| Age* | 30–44 | 14 | 6 |
| 45–54 | 43 | 18 | |
| 55–64 | 57 | 24 | |
| 65–74 | 76 | 32 | |
| ≥75 | 47 | 20 | |
| Barrett’s segment length (cm) | <3 | 69 | 29 |
| 3–6 | 101 | 42 | |
| 7–10 | 48 | 20 | |
| >10 | 21 | 9 |
No data for sex and age for 2 participants
The second type of clonal diversity index combines a measure of the total number of clones and their relative abundance. Most diversity measures in ecology fall into this category. Hill showed that, using Equation 1, number and abundance measures could be unified into a single framework with one parameter (q) that can be adjusted to achieve the various common diversity indices (27). The values of q scale the relative importance of rare vs. dominant clones in the measure (Figure 1) and are sometimes called Hill numbers (27). When q=0, Equation 1 simply counts the number of clones. This maximizes the impact of rare clones, since the smallest clone detected will increment the clone count by the same amount as the largest clone detected.
Figure 1.
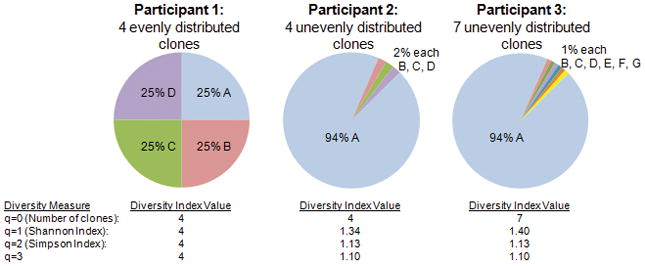
Example of effect of the distribution of clones on diversity indices for different Hill numbers (values of q) (27). Participants 1 and 2 have 4 clones (Diversity index=4 at q =0). In this hypothetical example, as q increases, a single dominant clone in Participants 2 and 3 has an increasingly strong negative effect on the total diversity. In other words, the rare clones in Particpants 2 and 3 have less impact on diversity as q increases. The addition of 3 more rare clones in Participant 3, relative to Participant 2, has a large impact on the q =0 diversity measure (number of clones), a small effect on the Shannon Index (q =1), and negligible effect on the q =2 (Simpson index) or q=3 diversity measures in Participant 3.
However, a neoplasm with four equally abundant clones (Figure 1, Participant 1) can be logically considered more diverse than a neoplasm with one dominant clone and three rare clones (Figure 1, Participant 2). Hill numbers > 0 capture this assumption by including relative abundance of each clone in the index along with number of clones. Equation 1 is undefined for q=1, but in the limit, as q approaches 1, Equation 1 is the exponential of the Shannon entropy index (10). When q=2, Equation 1 is the reciprocal of the Simpson index of diversity, which, like the Shannon index, is common in ecology for measuring species diversity (13, 29, 30). The addition of small clones has a substantial effect on diversity when q=0 but little impact on diversity indices with high values of q (Figure 1 Participant 2 vs. 3). The presence of numerous rare clones may have biological importance as a mechanism for generating variation for clonal expansion or in the evolution of resistance to interventions. We tested a variety of q values as they may provide different information about the risk of progression to cancer.
Some people have suggested that diversity should be measured in the functional differences between organisms (11, 31, 32). The best analogy to this functional diversity in a neoplasm may be to define clones based on selected alterations that affect the fitness of a clone. We have previously shown that alterations that inactivate an allele of CDKN2A or TP53 are associated with large clonal expansions and so appear to increase the fitness of a clone (20). For all our diversity indices (mean pairwise genetic divergence and entropy measures based on different q values), we tested four different sets of loci and alterations to define a clone. First, we used all genetic alterations measured. Second, we used only LOH in the microsatellite loci and DNA content abnormalities (10). Third, we used only selected alterations and fourth, we used only evolutionarily neutral alterations – those that have no effect on the fitness of a clone, as well as DNA content abnormalities. These different definitions of a clone and their effects on diversity measures are illustrated in an example of a Barrett’s esophagus segment from a single participant (Figure 2).
Figure 2.
