

Abstract
Stimulation of the prefrontal cortex by acetylcholine is critical for attention; however, the cellular mechanisms underlying its influence on attention pathways within the brain are not well understood. Pyramidal neurons in layer VI of the prefrontal cortex are believed to play an important role in this process because they are excited by acetylcholine and provide a major source of feedback projections to the thalamus. Here, we show using whole-cell electrophysiology that the relatively rare α5 subunit of the nicotinic acetylcholine receptor powerfully enhances nicotinic currents in layer VI pyramidal neurons in prefrontal cortical brain slices from adult mice. In addition, behavioral experiments using the five-choice serial reaction time test show that the presence of the nicotinic receptor α5 subunit also increases the accuracy of adult mice on this visual attention task under highly demanding conditions. Together, these findings demonstrate a novel and important role for the nicotinic receptor α5 subunit in adult brain circuitry required for attentional performance.
Introduction
Modulation of medial prefrontal cortex (mPFC) by acetylcholine (ACh) is critical for attention (Passetti et al., 2000; Dalley et al., 2004; Parikh et al., 2007), and stimulation of the nicotinic subclass of acetylcholine receptor (nAChR) within the mPFC appears to be particularly important for attentional performance under conditions of high attentional demand (Hahn et al., 2003; Newhouse et al., 2004; Lambe et al., 2005; Day et al., 2007). Pyramidal neurons within layer VI of the mPFC are likely central to this process because they are a major source of feedback projections to the thalamus (Alitto and Usrey, 2003; Gabbott et al., 2005; Zikopoulos and Barbas, 2006) and are directly excited by nAChRs (Kassam et al., 2008). These layer VI nicotinic receptors are pentameric ligand-gated cation channels of the α4β2* subtype (Kassam et al., 2008), which are composed of two α4 subunits, two β2 subunits, and a fifth accessory subunit (for review, see Albuquerque et al., 2009; Gotti et al., 2009). The accessory subunit does not participate in ligand binding but rather influences agonist efficacy at the receptor (Moroni et al., 2006; Kuryatov et al., 2008). Although an additional α4 or β2 subunit can occupy this accessory position, the α5 subunit functions exclusively as an accessory subunit (Ramirez-Latorre et al., 1996; Gotti et al., 2009). We are particularly interested in the α5 accessory subunit because it is densely expressed within layer VI of mPFC (Wada et al., 1990; Marks et al., 1992; Salas et al., 2003; Winzer-Serhan and Leslie, 2005) and may therefore influence nicotinic regulation of attention within this brain region. It is not known how the α5 accessory subunit affects the function of whole neurons of attention circuits and the performance of attentional tasks.
Here, we investigate the role of the α5 nicotinic subunit in nicotinic excitation of layer VI pyramidal neurons within the mPFC using a mouse line in which the α5 subunit has been genetically deleted (Salas et al., 2003). Using whole-cell electrophysiological recording in acute brain slices from adult mice, we found that the presence of the α5 subunit dramatically increased α4β2* nicotinic currents in layer VI pyramidal neurons. Because the α5 subunit changed the properties of nicotinic currents in a population of neurons thought to be important for attention, we next examined the role of the α5 subunit on the cognitive performance of mice on an attention task, the five-choice serial reaction time test (5-CSRTT). Under conditions of high attentional demand, we found that accuracy in this task was significantly greater in wild-type (WT) mice expressing the α5 subunit compared with mice that did not express the α5 subunit. Our results indicate that the α5 nicotinic subunit plays a distinct role in cholinergic modulation of the neurons in the major corticothalamic feedback layer and is required for normal attentional performance under challenging conditions.
Materials and Methods
Experimental animals.
Mice deficient for the nAChR α5 subunit were generated and described previously (Salas et al., 2003). This congenic line had been backcrossed into a C57BL/6J background for >10 generations and was the source for wild-type and nAChR α5 subunit knock-out (α5−/−) mice used in this current study. Mice were weaned at 3 weeks of age, separated based on sex, and housed in groups of two to four per cage at an ambient temperature of 22°C with a 12 h light/dark cycle with lights on at 7:00 A.M. Mice were given ad libitum access to food and water except when noted below for behavioral experiments. Male mice were used for all experiments. All experimental animals were cared for according to the principles and guidelines of the Canadian Council on Animal Care. The experimental protocol was approved by the University of Toronto Animal Care Committee, and the behavioral experiments were approved by the Centre for Addiction and Mental Health Animal Care Committee.
Brain slice preparation.
Coronal slices 400 μm thick of the mPFC were prepared from adult mice aged from postnatal days 60–180. Mice were killed by decapitation under chloral hydrate anesthesia (400 mg/kg) administered intraperitoneally in a dilute solution. Brains were excised as rapidly as possible and cooled in 4°C oxygenated sucrose artificial CSF (ACSF) (in mm: 254 sucrose, 10 d-glucose, 24 NaHCO3, 2 CaCl2, 2 MgSO4, 3 KCl, and 1.25 NaH2PO4, pH 7.4). Prefrontal slices were cut using a Dosaka Linear Slicer (SciMedia) from anterior to posterior using the appearance of white matter and the corpus callosum as anterior and posterior guides to target recording to the prelimbic region (Paxinos and Franklin, 2001; Gabbott et al., 2005). Slices were transferred to oxygenated ACSF (composition listed above except 128 mm NaCl was substituted for sucrose) maintained at 30°C in a prechamber (Automate Scientific) and allowed to recover for at least 2 h before the beginning of an experiment. For whole-cell recordings, slices were placed in a modified chamber (Warner Instruments) mounted on the stage of an Olympus BX50WI microscope. Regular ACSF was bubbled with 95% oxygen and 5% carbon dioxide and flowed over the slice at room temperature with a rate of 3–4 ml/min.
