

Abstract
Cancer development involves major alterations of cells’ metabolism. Enhanced glycolysis and de novo fatty acids synthesis are indeed characteristic features of cancer. Cell proliferation and metabolism are tightly linked cellular processes. Others and we have previously demonstrated a close relationship between metabolic responses and proliferative stimuli. In addition to trigger proliferative and survival signaling pathways, most oncoproteins also trigger metabolic changes in order to transform the cell. We present herein the view that participation of cell-cycle regulators and oncogenic proteins to cancer development extend beyond the control of cell proliferation, and discuss how these new functions may be implicated in metabolic alterations concomitant to the pathogenesis of human cancers.
Keywords: Animals; Cell Cycle; physiology; Cell Proliferation; Humans; Lipid Metabolism; physiology; Models, Biological; Neoplasms; metabolism; pathology; physiopathology; Signal Transduction; genetics; physiology; Transcription Factors; metabolism; physiology
Keywords: Cancer, Metabolism, Oncogenes, Cell-Cycle regulators, Glycolysis, de novo Lipogenesis
Introduction
Over the past decades, accumulating evidence of a metabolic reorganization during cancer development has emerged from studies on various tumor types. We present herein the view that participation of cell-cycle regulators and oncogenic proteins to cancer development extend beyond the control of cell proliferation, and discuss how these new functions may be implicated in metabolic alterations concomitant to the pathogenesis of human cancers. This is consistent with the observation that changes in metabolism are part of a coordinated response of the cell to distinct physiological and pathological conditions. In mammalian cells the metabolic response requires a permanent coordination of cell activity, including cell proliferation with nutrient availability, hormonal and stress signaling and with regulation of energy homeostasis. This links external signaling events with the activation or inhibition of particular metabolic pathways, such as oxidative or glycolytic metabolism, and biosynthetic processes. Nutrient availability enables nucleic acid, protein, and lipid synthesis to promote cell growth and proliferation. It is also correlated with increased glycolytic flux, and decreased oxidative metabolism (Jezek et al 2010). On the opposite, nutrient deprivation implicates a metabolic switch that triggers energy production pathways, such as oxidative phosphorylation that restrict cell proliferation. In general, cell proliferation and metabolism are tightly linked cellular processes. In non-proliferating cells from differentiated tissues, catabolic metabolism is predominantly used to fuel energetic needs, and is associated to preferential use of mitochondrial oxidative phosphorylation (OXPHOS) with oxidation of macromolecules. In sharp contrast, in tissues with high proliferative rate, bioenergetics resources and biosynthetic pathways are reorganized to allow cell cycle progression, leading to a shift toward an anabolic metabolism, with high glycolytic flux, lactate production and biosynthesis of macromolecules. Several physiopathological conditions are indeed characterized by a switch from an oxidative to a glycolytic metabolism. This is the case for developmental processes, tissue regeneration, intensive exercise, or immune response. In a pathological context, it is currently well established that cancer growth and invasion also involves major alterations of cells’ metabolism. A common regulatory pathway exists therefore between cell proliferation and metabolism to insure a coordinated cellular response. This supports the hypothesis, which we develop in this review that the same factors that trigger proliferation, such as oncogenes or cell cycle regulators are indeed coordinators of the metabolic response of the cells to the external stimuli.
Metabolic changes in cancer: the contribution of lipids metabolism
One of the first identified biochemical hallmark of cancer cells were changes in metabolism. Early in the last century, Otto Warburg (1930) observed that tumor cells have a higher rate of glucose metabolism than their normal counterparts and preferentially use glycolysis instead of OXPHOS even under appropriate oxygen concentrations (Warburg 1930, Warburg 1956a, Warburg 1956b). Since this first observation, the « aerobic » glycolysis switch has been detected in many tumor types and cumulating studies on various proliferating cells have led to the evidence of a global metabolic reorganization concomitant to cancer progression. Most tumor cells are characterized by higher rates of glycolysis, lactate production, and macromolecules and lipids biosynthesis (Kroemer and Pouyssegur 2008, Vander Heiden et al 2009). During aerobic glycolysis, glucose is converted to pyruvate, and the final sub-product of this reaction is lactate, which is exported out of the cell, contributing to extracellular acidification. The preferential use of aerobic glycolysis offers several advantages to highly proliferating cells, concerning both bioenergetics and biosynthetic requirements. First, it allows for the use of the most abundant extracellular nutrient, which is glucose. Second, the flux of ATP can exceed that produced during OXPHOS. Third, it is likely that one of the critical roles of aerobic glycolysis in cancer cells is to provide essential metabolic intermediates for biosynthesis of macromolecules (lipids, proteins and nucleic acids) in order to support increased proliferation (Bauer et al 2005).