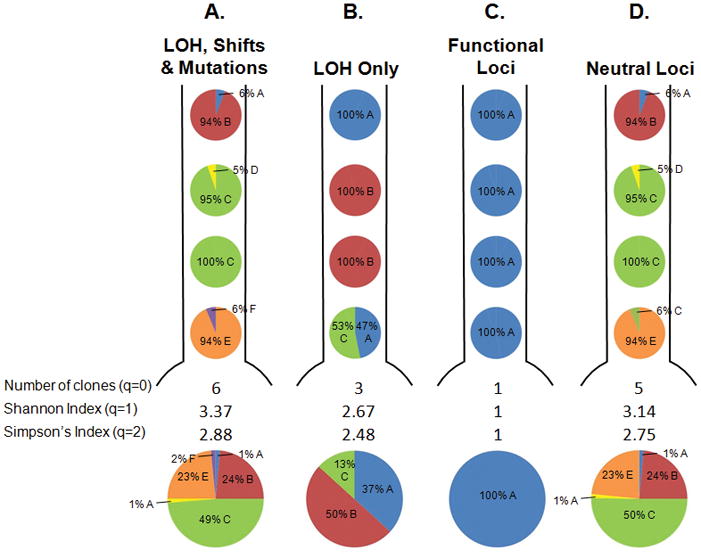
Map of the clones in the BE segment from a single participant from our cohort under different definitions of a clone. The pie charts below each segment represent total % of each clone in the BE segment. Defining clones in different ways alters the distribution and number of clones and the diversity indices. A. Both LOH and changes in microsatellite lengths (shifts) in all 18 microsatellites, as well as sequence mutations in CDKN2A (p16/INK4A) and TP53 (p53), and DNA content are used to define a clone. B. Only LOH lesions in the 18 microsatellites and DNA content are used to define a clone, while shifts and mutations are ignored. C. Only lesions that increase the fitness of a clone are used to define a clone. These lesions include LOH or sequence mutations in CDKN2A or TP53, or hypermethylation of the CDKN2A promoter (20). D. DNA content and lesions that have no detectable effect on the fitness of a clone were used, including shifts in any of the 19 microsatellites and LOH on the q arms of chromosomes 9 and 17 (20). This illustrative patient was chosen because the different definitions of clones lead to different diversity measures, though this was often not the case for other participants in the cohort.
Our results demonstrate that diversity is a strong predictor of progression from Barrett’s esophagus to esophageal adenocarcinoma regardless of whether divergence (Figure 3), number of clones, Shannon index, Simpson index or other q values are used (Figures 4, 5 and Table 2, Cox regression p<0.001 in all cases). Our results also show that diversity is a robust predictor of progression across all types of genetic alterations, whether LOH alone, selected alterations, neutral alterations, or all genetic alterations were used to define a clone (Table 2, pFigures 3–5, Cox regression <0.001 in all cases). We do find that LOH, alone, generates slightly lower p-values than the other categories of loci for entropy-based diversity measures (Figure 4 and 5) and may provide the best category of loci for measuring diversity. However, we did not have the power to distinguish significant differences between loci categories in this study, which would require larger numbers of individuals with Barrett’s esophagus and ideally, validation in a separate Barrett’s esophagus cohort. Our results do provide strong evidence that in endoscopic biopsies from Barrett's esophagus cohort participants obtained an average of 5.2 years prior to their last assessed clinical outcome, all diversity indices, measured in all categories of loci, were predictive of future progression to esophageal adenocarcinoma.
Figure 3.

Kaplan-Meier cancer incidence curves for mean pairwise genetic divergence based on four different definitions of clones (p<0.001 in all cases). Red=upper quartile, Black=bottom three quartiles. The (number of cancers/total number of participants) in the upper and lower 3 quartiles are given as numbers to right of the curves.
Figure 4.

Kaplan-Meier cancer incidence curves for q=0 based on four different definitions of clones (p<0.001 in all cases). Red=upper quartile, Black=bottom three quartiles. The (number of cancers/total number of participants) in the upper and lower 3 quartiles are given as numbers to right of the curves.
Figure 5.

Kaplan-Meier cancer incidence curves for q=2 based on four different definitions of clones (p<0.001 in all cases). ). Red=upper quartile, Black=bottom three quartiles. The (number of cancers/total number of participants) in the upper and lower 3 quartiles are given as numbers to right of the curves.
Table 2.