Electrophysiology.
Whole-cell patch electrodes (2–3 MΩ) contained 120 mm K-gluconate, 5 mm KCl, 2 mm MgCl, 4 mm K2-ATP, 400 μm Na2-GTP, 10 mm Na2-phosphocreatine, and 10 mm HEPES buffer, adjusted to pH 7.3 with KOH. Medial prefrontal layer VI pyramidal neurons were patched under visual control using infrared differential interference contrast microscopy. All recordings were made using an Axopatch 200B (Molecular Devices), acquired, and low-pass filtered at 2 kHz using a Digidata 1440A data acquisition system (Molecular Devices). Patched neurons were first recorded in current-clamp mode at their resting membrane potentials. Their membrane potential responses to positive and negative current steps were recorded to measure their electrophysiological properties. Pharmacological experiments were not performed on fast-spiking interneurons because they have been demonstrated to respond only to indirect effects of nicotinic stimulation (Kassam et al., 2008).
For pharmacological experiments, neurons were held at −75 mV under voltage-clamp mode, which is near the calculated equilibrium potential for chloride under these conditions. All agonists were applied in the bath in the presence of atropine (200 nm, continuous application) to block muscarinic receptors. For certain ACh concentration–response experiments only, slices were preexposed to 20 μm diisopropyl fluorophosphate (DFP) in the bath for 10 min to inactivate endogenous acetylcholinesterase and facilitate comparison of EC50 values with those obtained previously in reduced preparations. For all experiments, agonists were applied after a baseline period, and peak acetylcholine and nicotine currents were measured using Clampfit software (Molecular Devices) by subtracting the mean inward current at the peak of the response from the mean inward current at baseline. Acetylcholine was allowed to wash out for at least 5 min before reapplication (10 min for concentration–response experiments after exposure to DFP), and nicotine was only applied to one cell per slice. The effects of repeated nicotine applications (with no or specified washout periods) were recorded in certain experiments with recordings made in only one cell per exposed slice.
Recording of GABAergic spontaneous postsynaptic currents (sPSCs) was performed with patch electrodes that contained 50 mm K-gluconate, 75 mm KCl, 2 mm MgCl, 4 mm K2-ATP, 400 μm Na2-GTP, 10 mm Na2-phosphocreatine, and 10 mm HEPES buffer, adjusted to pH 7.3 with KOH. GABAergic sPSCs were recorded under the same conditions described above but with the addition of continuous application of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (20 μm) to block AMPA glutamate receptors. Under these conditions, the observed sPSCs were completely blocked by the addition of 10 μm bicuculline. Analysis of GABAergic sPSCs at baseline and during the application of nicotine (100–300 nm) was performed using Mini Analysis Program (Synaptosoft).
Tetrodotoxin (TTX) was obtained from Alomone Labs. Atropine, acetylcholine chloride, nicotine hydrogen bitartrate, dihydro-β-erythroidine hydrobromide (DHβE), CNQX, and bicuculline were obtained from Sigma-Aldrich or Tocris Cookson and stored in stock solutions at −20°C before being diluted and applied to the slice in oxygenated ACSF.
Five-choice serial reaction time test.
The 5-CSRTT was conducted in operant conditioning test chambers (Med Associates) measuring 21.6 × 17.8 × 12.7 cm. One curved, stainless steel wall of the chamber contained five round apertures measuring 1.3 cm in diameter that were 1 cm deep and located 0.8 cm above the floor. An infrared photodetector was located at the entrance to each aperture, and a yellow stimulus light diode was centered at the back of each aperture. The opposite wall of the chamber was also constructed of stainless steel. A reinforcer magazine, equipped with an infrared photodetector and a light, was centered in this wall 2.5 cm above the floor. A motor-driven dipper arm could be raised to deliver 0.01 ml of 10% (w/v) sucrose solution to the magazine. Each box was illuminated by a house light and was enclosed in a sound-attenuating chamber equipped with a ventilation fan. All boxes were controlled by a personal computer running Med-PC-IV software (Med Associates).
Thirteen wild-type and 13 α5−/− mice were housed individually with ad libitum access to water and were food restricted to maintain a body weight of ∼85% of their free-feeding body weight. Training was initiated by placing a mouse in the chamber with all five response apertures illuminated. A response in any of the apertures extinguished the lights, illuminated the magazine light, and resulted in dipper elevation presenting 0.01 ml of 10% (w/v) sucrose. The dipper remained elevated until the reward had been collected. Sessions lasted for 30 min or 100 trials. Subsequently, mice were placed in the chamber with only one aperture illuminated. A response in that aperture extinguished that light, illuminated the magazine light, and resulted in dipper elevation until the reward had been collected. Sessions lasted for 30 min or 100 trials. Once all mice had successfully acquired this task, training on the 5-CSRTT began.