Increased de novo fatty acids (FA) synthesis, or lipogenesis, is considered as another major metabolic alteration of cancer cells (Medes et al 1953, Sabine et al 1967). Figure 1 summarizes the participation of lipids and carbohydrates metabolism in cancer development and progression. Although de novo FA synthesis is very active during embryogenesis, most adult normal cells and tissues, even those with high cellular turnover, preferentially use circulating FA for the synthesis of new structural lipids. In contrast, various tumors and their precursor lesions, undergo exacerbated endogenous FA biosynthesis irrespective of the levels of extracellular lipids (Medes et al 1953). The increased lipogenesis in cancer is reflected in over expression and hyperactivity of lipogenic enzymes such as ATP citrate lyase (ACL), acetyl-CoA carboxylase (ACC), or the fatty acid synthase (FAS) (Kuhajda 2000). ACC carboxylates acetyl-CoA to form malonyl-CoA, which is further converted to long-chain FA by FAS. Interestingly, studies with chemical inhibitors have revealed that inhibition of FAS activity results in decreased proliferation and increased apoptosis of cancer cells (Kuhajda et al 1994, Kuhajda 2000). De novo FA biosynthesis is required for cancer cells to synthesize new membranes, which have a particular lipidic composition that facilitates the formation of lipid rafts for increased signaling of cell growth receptors. Some lipid synthesis intermediates, such as malonyl CoA participate in the transcriptional regulation of growth factor receptors (reviewed in (Menendez and Lupu 2007). Moreover, circulating lipids impact on cell cancer growth, migration, and invasion. This is the case for the lysophosphatidic acid (LPA), which signals through LPA receptors to stimulate cancer cell proliferation and survival (Bauer et al 2005). Enhanced lipogenesis in cancer cells has also been proposed to be required to balance the redox potential via the utilization of NADPH (Porstmann et al 2009). In addition, de novo FA synthesis in cancer cells facilitates the production of lipids that will regulate the activity of various oncogenes. Indeed, signaling lipids, such as phosphatidyl inositol, phosphatidyl serine, or phosphatidyl coline, are recognized to be important factors that activate and mediate proliferative and survival pathways, notably the PI3K/AKT, Ras, or Wnt pathways (Nadolski and Linder 2007). Finally, post-translational modification with lipid moieties is also a key process regulating transport and function of various cellular and secreted oncoproteins. Indeed, several studies have demonstrated that fatty acid synthesis facilitates the formation of particular fatty acids that modify the protein structure of a particular set of oncogenes, such as Ras, Src, or Wnt. This in turn facilitates the activation of their oncogenic pathways (Nadolski and Linder 2007, Xue et al 2004). These findings highlight the relevance of lipid synthesis in oncogenesis and fully support the investigation of lipid synthesis inhibitors as anti-cancer agents.
Figure 1. Cross-talk between glycolysis and de novo FA synthesis in cancer cells.
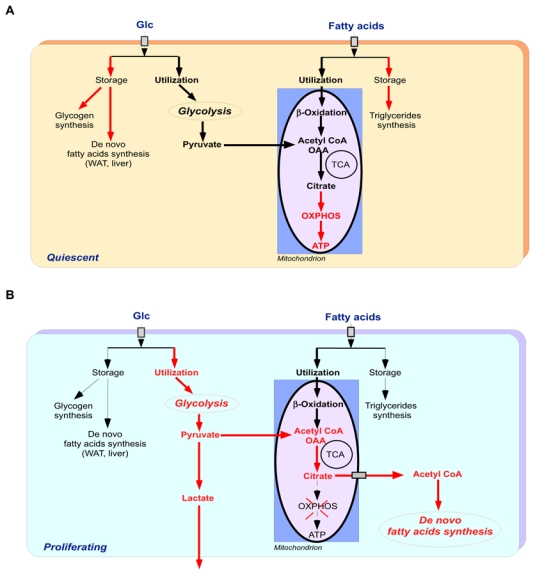
This model highlights the main differences in glucose and fatty acids utilization between quiescent (A) and highly proliferative cells (B). Red arrows indicate preferred metabolic pathway. In non-proliferating cells glucose can either be used for glycogen or fatty acids synthesis, or can be converted to pyruvate through basal rate of glycolysis that occurs in the cytoplasm. Pyruvate is then imported into the mitochondrion where it is decarboxylated to acetyl Coa or oxalacetate (OAA). Circulating fatty acids are also imported into non-proliferating cells were they are stored as triglycerides or oxidized into the mitochondrion to generate acetyl-CoA. Acetyl-CoA derived from both glucose and fatty acids is then oxidized through TCA cycle that produces intermediate citrate that is directed toward mitochondrial OXPHOS to generate ATP. In contrast, in proliferative cancer cells, the glycolytic flux is largely increased and most of the imported glucose is converted to the glycolytic end-product, pyruvate. The resulting pyruvate is mainly converted to lactate which is secreted from the cells, whereas the remaining pyruvate is converted to acetyl-CoA, which in turn is directed toward de novo FA synthesis. Circulating fatty acids are also mainly oxidized to generate acetyl-CoA. Acetyl-CoA derived from both glucose and fatty acids is then oxidized through TCA cycle to produced intermediate citrate. The resulting citrate is not directed toward OXPHOS but rather is exported to the cytoplasm where it is re-converted to acetyl-CoA, which, in turn, is used for de novo fatty acids biosynthesis.
Abbreviations: Glc, glucose; FA, fatty acids; OAA, oxaloacetate; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation.