Relative Risk for each unit of the diversity measure for progression to EA, evaluated for mean pairwise genetic divergence and the entropy based measures with varying q values (Equation 1) and different alterations used to define a clone.
| LOH, Microsatellite Shifts and mutations | LOH only | Selective Loci | Neutral Loci | |
|---|---|---|---|---|
| RR (95% CI) | RR (95% CI) | RR (95% CI) | RR (95% CI) | |
| Divergence, per 10% | 1.46 (1.20–1.77) | 1.87 (1.38–2.54) | 1.41 (1.23–1.61) | 1.58 (1.22–2.05) |
| q=0 | 1.68 (1.46 – 1.94) | 1.71 (1.48 – 1.98) | 2.17 (1.67 – 2.82) | 1.86 (1.57 – 2.2) |
| q=0.2 | 1.77 (1.52 – 2.06) | 1.77 (1.52 – 2.07) | 2.29 (1.7 – 3.09) | 1.91 (1.6 – 2.29) |
| q=0.4 | 1.84 (1.56 – 2.16) | 1.81 (1.54 – 2.13) | 2.35 (1.7 – 3.26) | 1.95 (1.61 – 2.36) |
| q=0.6 | 1.88 (1.59 – 2.23) | 1.84 (1.56 – 2.18) | 2.36 (1.67 – 3.34) | 1.98 (1.62 – 2.41) |
| q=0.8 | 1.91 (1.61 – 2.28) | 1.87 (1.57 – 2.22) | 2.34 (1.63 – 3.37) | 2 (1.62 – 2.46) |
| q=1 | 1.93 (1.62 – 2.31) | 1.88 (1.58 – 2.25) | 2.32 (1.59 – 3.37) | 2.02 (1.62 – 2.51) |
| q=1.2 | 1.95 (1.62 – 2.34) | 1.9 (1.58 – 2.28) | 2.28 (1.55 – 3.36) | 2.03 (1.62 – 2.56) |
| q=1.4 | 1.96 (1.63 – 2.37) | 1.91 (1.59 – 2.3) | 2.25 (1.52 – 3.34) | 2.05 (1.62 – 2.6) |
| q=1.6 | 1.97 (1.63 – 2.38) | 1.92 (1.59 – 2.32) | 2.22 (1.49 – 3.33) | 2.06 (1.61 – 2.63) |
| q=1.8 | 1.98 (1.63 – 2.4) | 1.93 (1.59 – 2.34) | 2.2 (1.46 – 3.31) | 2.08 (1.61 – 2.67) |
| q=2 | 1.98 (1.63 – 2.41) | 1.94 (1.6 – 2.36) | 2.18 (1.44 – 3.3) | 2.09 (1.61 – 2.7) |
| q=2.2 | 1.99 (1.63 – 2.43) | 1.95 (1.6 – 2.38) | 2.16 (1.41 – 3.29) | 2.1 (1.61 – 2.74) |
| q=2.4 | 1.99 (1.63 – 2.44) | 1.96 (1.6 – 2.4) | 2.14 (1.4 – 3.28) | 2.11 (1.61 – 2.77) |
| q=2.6 | 2 (1.63 – 2.45) | 1.97 (1.6 – 2.41) | 2.13 (1.38 – 3.28) | 2.12 (1.61 – 2.79) |
| q=2.8 | 2.01 (1.63 – 2.46) | 1.97 (1.61 – 2.43) | 2.11 (1.37 – 3.27) | 2.13 (1.61 – 2.82) |
| q=3 | 2.01 (1.63 – 2.48) | 1.98 (1.61 – 2.44) | 2.1 (1.35 – 3.27) | 2.14 (1.61 – 2.84) |
p<0.001 for all diversity measures. q=0 measures the number of clones, the RR is per clone.
q=1 is the Shannon index and q=2 is the Simpson index.
Discussion
During neoplastic evolution, the genome develops a variety of selective and neutral alterations that create diversity in evolving cell populations. Methods developed for ecological studies provide a general framework for analyzing genomic and epigenomic abnormalities that develop during progression to cancer. In this study, we applied different diversity indices from the ecology literature for cancer risk prediction in Barrett’s esophagus using microsatellite, DNA content, sequence mutation and promoter hypermethylation data. All diversity measures, including mean pairwise genetic divergence and the entropy-based measures, are highly significant predictors of future progression to esophageal adenocarcinoma in this data set (Table 2, Figures 3–5). Interestingly, the type of alteration used to characterize diversity appears to make little difference, though there is some evidence that with only 5 loci, the diversity measures based on selective loci lose some sensitivity (Table 2, Figures 3–5). The consistency of our results with respect to the type of diversity measure and alterations used to define clones suggests that diversity measures are robust biomarkers for risk stratification.