The training session began with illumination of the house light and the magazine light and elevation of the dipper. A nose poke in the magazine initiated the first trial. After a fixed intertrial interval (ITI) of 5 s, one of the five light stimuli was illuminated for a brief period; a response in that hole while the light was on or during a short limited-hold period of 5 s resulted in the elevation of the dipper and illumination of the magazine light. A nose poke into the magazine to collect the reinforcer initiated the ITI to the next trial. Incorrect responses or failures to respond were not reinforced and were followed by a 5 s timeout period of darkness. A timeout period also followed perseverative responding, defined as additional responses made in any of the five holes before reinforcer collection. At the end of timeout periods, the magazine light was turned on, and a nose poke in the magazine began the next trial. Responses during the ITI were recorded as premature responses and were followed by a timeout. Magazine responses at the end of the timeout periods restarted the same trial. Sessions lasted for 40 min or 100 trials; each stimulus was presented up to 20 times in a random order. Training began with the stimulus duration set at 30 s, which was gradually reduced depending on performance to a final training stimulus duration of 1 s. Accuracy of responding was measured by calculating the percentage of correct responses [correct responses/(correct responses + incorrect responses) × 100] and the percentage of omissions was calculated as (omissions/total number of trials) × 100. The number of trials performed, number of premature responses, latency to respond correctly, and latency to collect the reinforcer also were recorded.
Testing at different stimulus durations commenced once all mice had successfully acquired the task with the stimulus duration set at 1 s. Each mouse was tested at all four of the following stimulus durations: 0.125, 0.25, 0.5, and 1 s. The order for stimulus duration testing was determined from Latin squares, with mice being tested on Tuesdays and Saturdays. On intervening weekdays, mice were run on the basal training task with the stimulus duration set at 1 s. For all testing and training conditions, the ITI and limited hold were both set to 5 s.
Testing for the effects of nicotine took place 2 months after the testing of baseline attentional performance described above. The stimulus duration for this pharmacological experiment was set at 0.063 s with the ITI and limited hold, both set at 5 s. Each mouse was tested with 0.03 mg/kg nicotine and saline (0.9% NaCl) vehicle control in a random-balanced order on a Tuesday and Friday of the same week. On nontest weekdays before and during the experiment, mice were run on the basal training task with the stimulus duration set at 1 s. We used (−)-nicotine bitartrate and calculated the dose according to the free base, and all solutions were adjusted to pH 7.0. Treatment was administered subcutaneously at a volume of 1 ml/100 g body weight, and the testing session began 5 min after each injection, at which time plasma nicotine concentration is near maximum for C57BL/6 mice after this route of administration (Siu and Tyndale, 2007). We calculated that this subcutaneous dose of 0.03 mg/kg likely results in peak mouse brain nicotine concentrations near 117 nm, which is relevant to the levels of nicotine observed in the human brain during cigarette smoking (Rose et al., 2010). Our calculations were based on a previous study that examined the plasma and brain concentrations of nicotine in mice after intraperitoneal injections (Petersen et al., 1984) and one that examined the plasma concentration of nicotine in mice after subcutaneous injections (Siu and Tyndale, 2007). For our calculations, we assumed a mouse cerebral cortex density of 1.05 g/ml (Thumwood et al., 1988).
Because systemic nicotine at 0.5 mg/kg (more than an order of magnitude above the dose we used in the 5-CSRTT) has been shown to inhibit mouse locomotor ability (Salas et al., 2003), we tested the effect of saline and two concentrations of nicotine on locomotor activity in our mice. Motor activity experiments were conducted 3 weeks after the completion of the nicotine 5-CSRTT experiment in a custom-built monitor system, consisting of clear polycarbonate cages measuring 25 cm wide, 20 cm high, and 45 cm in length. An array of 12 infrared photocells was attached outside the longer sides of the cages. The photocells were spaced 3.75 cm apart and were located 2 cm above the floor of the cage. Mice were administered each of saline vehicle, 0.03 mg/kg nicotine, and 0.3 mg/kg nicotine in a balanced order based on Latin squares on 3 testing days that were each separated by four nontesting days. Solutions were made as described above and administered subcutaneously. Mice were immediately placed singly into the testing cages, and locomotor testing began 5 min after each injection for a total of 40 min.
Statistical analysis.
All data are presented as mean ± 1 SEM. All datasets analyzed were found to be normally distributed by the Shapiro–Wilk normality test. Genotype analysis of electrophysiology experiments comparing the response to acetylcholine and nicotine across the genotypes were performed using unpaired two-tailed t tests. The electrophysiological experiments to assess the inward currents and changes in root mean square (RMS) current noise elicited by different concentrations of nicotine were analyzed by repeated-measures two-way ANOVA, followed by Bonferroni's-corrected post hoc tests, as were the effects of nicotine on the GABAergic sPSC frequency and amplitude. When significant effects were found, the influence of nicotine within each genotype was tested by one-way ANOVA. The desensitization experiment was analyzed by two-way ANOVA, followed by Bonferroni's-corrected post hoc tests. The behavioral experiments to assess baseline attention and the effects of nicotine on locomotor activity were also analyzed by repeated-measures two-way ANOVA, followed by Bonferroni's-corrected post hoc tests. Analysis to determine the effects of nicotine in the 5-CSRTT was performed using a paired two-tailed t test within each genotype group. All statistical analyses were performed using GraphPad Software Prism 5.