Cell cycle, transcription factors, and oncogenes, as dual proliferation-metabolism regulators
Increasing evidence support the symmetrical effects of oncogenes and cell cycle regulators on metabolic pathways. Uncontrolled cell growth is an invariable characteristic of human cancer that implies well-documented genetic alterations of the cell cycle regulators, particular transcription factors and oncogenes. Importantly, beside their well-known role in controlling cell proliferation increasing evidence points to these factors as crucial regulators of cell metabolism. It is likely that oncogenic and proliferative factors not only control proliferation and spread of cancer cells, but also concomitantly trigger an adapted metabolic response of these cells. This is illustrated in figure 2, and will be discussed below.
Figure 2. Oncogenic regulatory pathways in the control of the cancer cells metabolism.
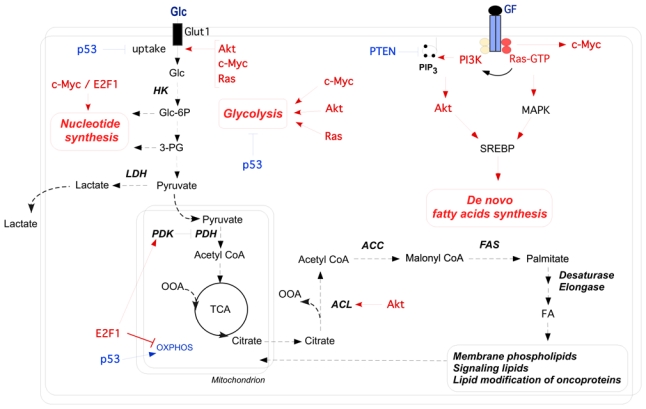
This scheme represents the main implications of oncogenic pathways (in red) in the regulation of glycolysis and de novo lipogenesis in cancer cells. Metabolic effects of the PI3K/Akt, Ras and c-Myc oncogenic pathways include enhanced glucose uptake through increased surface expression of glucose transporter. Hence, they enhance the glycolytic flux, including lactate production, by stimulating glycolytic enzymes activity. Moreover, both Akt and Ras signaling promote de novo FA synthesis through increased SREBP-1 mediated transcription of lipogenic enzymes, and direct stimulation of ACL activity by Akt channels glucose-derived metabolites towards de novo FA biosynthesis. In cooperation with E2F1, c-Myc promotes the transcription of enzymes necessary to support nucleotides biosynthesis from intermediates of the glycolysis. In the case of p53 or PTEN tumor suppressors (in blue), loss of function mutations in cancer cells has the opposite of the effects shown here. Abbreviations: Glc, Glucose; HK, hexokinase; Glc-6-P, glucose-6-phosphate; 3-PG, 3-phosphoglycerate; GF, growth factors; PI3K, phosphatidylinositol 3-Kinase; PIP3, phosphatidylinositol tri-phosphate; PTEN, phosphatase and tensin homolog; MAPK, mitogen-activated protein kinase; SREBP, sterol regulatory element-binding protein; LDH-A, lactate dehydrogenase-A; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; OAA, oxaloacetate; ACL, ATP citrate lyase; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase; FA, fatty acids; TCA, tricarboxylicacid; OXPHOS, oxidative phosphorylation
Cell cycle regulators
Cell cycle progression is a tightly regulated process that support cell proliferation during normal growth and development. It involves the coordinated action of many regulatory proteins that comprises the retinoblastoma tumor suppressor family (pRB, p107, p130), the cyclin dependent kinases (cdks) and their regulatory partners the cyclins (cyc), as well as the family of the cdk inhibitors (CKI). Members of E2F transcription factors (E2F1-8) family play pivotal role in regulating survival and proliferation, by conferring cell-cycle specific expression to promoters containing E2F-binding sites, which concern many genes involved in cell-cycle progression (Helin 1998). Indeed, E2F activity is essential for transactivation of genes that regulate the onset of S phase, such as c-Myc and cyclin E, DNA replication and mitosis. When they do not function as free heterodimers with DP1 or DP2, E2Fs exist in repressive association with members of the pRB family that actively inhibit E2F transactivation through the recruitment of histones deacetylases (Brehm et al 1998, Magnaghi-Jaulin et al 1998) and lysine/Arginine methyl-transferases (Fabbrizio et al 2002). The subsequent phosphorylation of retinoblastoma proteins by the cyclins-cdks complexes releases E2F, allowing transcription of its target genes and cell cycle progression trough the G1/S phase (Dimova and Dyson 2005).
E2F activity is commonly increased in numerous human cancers, including glioblastoma, lung, ovarian, breast, stomach and colon cancers (Chen et al 2009) and much evidence support an oncogenic role for E2F1-3. It is clear that the increased expression of E2Fs contributes to the uncontrolled proliferation of cancer cells, although a specific role of E2Fs in the deregulation of cancer cell metabolism cannot be ruled out. There is compelling evidence for an E2F-RB specific effect in metabolism. This includes a recent observation implicating RB in the control of oxidative metabolism in adipose tissue (Dali-Youcef et al 2007). In addition, we previously demonstrated that cell cycle regulators participate in lipids metabolism. We showed that E2Fs regulate adipogenesis through modulating the expression of the nuclear receptor PPARγ, which is a master regulator of adipogenesis (Fajas et al 2002b). Similarly, we have documented the adipogenic role of Cyclin D3 (Sarruf et al 2005), cdk4 (Abella et al 2005) and cdk9 (Iankova et al 2006) through their positive regulation of PPARγ activity. At the opposite, we found that PPARγ activity and adipocyte differentiation are repressed by RB through the recruitment of HDAC3 (Fajas et al 2002a).