Measures of diversity are expected to be correlated with genetic instability, but are not equivalent to genetic instability. Clonal diversity is a function of both the generation of mutations and selection upon those mutations. Our assays only measure the majority clone of a biopsy sample, and thus a clone with a new genetic variant would need to expand to at least thousands of cells before we could detect it. Future work may use alternative assays to provide measurements of minority clones in a biopsy and potentially measure mutation rates to provide information about the level of genetic instability in a tumor. We expect that many of the smaller clones detected with these assays may not have a selectively advantageous mutation and thus may only provide indirect information about risk of progression, but this remains to be tested.
Since mean pairwise genetic divergence and all the entropy-based diversity measures are significant predictors of progression, and since it mattered little which loci were used to define the clones for the given data, our results suggest that use of clonal diversity as a biomarker could be robust with respect to changes in the assay used to detect diversity in a neoplasm. However, we expect that these measures are unlikely to be adequate for clinical use as single biomarkers, and may need to be combined with other measures of risk in a biomarker panel. Theoretically, this is because all the indices evaluated in this study were the results of the compression of a large amount of genomic and spatial information into a single value. Therefore, the indices may give some reasonable quantification of genome instability and long-term coexistence of clones, but may lose some ‘resolution’ or accuracy for cancer risk prediction as a single marker. Measures of diversity do provide independent information on the evolutionary dynamics of progression and thus may be useful for consideration in a panel of markers used to assess risk of progression.
Our method of measuring genetic diversity specifically measures only the viable clones that are capable of expanding to detectable sizes and does not require single cell assays. We find that at this scale even very simple entropy measures of diversity such as counting number of clones (Hill number q = 0), performed on multiple biopsies, predict risk of progression and that the subset of loci used does not matter for the clones described here. The process of neoplastic evolution is likely driven by minority clones that acquire selectively advantageous mutations. Lower q values account for these rare clones and thus may be superior to diversity indices based on higher q values. However, all entropy-based measures, particularly those at lower q values, are unstable with respect to the number of biopsies taken and assay used. We are interested in evaluating whether a good performance of the indices for cancer risk stratification could be achieved with genome wide data using a different platform such as a SNP array. Use of a subset of loci will likely be important for the translation of these entropy-based clonal diversity measures to whole genome assays. With millions of loci, genomic assays could identify unique alterations in every sample. This would make the number of clones, and the other measures based on Equation 1, simply a function of the number of samples, thus rendering them uninformative for risk stratification. In such a case, a subset of the loci should be used to define the clones. The genetic divergence measure is not expected to suffer from the same potential bias based on the number of samples and so should be generalizable to whole genome measures. We thus anticipate that divergence measurements may be superior for clinical use since they can be compared with different sampling techniques and assay platforms.
There are limitations to this study. We have only measured alterations on chromosomes 9 and 17, which are known to be important in BE progression. Though we have shown that some of those alterations are evolutionarily neutral (20), we do not yet know if alterations on other chromosomes would behave similarly in diversity assays. The fact that the different entropy-based measures perform similarly, regardless of the weighting placed on small or large clones, suggests that relative abundances of clones are not particularly important for risk stratification in this cohort. However, small clones (e.g., < 5,000 cells in a flow sorted fraction) would not be detectable in our LOH assays. Since small Hill numbers (q values) increase the sensitivity of the diversity index to rare clones, some q values might show significant differences as biomarkers of progression if used with an assay that could detect very small clones. Another limitation of these diversity measures is that they require assays of multiple independent samples (or some other way to separate clones) within a Barrett’s segment, which is not always feasible.
Barrett’s esophagus segments have many molecular similarities to other conditions that can predispose to cancer (CDKN2A LOH, TP53 LOH, and aneuploidy), including oral leukoplakia, dysplasia and carcinoma in situ precursor alterations of the bladder, and metaplastic and dysplastic regions of lung tissue associated with lung cancer (8, 33–38). This implies that the relationship between diversity and progression may be generalizable to other alterations that predispose to progression to cancer, particularly those associated with chronic inflammation. Since clonal diversity is a fundamental property of clonal evolution, it is likely to be relevant to most conditions that predispose to progression to cancer, in contrast to specific molecular alterations that characterize neoplastic progression in different organs. We thus predict that measures of diversity will be applicable to other conditions that predispose to cancer. Recent work also suggests that diversity measurements may be associated with clinical variables in breast cancer (39), potentially expanding the utility of these diversity measurements beyond analysis of progression risk in premalignant conditions. In addition, because acquired therapeutic resistance is often generated by selection on the (epi)genetic diversity within a neoplasm (40), we predict that measures of diversity will be useful in therapeutic prognosis, as well as neoplastic progression. This remains to be tested.