Results
The α5 nicotinic subunit boosts the amplitude of nicotinic currents in layer VI
To examine the role of the α5 subunit in the ACh current response in layer VI pyramidal neurons of mPFC, we prepared acute brain slices from adult male C57BL/6 mice that were either wild type or constitutively deleted for the α5 subunit (α5−/−) (Salas et al., 2003). We blocked muscarinic acetylcholine receptors using 200 nm atropine in the bath and performed whole-cell electrophysiological recordings of neurons that were held at −75 mV under voltage clamp. As illustrated in Figure 1, the inward currents elicited by ACh (1 mm, 10 s) were significantly greater in neurons from wild-type mice (40.2 ± 4.7 pA; n = 18) compared with α5−/− mice (14.0 ± 1.3 pA; n = 33) (two-tailed unpaired t test, p < 0.0001). We observed no genotype effect on the basic electrophysiological properties of these same neurons, including resting membrane potential (wild type, −72.3 ± 1.4 mV; α5−/−, −72.7 ± 0.8 mV; two-tailed unpaired t test, p = 0.8), spike amplitude (wild type, 104.9 ± 2.5 mV; α5−/−, 105.6 ± 1.6 mV; p = 0.8), or input resistance (wild type, 171.9 ± 15.2 MΩ; α5−/−, 186.2 ± 11.5 MΩ; p = 0.5). The magnitude of current responses to ACh in neurons from both wild-type and α5−/− mice was resistant to blocking action potentials with TTX (2 μm, 10 min) before ACh application (two-tailed paired t test, p = 0.1; n = 5) (exemplary traces shown in Fig. 1B) and, in fact, showed a small increasing trend under these conditions. The nicotinic currents in layer VI neurons of both genotypes were significantly attenuated when we applied the α4β2* nAChR-selective competitive inhibitor DHβE (3 μm, 10 min) before ACh (two-tailed paired t test, p = 0.02; n = 5) (exemplary traces shown in Fig. 1B). These results using TTX and DHβE are consistent with the previous results obtained in rat (Kassam et al., 2008) and mice (Alves et al., 2010) and suggest that somatodendritic α4β2* nAChRs mediate the nicotinic inward currents on the recorded layer VI pyramidal neurons.
Figure 1.

Direct excitation of medial prefrontal layer VI pyramidal neurons by acetylcholine is dependent on the nicotinic receptor α5 subunit. A, The peak inward current response to 10 s bath application of 1 mm acetylcholine was significantly greater in neurons from wild-type mice expressing the α5 subunit (WT) compared with neurons from mice in which the α5 subunit had been genetically deleted (α5−/−) (two-tailed unpaired t test, *p < 0.0001). Values are mean ± 1 SEM. B, Voltage-clamp traces showing typical responses in one neuron each from a WT and an α5−/− mouse. For neurons of both genotypes, inward current responses to acetylcholine were resistant to 10 min pretreatment with TTX (2 μm) to block action potentials and were significantly reduced by 10 min pretreatment with the α4β2* nicotinic receptor competitive inhibitor DHβE (3 μm), indicating that responses were mediated by α4β2* receptors located directly on the recorded neurons.
We next sought to determine whether the larger ACh currents observed in the presence of α5-subunit-containing nAChRs resulted from a left shift in the ACh concentration–response curve. We first measured the current response to 30 s bath application of ACh at concentrations ranging from 300 nm to 3 mm and found that wild-type neurons showed a greater efficacy of response to ACh compared with α5−/− neurons (Fig. 2A,C). Expressing the same data as a function of the maximal ACh response within each individual neuron, shown in Figure 2B, allows differences in ACh sensitivity between the genotypes to be visualized more clearly. Because the concentration–response curves did not show a sigmoidal relationship, we hypothesized that the exogenously applied ACh was being actively metabolized by the endogenous acetylcholinesterase present in the brain slice preparation. To test this hypothesis and facilitate comparison with EC50 values obtained in cell system (Ramirez-Latorre et al., 1996; Tapia et al., 2007; Kuryatov et al., 2008) and synaptosome studies (McClure-Begley et al., 2009; Grady et al., 2010), a separate set of concentration–response experiments were conducted after inactivation of acetylcholinesterase in the brain slices with DFP at 20 μm for 10 min. The irreversible inactivation of acetylcholinesterase resulted in a striking left shift in the monophasic ACh concentration–response curve for neurons from wild-type mice (Fig. 2, compare D,E with A,B in untreated slices), in which the EC50 was measured to be 8 μm [n = 5; 95% confidence interval (CI), 6–10 μm]. In contrast, the concentration–response curve generated from α5−/− mice was best fit to a two-component nonlinear regression model (extra sum-of-squares F test, F(3,39) = 3.45; p = 0.03). The high-affinity component of this model contributed to 26% of the total response and had an EC50 of 0.9 μm (n = 5; 95% CI, 0.2–3 μm), whereas the low-affinity component contributed to 74% of the total response and was characterized by an EC50 of 91 μm (n = 5; 95% CI, 60–137 μm). The irreversible inhibition of acetylcholinesterase results in prolonged nicotinic currents, as shown in the exemplary traces in Figure 2F, demonstrating that acetylcholinesterase normally contributes to the rapid removal of ACh from the brain slices during each washout period.