Although no gross abnormalities were originally observed in E2F1−/− mice (Field et al 1996, Yamasaki et al 1996), a first evidence for a role of E2F1 in regulating glucose homeostasis came from the observation that E2F1−/− mice present impaired postnatal pancreatic growth and dysfunctional β-cells associated with defective insulin secretion and glucose intolerance (Fajas et al 2004). Furthermore, the observation that E2F1 was robustly expressed in non-proliferating β-cells suggested a role for E2F1 in pancreatic β-cells, independent to the control of proliferation. This hypothesis was demonstrated showing that the CDK4-pRB-E2F1 pathway mediates the transcriptional response to glucose regulating the expression of genes such as Kir6.2, a key component of the KATP channel involved in the regulation of glucose-induced insulin secretion (Annicotte et al 2009). This role of E2F1 seems to be specific of this E2F-family member, since no major metabolic phenotype has been observed in other E2F−/− mice. E2F2 could however partially compensate E2F1 deficiency. Indeed E2F1-2−/− mice are diabetic, but contribution of E2F2 is likely related to its role in bone marrow-derived cells (Iglesias et al 2004, Li et al 2003). Recent data suggest, in addition, that E2F1 is involved in the control of several other metabolic processes. In a genome-wide studies approach, a cohort of genes that are involved in mitochondrial function were identified among E2F targets, indicating a potential role for E2Fs in linking the metabolic state of the cell to cell cycle status (Cam et al 2004). E2F1 transcriptional activation of the pyruvate dehydrogenase kinase 4 (PDK4) gene, a key nutrient sensor that is constitutively highly expressed in diabetes, was also demonstrated to regulate glucose homeostasis in muscle through inhibition of glucose oxidation (Hsieh et al 2008). More recently, we also evidenced that E2F1 is a negative regulator of oxidative metabolism in skeletal muscle through the repression of PGC-1 α (unpublished results), suggesting that E2F1 may participate in the metabolic switch from OXPHOS to aerobic glycolysis in proliferative cells. Taken together these results support a dual role for the E2F pathway in the control of both cell proliferation and the metabolic response (Figure 3).
Figure 3. Dual role for the E2F pathway in the control of both cell proliferation and the metabolic response.
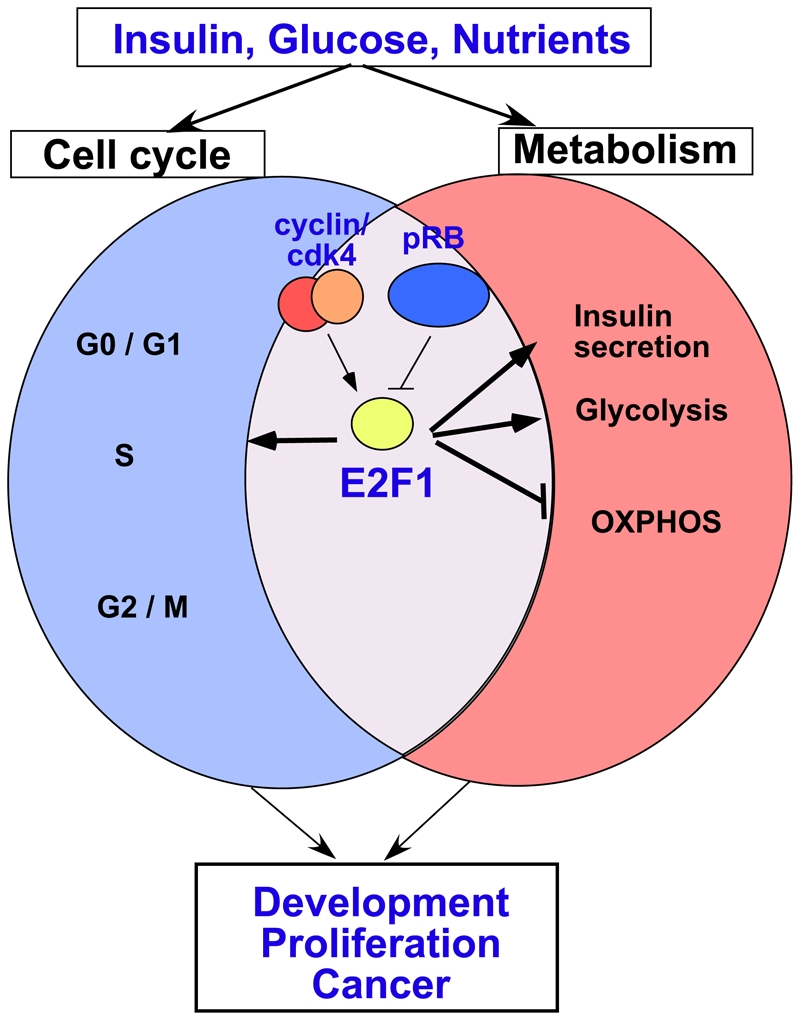
In response to proliferative stimuli such as insulin, glucose and nutrients, the cyclin/cdk4 complex is activated and phosphorylates the retinoblastoma protein pRB, which represses transcription when associated with E2F transcription factors. Activated E2F1 stimulates the expression of target genes implicated in cell cycle progression (blue circle). In addition, E2F1 triggers an adapted metabolic transcriptional response, depending on the cell type. In beta cells, E2F1 will stimulate the expression of Kir6.2, thus facilitating insulin secretion. In other cell types, such as muscle or cancer cells E2F1 facilitates glycolysis and represses, by still unknown mechanisms oxidative phosphorylation. This coordinated response is essential to sustain normal proliferation and development. It is conceivable that changes in this coordinated response might lead to abnormal metabolic changes during tumor development and cancer progression. Abbreviations: OXPHOS, oxidative phosphorylation.