Acknowledgments
We would like to thank Dennis Chao, Carissa Sanchez, Patricia C. Galipeau, and Alan Kristal for helpful advice and thoughtful comments on the manuscript. We would also like to thank Carissa Sanchez for providing the flow cytometry and cell cycle analysis data necessary to calculate the frequency of clones and Patricia Galipeau for generating the microsatellite, sequence and methylation data used to define the clones. This work was funded in part by NIH grants P01 CA91955, R01 CA119224, R03 CA137811, P30 CA010815, R01 CA14065 and F32 CA132450, the American Cancer Society, a Landon AACR Innovator Award for Cancer Prevention, the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health, and the Pew Charitable Trust.
References
- 1.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 2.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–35. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 3.Kerkhof M, Kusters JG, van Dekken H, Kuipers EJ, Siersema PD. Biomarkers for risk stratification of neoplastic progression in Barrett esophagus. Cell Oncol. 2007;29:507–17. doi: 10.1155/2007/814950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin CM, Kehoe L, Spillane CO, O'Leary JJ. Gene discovery in cervical cancer : towards diagnostic and therapeutic biomarkers. Mol Diagn Ther. 2007;11:277–90. doi: 10.1007/BF03256249. [DOI] [PubMed] [Google Scholar]
- 5.Tabor MP, Braakhuis BJ, van der Wal JE, et al. Comparative molecular and histological grading of epithelial dysplasia of the oral cavity and the oropharynx. J Pathol. 2003;199:354–60. doi: 10.1002/path.1285. [DOI] [PubMed] [Google Scholar]
- 6.Kawaguchi H, El-Naggar AK, Papadimitrakopoulou V, et al. Podoplanin: a novel marker for oral cancer risk in patients with oral premalignancy. J Clin Oncol. 2008;26:354–60. doi: 10.1200/JCO.2007.13.4072. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava A, Hornick JL, Li X, et al. Extent of low-grade dysplasia is a risk factor for the development of esophageal adenocarcinoma in Barrett's esophagus. Am J Gastroenterol. 2007;102:483–93. doi: 10.1111/j.1572-0241.2007.01073.x. quiz 694. [DOI] [PubMed] [Google Scholar]
- 8.Lee JJ, Hong WK, Hittelman WN, et al. Predicting cancer development in oral leukoplakia: ten years of translational research. Clin Cancer Res. 2000;6:1702–10. [PubMed] [Google Scholar]
- 9.Mao L, Lee JS, Fan YH, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nature Medicine. 1996;2:682–5. doi: 10.1038/nm0696-682. [DOI] [PubMed] [Google Scholar]
- 10.Maley CC, Galipeau PC, Finley JC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 11.Magurran AE. Measuring biological diversity. Malden, Ma: Blackwell Pub; 2004. [Google Scholar]
- 12.Clarke KR, Warwick RM. A Taxonomic Distinctness Index and its Statistical Properties. Journal of Applied Ecology. 1998;35:523–31. [Google Scholar]
- 13.Jost L. Entropy and Diversity. Oikos. 2006;113:363–75. [Google Scholar]
- 14.Maley CC. Multistage carcinogenesis in Barrett's esophagus. Cancer Lett. 2006 doi: 10.1016/j.canlet.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Thomas T, Abrams KR, De Caestecker JS, Robinson RJ. Meta analysis: Cancer risk in Barrett's oesophagus. Aliment Pharmacol Ther. 2007;26:1465–77. doi: 10.1111/j.1365-2036.2007.03528.x. [DOI] [PubMed] [Google Scholar]
- 16.Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst. 2008;100:1184–7. doi: 10.1093/jnci/djn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birkmeyer JD, Siewers AE, Finlayson EV, et al. Hospital volume and surgical mortality in the United States. N Engl J Med. 2002;346:1128–37. doi: 10.1056/NEJMsa012337. [DOI] [PubMed] [Google Scholar]
- 18.Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett's oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer. 2010;10:87–101. doi: 10.1038/nrc2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang KK, Sampliner RE. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett's esophagus. Am J Gastroenterol. 2008;103:788–97. doi: 10.1111/j.1572-0241.2008.01835.x. [DOI] [PubMed] [Google Scholar]