Figure 2.
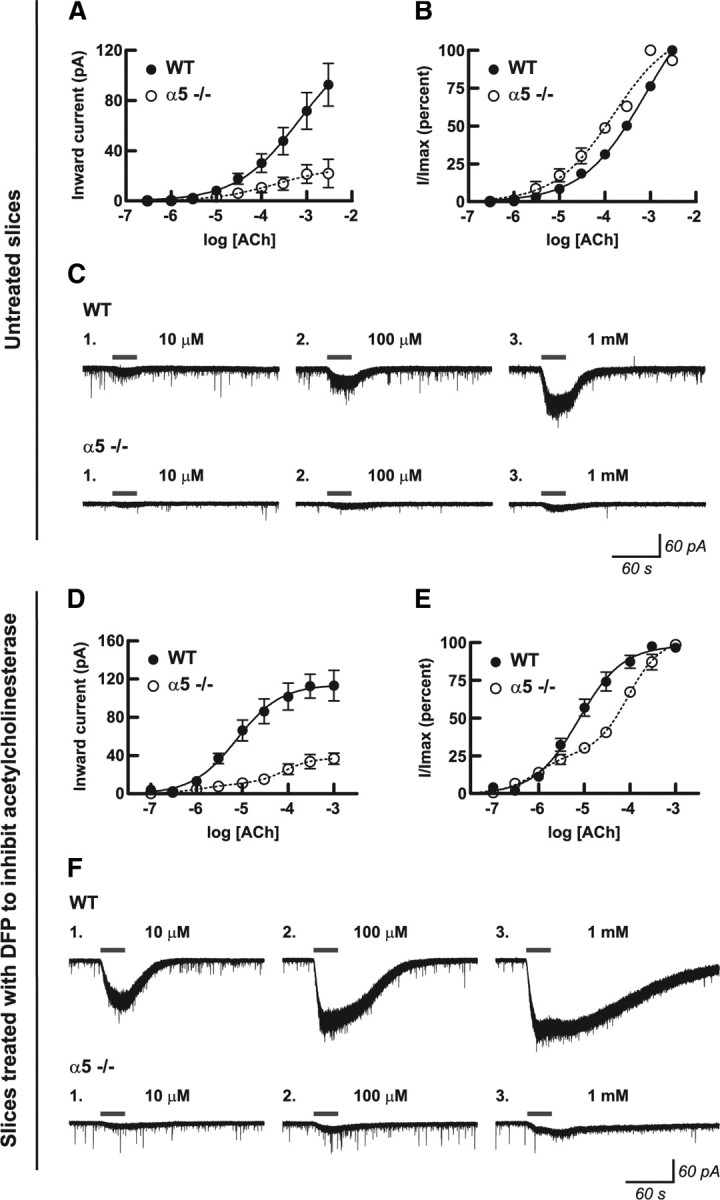
Presence of the α5 subunit alters the efficacy and sensitivity of the nicotinic receptor response to acetylcholine in medial prefrontal layer VI pyramidal neurons. Concentration–response curves for 30 s bath application of acetylcholine in untreated slices (A–C) and after inhibition of acetylcholinesterase with DFP (D–F) are shown for the absolute peak inward current response (A, D) and for the percentage of the maximal response within each neuron (B, E) for neurons from WT mice and for neurons from mice in which the α5 subunit had been genetically deleted (α5−/−). Values are shown as mean ± 1 SEM. Nonlinear curve fit analysis of the percentage maximal response after inhibition of acetylcholinesterase (E) finds that neurons from WT mice show one high-affinity response site, whereas neurons from α5−/− mice show both high-affinity and low-affinity response sites, with the high-affinity component being responsible for 26% of the total response. Exemplary voltage-clamp traces showing inward current responses at three acetylcholine concentrations are shown for one neuron each from a WT and an α5−/− mouse in untreated slices (C) and in slices after inhibition of acetylcholinesterase (F). Because acetylcholinesterase normally contributes to the rapid removal of acetylcholine from the slice during the washout period (C), irreversibly inhibiting this enzyme with DFP results in prolonged nicotinic currents (F).