Sp family of transcription factors
Members of the Sp/KLF family of transcription factors Sp1, Sp3 and Sp4, which are over expressed in a variety of cancers are known to regulate cell proliferation by modulating the expression of genes containing GC-rich Sp1 binding sites, including but not limited to several cell cycle regulatory proteins (Black et al 2001). Interestingly, Sp1 sites were also found in the promoters of lipogenic enzymes, such as ACL, ACC and FAS (Daniel and Kim 1996, Moon et al 1999, Xiong et al 2000) and it was previously showed that both Sp1 and Sp3 can activate ACL in response to glucose in hepatoma cells (Moon et al 1999). In a recent report, the role of Sp in coordinating proliferation and lipogenesis in the context of cancer was investigated. In this work, the authors demonstrated that Sp1 regulates both proliferation and FAS expression in human MCF-7 breast, HCT116 colon and LNCaP prostate-cancer cells (Lu and Archer 2009). Stimulation of proliferation by Sp1 is mediated by the regulation of specific cell cycle proteins, such as CDC25A. This dual regulation of metabolic and proliferative genes by a single transcription factor supports the hypothesis that common regulatory pathways exist between proliferative and metabolic factors.
ChREBP
One of the important advantages of aerobic glycolysis common regulatory pathways relies on the possibility to redirect glucose carbon flux into anabolic pathways such as de novo lipogenesis and nucleotide biosynthesis. Sensing increased glucose input is therefore of utmost importance for cancer cells to trigger an adequate transcriptional response. This relies, at least partially on the carbohydrate responsive element binding protein (ChREBP), which is a glucose-responsive transcription factor. The critical role of ChREBP in mediating glucose-dependent induction of glycolytic and lipogenic genes has been extensively explored in non-proliferating hepatocytes (Denechaud et al 2008, Dentin et al 2004, Yamashita et al 2001). More recently, a role for ChREBP in proliferating cells has been described, showing its contribution in cancer cell proliferation and tumorigenesis through attenuation of p53 activity (Tong et al 2009). One important finding in this study is the demonstration that ChREBP suppression in HCT116 colorectal and HepG2 hepatic human cancer cell lines resulted in decreased glycolysis, associated with diminished de novo lipogenesis and nucleotide biosynthesis, although mitochondrial respiration was increased. These data strongly suggest the participation of ChREBP in the metabolic switch from OXPHOS to aerobic glycolysis in cancer cells, a hypothesis that will require further investigation.
The oncogenic pathways: from Akt to c-Myc and Ras
The serine threonine kinase Akt (or PKB) is a paradigm of the dual and integrative regulation of metabolism and proliferation. Possibly, is the most versatile factor that controls the metabolic response to both metabolic and oncogenic stimuli, such as insulin and Ras signaling respectively. The function of Akt in metabolism covers from glucose homeostasis to protein and fatty acid synthesis. It is also a component of the growth factor signaling transduction cascade regulating a wide range of cellular functions including growth, survival and proliferation (reviewed in (Manning and Cantley 2007)). Since Akt is known to be a major mediator of glucose uptake and utilization it is thereby not surprising that Akt activity is correlated with high glycolytic rates in cancer cells (Elstrom et al 2004). Several studies have brought important clues to understand the participation of Akt in the glycolytic cancer phenotype. Indeed, growth factor-induced Akt activation was shown to result in increased expression and membrane distribution of the GLUT1 glucose transporter (Barthel et al 1999, Wieman et al 2007, Wofford et al 2008), association of hexokinase I and II to the mitochondria (Gottlob et al 2001, Majewski et al 2004, Robey and Hay 2006), and stimulation of phospho-fructokinase activity, thereby increasing glycolysis. In addition to its well-documented function in regulating glucose homeostasis, and cell proliferation there is also growing evidence that Akt plays important role in activating de novo FA synthesis. Activation of Akt was shown to induce gene expression of numerous enzymes involved either in cholesterol or fatty acid biosynthesis, including but not limited to the HMG-CoA synthase, HMG-CoA reductase, ACL, FAS, and stearoyl-CoA desaturase (SCD) (Porstmann et al 2005). Transcription of these enzymes in regard to Akt pathway has been shown to require the activity of the sterol regulatory element-binding protein (SREBP) family of transcription factors, which synthesis and nuclear accumulation are also induced by activated Akt (Porstmann et al 2005). Of particular importance is the observation that Akt also directly phosphorylates and activates the ATP-citrate lyase (ACL) (Bauer et al 2005, Berwick et al 2002, Buzzai et al 2005), which is a key enzyme integrating glucose and lipid metabolism. Akt participates therefore in the first committed step in channeling glucose-derived metabolites towards a lipid biosynthetic fate.