- 20.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett's esophagus. Cancer Res. 2004;64:3414–27. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 21.Barrett MT, Sanchez CA, Prevo LJ, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nature Genetics. 1999;22:106–9. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galipeau PC, Li X, Blount PL, et al. NSAIDs modulate CDKN2A, TP53, and DNA content risk for progression to esophageal adenocarcinoma. PLoS Med. 2007;4:e67. doi: 10.1371/journal.pmed.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett's esophagus III: baseline flow cytometric variables. American Journal of Gastroenterology. 2001;96:3071–83. doi: 10.1111/j.1572-0241.2001.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kissel HD, Galipeau PC, Li X, Reid BJ. Translation of an STR-based biomarker into a clinically compatible SNP-based platform for loss of heterozygosity. Cancer Biomark. 2009;5:143–58. doi: 10.3233/CBM-2009-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 26.Levine DS, Rabinovitch PS, Haggitt RC, et al. Distribution of aneuploid cell populations in ulcerative colitis with dysplasia or cancer. Gastroenterology. 1991;101:1198–210. doi: 10.1016/0016-5085(91)90068-v. [DOI] [PubMed] [Google Scholar]
- 27.Hill MO. Diversity and Evenness: A Unifying Notation and its Consequences. Ecology. 1973;54:427–32. [Google Scholar]
- 28.Harrington DP, Fleming T. A class of rank test procedures for censored survival data. Biometrika. 1982;69:553–66. [Google Scholar]
- 29.Chalmers G, Martin SW, Prescott JF, Boerlin P. Typing of Clostridium perfringens by multiple-locus variable number of tandem repeats analysis. Vet Microbiol. 2007 doi: 10.1016/j.vetmic.2007.09.018. in press. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Stohlgren TJ, Chong GW. Spatial heterogeneity influences native and nonnative plant species richness. Ecology. 2006;87:3186–99. doi: 10.1890/0012-9658(2006)87[3186:shinan]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 31.Tilman D, Reich PB, Knops J, Wedin D, Mielke T, Lehman C. Diversity and productivity in a long-term grassland experiment. Science. 2001;294:843–5. doi: 10.1126/science.1060391. [DOI] [PubMed] [Google Scholar]
- 32.Loreau M. Are Communities Saturated? On the relationship between alpha, beta and gamma diversity. Ecology Letters. 2000;3:73–6. [Google Scholar]
- 33.Chaturvedi V, Li L, Hodges S, et al. Superimposed histologic and genetic mapping of chromosome 17 alterations in human urinary bladder neoplasia. Oncogene. 1997;14:2059–70. doi: 10.1038/sj.onc.1201044. [DOI] [PubMed] [Google Scholar]
- 34.Czerniak B, Chaturvedi V, Li L, et al. Superimposed histologic and genetic mapping of chromosome 9 in progression of human urinary bladder neoplasia: implications for a genetic model of multistep urothelial carcinogenesis and early detection of urinary bladder cancer. Oncogene. 1999;18:1185–96. doi: 10.1038/sj.onc.1202385. [DOI] [PubMed] [Google Scholar]
- 35.Franklin WA, Gazdar AF, Haney J, et al. Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest. 1997;100:2133–7. doi: 10.1172/JCI119748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braakhuis BJ, Leemans CR, Brakenhoff RH. A genetic progression model of oral cancer: current evidence and clinical implications. J Oral Pathol Med. 2004;33:317–22. doi: 10.1111/j.1600-0714.2004.00225.x. [DOI] [PubMed] [Google Scholar]
- 37.Mao L, Lee JS, Kurie JM, et al. Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst. 1997;89:857–62. doi: 10.1093/jnci/89.12.857. [DOI] [PubMed] [Google Scholar]
- 38.Cairns P, Polascik TJ, Eby Y, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995;11:210–2. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 39.Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120:636–44. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pepper J, Scott Findlay C, Kassen R, Spencer S, Maley C. Cancer research meets evolutionary biology. Evolutionary Applications. 2009;2:62–70. doi: 10.1111/j.1752-4571.2008.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]