The α5 nicotinic subunit increases the current elicited by nicotine
Nicotine binds with high affinity to α4β2* nAChRs in human brain (Brody et al., 2006) and within the rodent prefrontal cortex (Gozzi et al., 2006). We tested the ability of nicotine to activate α4β2* nAChRs on layer VI mPFC neurons to assess how the α5 subunit affects activation by nicotine in an intact neuronal system. Application of 300 nm nicotine, a level consistent with that seen in the blood of smokers (Henningfield et al., 1993; Rose et al., 2010), resulted in an inward current that had a slow onset and persisted with continued application of nicotine (Fig. 3). The steady-state amplitude was equivalent to the current elicited by a low level of ACh. The mean inward current in response to nicotine was approximately three times greater in wild-type mice (16.2 ± 1.7 pA; n = 10) compared with α5−/− mice (6.1 ± 1.2 pA; n = 7) (two-tailed unpaired t test, p = 0.0006). Exemplary traces are shown in Figure 3A. Next, we examined the ability of increasing concentrations of nicotine to elicit inward currents, to mimic the conditions the brain experiences as someone smokes a cigarette (Rose et al., 1999, 2010). As illustrated in Figure 3B, applications of 100, 300, and 500 nm nicotine in consecutive 5 min segments resulted in successive increases in the magnitude of the nicotine-induced inward current in neurons from wild-type mice (five of five neurons) but did not result in successive increases in neurons from α5−/− mice (zero of five neurons), suggesting potential desensitization of their nicotinic receptors by the first concentration of nicotine applied (100 nm). For both genotypes, the mean inward currents elicited by the increasing concentrations of nicotine are shown in Figure 3C. Two-way repeated-measures ANOVA found effects of nicotine concentration (F(2,16) = 15.90; p = 0.0002) and genotype (F(1,16) = 31.41; p = 0.0005), with an interaction between the two (F(2,16) = 17.63; p < 0.0001), and Bonferroni's post hoc analysis found genotype differences at each nicotine concentration (p < 0.05). Interestingly, separate analysis of each genotype by one-way repeated-measures ANOVA found that nicotine significantly influenced current amplitude in neurons from wild-type mice (F(2,8) = 17.76; p = 0.001) but not in neurons from α5−/− mice (F(2,8) = 0.68; p = 0.5). The opening of ionotropic nicotinic receptor channels by nicotine would be expected to increase the RMS current noise of the voltage-clamp recordings. The current noise analysis for this experiment is shown in Figure 3D, in which two-way repeated-measures ANOVA found significant effects of nicotine (F(3,21) = 37.85; p < 0.0001) and genotype (F(1,21) = 14.27; p = 0.007), with an interaction between the two (F(3,21) = 17.95; p < 0.0001). Figure 3D shows that RMS current noise was comparable between the genotypes at baseline, and Bonferroni's post hoc analysis found that the nicotine-induced increase in RMS current noise was significantly greater in neurons from wild-type mice compared with neurons from α5−/− mice at each concentration of nicotine (p < 0.05). Separate analysis of each genotype by one-way repeated-measures ANOVA found that nicotine significantly increased RMS current noise in neurons from both wild-type (F(3,12) = 23.41; p = 0.0001) and α5−/− (F(3,12) = 11.14; p = 0.0009) mice.
Figure 3.

Excitation of medial prefrontal layer VI pyramidal neurons by nicotine is dependent on the nicotinic receptor α5 subunit. A, Example voltage-clamp traces showing the inward current response to 300 nm nicotine in one neuron each from a WT mouse expressing the α5 subunit and a mouse in which the α5 subunit had been genetically deleted (α5−/−). The peak inward currents were significantly greater in neurons from WT mice (16.2 ± 1.7 pA; n = 10) compared with neurons from α5−/− mice (6.1 ± 1.2 pA; n = 7) (two-tailed unpaired t test, p = 0.0006). Nicotine applied in the bath at 100, 300, and 500 nm in consecutive 5 min intervals (exemplary traces shown in B) resulted in successive increasing current responses in neurons from WT mice but resulted in small currents in neurons from α5−/− mice that did not appreciably increase in response to greater nicotine concentrations. Mean inward currents from this experiment (n = 5 for each genotype) are shown in C, in which there were significant effects of nicotine (two-way repeated-measures ANOVA, p = 0.0002) and genotype (p = 0.0005), with a significant interaction between the two (p < 0.0001). Neurons from WT mice showed greater inward current compared with neurons from α5−/− mice at each nicotine concentration (Bonferroni's post hoc test, *p < 0.05). There were significant effects of nicotine in WT mice (one-way repeated-measures ANOVA, p = 0.001) but not in α5−/− mice (p = 0.5). RMS current noise analysis is shown in D, in which there were similar effects of nicotine (two-way repeated-measures ANOVA, p < 0.0001), genotype (p = 0.007), and an interaction between the two (p < 0.0001). Current noise was not different at baseline, but it was greater in WT mice compared with α5−/− mice at each nicotine concentration (Bonferroni's post hoc test, *p < 0.05). There were significant effects of nicotine on the magnitude of current noise in both WT mice (one-way repeated-measures ANOVA, p = 0.0001) and α5−/− mice (p = 0.0009). Values are shown as mean ± 1 SEM.