Myc or Ras oncogenes can be also considered as coordinators of the proliferative and metabolic response of transformed cells (DeBerardinis et al 2008). Altered expression or mutations in the Myc or Ras oncogenes respectively, have been associated to the genesis of most common human cancers. Apart from its role in the regulation of cell proliferation and cell cycle, there is compelling evidence that c-Myc and Ras also participate to the control of cancer cell metabolism. Since the discovery that c-Myc regulates the lactate dehydrogenase A (LDH-A) (Lewis et al 2000, Shim et al 1997), which converts pyruvate to lactate and contributes to the Warburg effect, c-Myc has been found to directly regulate many enzymes from the glycolytic pathway, therefore promoting glucose uptake and utilization (Kim et al 2004, Kim et al 2007, Osthus et al 2000). Furthermore, many studies have evidenced that c-Myc also plays a pivotal role in regulating mitochondrial biogenesis and function. This was supported by the observations that many genes involved in mitochondrial biogenesis and function are over-expressed among c-Myc direct transcriptional targets (Kim et al 2008, Li et al 2005, Morrish et al 2008). Since one important issue discussed in this review is the participation of cell cycle regulators in the metabolic control of cancer cells it is worth mentioning the link between Myc and E2F1 pathway. Both c-Myc and E2F1 are important regulators of cell cycle, and it has been suggested that c-Myc metabolic gene targets are regulated in a cell cycle dependent way. Indeed, after induction of energy metabolism and ribosomal biogenesis-related genes during the G1 phase, c-Myc induces E2F1 expression as cells entry into the S-phase, that together with c-Myc activate genes involved in nucleotide biosynthesis and DNA replication (Liu et al 2008). There is no need to discuss the importance of Ras oncogenes in the control of cell proliferation and tumor development (Drosten et al 2010). Interestingly, Ras-mediated cell transformation is coupled to profound changes in metabolism, including enhanced glucose uptake and glycolysis, mitochondrial dysfunction, and increased lactic acid production. This is the result of increased expression of several genes of the aerobic glycolytic pathway and lactate dehydrogenase (Chiaradonna et al 2006, Mazurek et al 2001, Ramanathan et al 2005). Similar to other oncogenes, Ras also participates in the control of de novo lipid synthesis, mainly through regulation of the activity of the transcription factor SREBP mediated by the MAPK pathway (Menendez et al 2005, Yang et al 2002). Most interestingly, recent data from our lab show that Ras transformation is abrogated when lipid synthesis pathways are inhibited (unpublished results). Taken together, the available literature suggests that oncogenes are at the origin of the metabolic transformations observed during cell transformation. These metabolic changes contribute to sustained proliferation of cancer cells, creating a positive feed-back loop. This is supported by the observation that changes in metabolism are also required to sustain the activation of oncogenes. Notably, oncogene-induced fatty acid synthesis, such as described above facilitates the formation of a subset of fatty acids, such as palmitate and palmitoleate, which impact on the traffic and cellular localization of several oncogenes, such as Ras, facilitating their anchorage to membranes required for the activation of their oncogenic pathways (Nadolski and Linder 2007). From these findings we can argue that inhibition of key enzymes involved in lipid synthesis will abrogate activation of oncogenic signaling, and therefore will result in tumor growth arrest. This has been shown for ACL (Bauer et al 2005), FAS (Kuhajda et al 1994, Kuhajda 2000), or SCD1 (Scaglia and Igal 2008) inhibition.
The counteracting tumor suppressor pathway
It is not surprising that tumor suppressors not only have opposite effects than oncogenes on the control of cell proliferation and cell cycle, but they also directly participate in the reversal of the glycolytic phenotype of the cells. Several examples are illustrative of this striking observation. The tumor suppressor p53, in addition to its well-known function in the control of cell cycle and apoptosis have an important role in the control of glycolytic and oxidative metabolism. It was shown that p53 participates in mitochondrial biogenesis and function, oxygen consumption, and decrease in glycolysis (Matoba et al 2006). These effects were mediated, at least in part through direct transcriptional regulation of the synthesis of cytochrome c oxidase 2, and apoptosis-inducing factor, which are essential genes in the control of mitochondrial function (Matoba et al 2006). Furthermore, opposite to oncogenes, such as Ras or Akt, p53 represses the expression of glucose transporters GLUT-1 and GLUT-4, thereby inhibiting glycolysis (Schwartzenberg-Bar-Yoseph et al 2004). In addition, p53 also senses the metabolic stress of the cell. Interestingly, under hypoxia or nutrient depletion, which are conditions characteristic of cancer cell environment, p53 appears to inhibit autophagy, which is required, under these conditions, to facilitate cancer cell growth (Tasdemir et al 2008). PTEN is another major tumor suppressor. Most of its effects are mediated through inactivation of the PI3K/Akt pathway. It is indeed a phosphatase of phosphatidyl inositol tri-phosphate (PIP3), which is a major effector of Akt. Loss of function or mutations in PTEN is not only related to cancer development, but also exhibits dramatic consequences for metabolism homeostasis. For instance, Pten-deleted muscle, liver or fat tissue exhibits increased glucose sensitivity indicating that PTEN modulates glucose uptake and utilization in the cells (Li et al 1997, Stiles et al 2004, Wijesekara et al 2005). This is of particular relevance in cancer cells, which are fully dependent on increased glycolytic flux. PTEN is also a negative regulator of the insulin pathway, and therefore has negative effects on lipogenesis, another hallmark of cancer cells. Paradoxically pRB is, in contrast to the other tumor suppressors, a major modulator of oxidative metabolism. RB specific deletion in adipose tissue results in increased mitochondrial activity and reversal from anabolic white adipose tissue to catabolic oxidative brown adipose tissue (Dali-Youcef et al 2007). In this particular situation RB would facilitate a cancer-type metabolism. More studies are required, however, to better understand the role of pRB in the metabolic control in cancer cells.
Concluding remarks
It is well established that the process of cancer development and growth involves major alterations of cells’ metabolism. In addition to triggering signaling cascades involved in proliferation and survival, oncogenes such as Ras, c-Myc or Akt, and most importantly cell cycle regulators such as the cdk4-E2F1 axis, also trigger concomitant metabolic changes. The majority of oncoproteins have been shown to stimulate aerobic glycolysis in transformed cells, including Akt, c-Myc, Ras and Src. Moreover, there is compelling evidence that in addition to trigger proliferation, oncogenes require the modification of metabolism, notably the increase in de novo FA synthesis in order to transform the cell. This modification in lipid synthesis results, in addition to other effects, in the activation of oncoproteins through protein lipidation. This creates a positive feed-back loop that contributes to sustained cancer cell growth and proliferation.
Most efforts have been directed towards the inhibition of the proliferative and invasive effects of oncogenes in cancer cells, whereas little attention has been paid to the metabolic changes in these cells. Further identification of the particular metabolic pathways regulated by oncogenic factors will certainly contribute to broaden the therapeutic options for cancer treatment.
Acknowledgments
Members of the Fajas’ lab are acknowledged for support and discussions. This work was supported by grants from Agence Nationale pour la Recherche (ANR physio), Association pour la Recherche contre le Cancer, Institut National du Cancer (INCA), and Fondation pour la Recherche Médicale. VF is supported by a grant of the INCA.
References
- Abella A, Dubus P, Malumbres M, Rane SG, Kiyokawa H, Sicard A, et al. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab. 2005;2:239–249. doi: 10.1016/j.cmet.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Annicotte JS, Blanchet E, Chavey C, Iankova I, Costes S, Assou S, et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol. 2009;11:1017–1023. doi: 10.1038/ncb1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr, et al. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277:33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, et al. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- Cam H, Balciunaite E, Blais A, Spektor A, Scarpulla RC, Young R, et al. A common set of gene regulatory networks links metabolism and growth inhibition. Mol Cell. 2004;16:399–411. doi: 10.1016/j.molcel.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaradonna F, Sacco E, Manzoni R, Giorgio M, Vanoni M, Alberghina L. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene. 2006;25:5391–5404. doi: 10.1038/sj.onc.1209528. [DOI] [PubMed] [Google Scholar]
- Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, et al. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A. 2007;104:10703–10708. doi: 10.1073/pnas.0611568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel S, Kim KH. Sp1 mediates glucose activation of the acetyl-CoA carboxylase promoter. J Biol Chem. 1996;271:1385–1392. doi: 10.1074/jbc.271.3.1385. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Denechaud PD, Dentin R, Girard J, Postic C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2008;582:68–73. doi: 10.1016/j.febslet.2007.07.084. [DOI] [PubMed] [Google Scholar]
- Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010 doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Fabbrizio E, El Messaoudi S, Polanowska J, Paul C, Cook JR, Lee JH, et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002;3:641–645. doi: 10.1093/embo-reports/kvf136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002a;3:903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell. 2002b;3:39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Fajas L, Annicotte JS, Miard S, Sarruf D, Watanabe M, Auwerx J. Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1 (−/−)mice. J Clin Invest. 2004;113:1288–1295. doi: 10.1172/JCI18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr, Livingston DM, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K. Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev. 1998;8:28–35. doi: 10.1016/s0959-437x(98)80058-0. [DOI] [PubMed] [Google Scholar]
- Hsieh MC, Das D, Sambandam N, Zhang MQ, Nahle Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem. 2008;283:27410–27417. doi: 10.1074/jbc.M802418200. [DOI] [PubMed] [Google Scholar]
- Iankova I, Petersen RK, Annicotte JS, Chavey C, Hansen JB, Kratchmarova I, et al. Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol Endocrinol. 2006;20:1494–1505. doi: 10.1210/me.2005-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias A, Murga M, Laresgoiti U, Skoudy A, Bernales I, Fullaondo A, et al. Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. J Clin Invest. 2004;113:1398–1407. doi: 10.1172/JCI18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek P, Plecita-Hlavata L, Smolkova K, Rossignol R. Distinctions and similarities of cell bioenergetics and the role of mitochondria in hypoxia, cancer, and embryonic development. Int J Biochem Cell Biol. 2010;42:604–622. doi: 10.1016/j.biocel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee JH, Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One. 2008;3:e1798. doi: 10.1371/journal.pone.0001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, et al. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol. 2004;24:5923–5936. doi: 10.1128/MCB.24.13.5923-5936.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A. 1994;91:6379–6383. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–208. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
- Lewis BC, Prescott JE, Campbell SE, Shim H, Orlowski RZ, Dang CV. Tumor induction by the c-Myc target genes rcl and lactate dehydrogenase A. Cancer Res. 2000;60:6178–6183. [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FX, Zhu JW, Tessem JS, Beilke J, Varella-Garcia M, Jensen J, et al. The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc Natl Acad Sci U S A. 2003;100:12935–12940. doi: 10.1073/pnas.2231861100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One. 2008;3:e2722. doi: 10.1371/journal.pone.0002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Archer MC. Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. Int J Cancer. 2009;126:416–425. doi: 10.1002/ijc.24761. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Mazurek S, Zwerschke W, Jansen-Durr P, Eigenbrodt E. Metabolic cooperation between different oncogenes during cell transformation: interaction between activated ras and HPV-16 E7. Oncogene. 2001;20:6891–6898. doi: 10.1038/sj.onc.1204792. [DOI] [PubMed] [Google Scholar]
- Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13:27–29. [PubMed] [Google Scholar]
- Menendez JA, Colomer R, Lupu R. Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids? Med Hypotheses. 2005;64:342–349. doi: 10.1016/j.mehy.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Moon YA, Kim KS, Cho UH, Yoon DJ, Park SW. Characterization of regulatory elements on the promoter region of human ATP-citrate lyase. Exp Mol Med. 1999;31:108–114. doi: 10.1038/emm.1999.18. [DOI] [PubMed] [Google Scholar]
- Morrish F, Neretti N, Sedivy JM, Hockenbery DM. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008;7:1054–1066. doi: 10.4161/cc.7.8.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadolski MJ, Linder ME. Protein lipidation. Febs J. 2007;274:5202–5210. doi: 10.1111/j.1742-4658.2007.06056.x. [DOI] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Lewis C, Griffiths B, Schulze A. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem Soc Trans. 2009;37:278–283. doi: 10.1042/BST0370278. [DOI] [PubMed] [Google Scholar]
- Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci U S A. 2005;102:5992–5997. doi: 10.1073/pnas.0502267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- Sabine JR, Abraham S, Chaikoff IL. Control of lipid metabolism in hepatomas: insensitivity of rate of fatty acid and cholesterol synthesis by mouse hepatoma BW7756 to fasting and to feedback control. Cancer Res. 1967;27:793–799. [PubMed] [Google Scholar]
- Sarruf DA, Iankova I, Abella A, Assou S, Miard S, Fajas L. Cyclin D3 promotes adipogenesis through activation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2005;25:9985–9995. doi: 10.1128/MCB.25.22.9985-9995.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglia N, Igal RA. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int J Oncol. 2008;33:839–850. [PubMed] [Google Scholar]
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–2633. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci U S A. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Natl Acad Sci U S A. 2009;106:21660–21665. doi: 10.1073/pnas.0911316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. Metabolism of Tumors. Arnold Constable; London: 1930. [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956a;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956b;124:269–270. [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesekara N, Konrad D, Eweida M, Jefferies C, Liadis N, Giacca A, et al. Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol Cell Biol. 2005;25:1135–1145. doi: 10.1128/MCB.25.3.1135-1145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood. 2008;111:2101–2111. doi: 10.1182/blood-2007-06-096297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong S, Chirala SS, Wakil SJ. Sterol regulation of human fatty acid synthase promoter I requires nuclear factor-Y- and Sp-1-binding sites. Proc Natl Acad Sci U S A. 2000;97:3948–3953. doi: 10.1073/pnas.040574197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Gollapalli DR, Maiti P, Jahng WJ, Rando RR. A palmitoylation switch mechanism in the regulation of the visual cycle. Cell. 2004;117:761–771. doi: 10.1016/j.cell.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YA, Han WF, Morin PJ, Chrest FJ, Pizer ES. Activation of fatty acid synthesis during neoplastic transformation: role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Exp Cell Res. 2002;279:80–90. doi: 10.1006/excr.2002.5600. [DOI] [PubMed] [Google Scholar]