A number of studies have shown that nicotinic agonists can directly excite a subset of interneurons within layers I–V of the rat cerebral cortex (Xiang et al., 1998; Porter et al., 1999; Christophe et al., 2002; Couey et al., 2007) and that this excitation can augment inhibitory signaling to pyramidal neurons within layer V of rat mPFC (Couey et al., 2007). We next sought to test whether nicotinic excitation of interneurons within mouse mPFC influences the inhibition of layer VI pyramidal neurons. Because the nAChR α5 subunit mRNA has been identified in only a subset of cortical interneurons directly excited by nicotinic agonists (Porter et al., 1999; Christophe et al., 2002), we also sought to test whether the presence of this subunit alters any changes in inhibitory signaling that are observed. To this end, we measured GABAergic sPSCs in layer VI pyramidal neurons of the mPFC under baseline conditions and in response to bath application of 100 nm nicotine for 5 min, followed immediately by 300 nm nicotine for 5 min. These experiments were performed with high chloride patch solution and glutamate receptor blockade. In wild-type animals, one of six neurons tested showed increases in sPSC frequency and amplitude when exposed to both 100 and 300 nm nicotine (Kolmogorov–Smirnov test comparing nicotine with baseline, p < 0.01), and another neuron showed an increase in sPSC amplitude at 300 nm nicotine only (p < 0.01). Similarly in the α5−/− mice, one of six neurons tested showed increased sPSC frequency with 300 nm nicotine (p < 0.01). Two-way repeated-measures ANOVA found no effect of genotype (F(1,20) = 1.17; p = 0.3) or nicotine (F(2,20) = 2.45; p = 0.1) on GABAergic sPSC frequency (means at baseline: wild type, 0.3 ± 0.1 Hz, n = 6; α5−/−, 0.5 ± 0.2 Hz, n = 6). Similarly, there were no effects of genotype (F(1,20) = 0.70; p = 0.4) or nicotine (F(2,20) = 0.49; p = 0.6) on GABAergic sPSC amplitude (means at baseline: wild type, 29.9 ± 3.5 pA, n = 6; α5−/−, 30.8 ± 2.6 pA, n = 6). Thus, concentrations of nicotine that consistently activate layer VI pyramidal neurons only activate their GABAergic afferents in a small proportion of instances, and no role was evident for the nAChR α5 subunit in this signaling.
The α5 nicotinic subunit protects against desensitization
Nicotine exhibits a complex pharmacology at nicotinic receptors in which concentrations sufficient to activate the receptor may convert active nAChRs via conformational change to a desensitized state that is unresponsive to subsequent agonist exposure (Pidoplichko et al., 1997; Quick and Lester, 2002). To test the hypothesis that nicotine has different desensitizing effects depending on genotype, we tested the layer VI neuronal response to ACh before and after prolonged exposure to nicotine. We first recorded the current response to ACh (1 mm for 30s). Next, we applied either 100 or 300 nm nicotine for 10 min, allowed for 10 min of washout to permit the inward current elicited by nicotine to return to baseline (to avoid interpretation problems associated with summation of currents), and then repeated the ACh application. We then calculated the percentage desensitization induced by nicotine within each individual neuron as [(ACh current before nicotine − ACh current after nicotine)/ACh current before nicotine] × 100. Desensitization was indeed greatly affected by the presence of the α5 subunit, because the neurons from wild-type mice showed approximately half as much desensitization at each concentration of nicotine as those from α5−/− mice. Two-way ANOVA of the desensitization data shown in Figure 4A found significant effects of genotype (F(1,25) = 47.94; p < 0.0001) and nicotine concentration (F(1,25) = 6.12; p = 0.02). Moreover, Bonferroni's post hoc test found significant effects of genotype at 100 nm nicotine (wild type, 29 ± 7% desensitization, n = 6; α5−/−, 57 ± 3% desensitization, n = 6; p < 0.01) and at 300 nm nicotine (wild type, 36 ± 4% desensitization, n = 9; α5−/−, 73 ± 4% desensitization, n = 8; p < 0.001).
Figure 4.
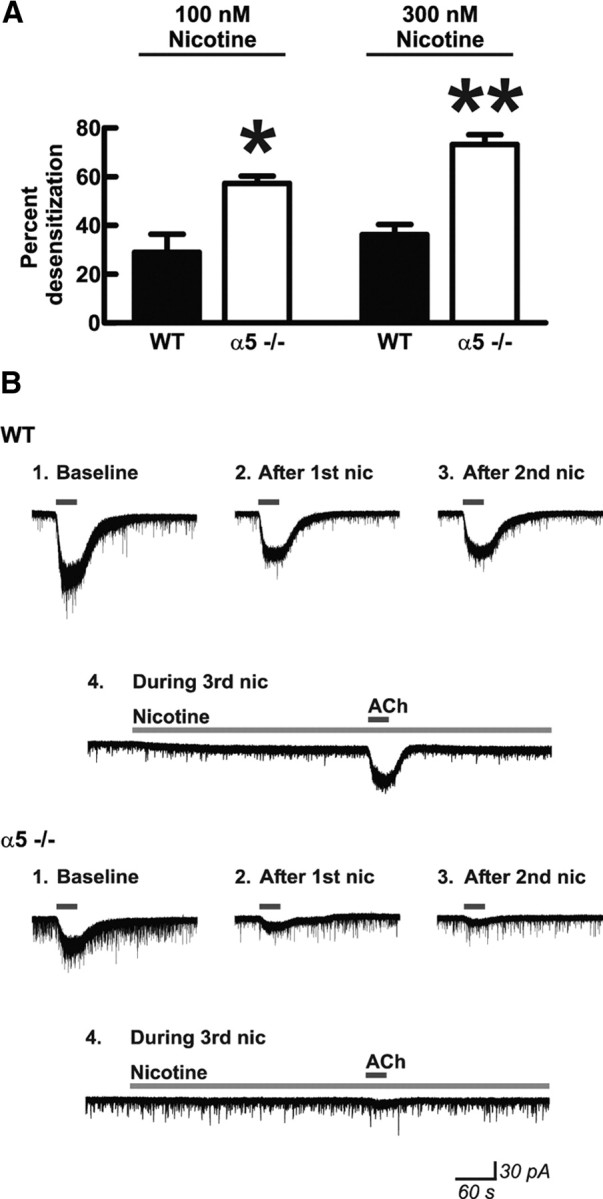
Presence of the nicotinic receptor α5 subunit protects medial prefrontal layer VI pyramidal neurons from nicotine-induced receptor desensitization. A, Nicotinic receptor desensitization by nicotine within individual neurons was calculated by measuring peak inward current responses to bath application of 1 mm acetylcholine (30 s) before and after exposure to either 100 or 300 nm nicotine for 10 min. There were significant effects of nicotine concentration (two-way ANOVA, p = 0.02) and genotype (p < 0.0001) on percentage desensitization after nicotine exposure. Neurons from mice in which the α5 subunit had been genetically deleted (α5−/−) showed greater desensitization compared with neurons from WT mice expressing the α5 subunit after exposure to both 100 nm nicotine (Bonferroni's post hoc test, *p < 0.01) and 300 nm nicotine (**p < 0.001). Values are mean ± 1 SEM. B, Example voltage-clamp traces in response to acetylcholine are shown before (Baseline) and after (After 1st nic) an initial 10 min exposure to 300 nm nicotine for one neuron each from a mouse of each genotype. Nicotine was washed out of the slices for 10 min, during which time current signals returned to baseline. Although there was an initial desensitization in neurons from WT mice, the resulting acetylcholine current response was preserved after a second 10 min exposure to nicotine (After 2nd nic) and was also preserved when acetylcholine was applied during a third 10 min exposure to nicotine (During 3rd nic). In contrast, the acetylcholine response was diminished or even abolished in neurons from α5−/− mice after subsequent 10 min exposures to nicotine. Note that nicotine continues to show activational effects (increase in inward current and current noise) even on the third application in the WT neuron but does not show these effects in the α5−/− neuron.
We further examined the protective properties of the α5 subunit against receptor desensitization by repeating the succession of nicotine and ACh applications. As shown for an example wild-type neuron in Figure 4B, the inward current resulting from 1 mm ACh (30s) was reduced after the first 10 min exposure to 300 nm nicotine but was not additionally reduced by a second exposure to nicotine. Even coapplication of ACh during a third nicotine application resulted in a cumulative inward current similar to that seen during the previous two ACh applications, suggesting that a component of the layer VI nicotinic current in wild-type mice is resistant to acute desensitization by 300 nm nicotine. In α5−/− mice, in contrast, the layer VI nicotinic currents appear more vulnerable to desensitization by exposure to this level of nicotine. As shown in an exemplary neuron in Figure 4B, the current elicited by ACh appeared to decrease after each successive nicotine application and the ACh current elicited in the presence of nicotine was negligible. Results from this series of experiments demonstrate that the presence of the α5 subunit within α4β2* nAChRs on mPFC layer VI pyramidal neurons increases the agonist efficacy of nicotine and appears to protect these receptors from desensitization induced by exposure to nicotine.
The α5 nicotinic subunit is required for normal attentional performance in mouse
Because ACh levels in mPFC play an important role in attention gating (Dalley et al., 2004; Parikh et al., 2007) and layer VI pyramidal neurons serve as a major output from this region to the thalamus (Gabbott et al., 2005), we next sought to determine whether the presence of the nicotinic α5 subunit influences attentional performance using the 5-CSRTT (Robbins, 2002). Mice (n = 26; 13 of each genotype) were trained to detect and respond to a brief light stimulus randomly presented in one of five spatial locations to receive a sucrose reward. The duration of the stimulus presentation in this task can be varied to alter demands on attentional resources, with a shorter stimulus duration requiring greater attentional demand. Because nicotinic receptor function is thought to be a limiting factor in attentional performance only under challenging conditions (Newhouse et al., 2004; Lambe et al., 2005), we examined the effect of α5 nicotinic subunit expression in our mice on attentional performance under challenging conditions characterized by short stimulus durations. For detailed description of the training and testing procedure, see Materials and Methods. Results from this experiment are shown in Table 1 and Figure 5A.
Table 1.
Performance on the 5-CSRTT at varying stimulus durations
| Stimulus duration (s) | Number of trials** |
Percentage accuracy** |
Percentage omissions** |
Premature** |
Correct latency (s)** |
Reinforcer latency (s) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | α5−/− | WT | α5−/− | WT | α5−/− | WT | α5−/− | WT | α5−/− | WT | α5−/− | |
| 1.0 | 98.2 ± 1.8 | 100 ± 0.0 | 97.6 ± 0.6 | 97.0 ± 0.9 | 24.3 ± 3.0 | 31.5 ± 3.6 | 1.3 ± 0.4 | 1.0 ± 0.3 | 0.75 ± 0.02 | 0.76 ± 0.03 | 1.45 ± 0.04 | 1.50 ± 0.04 |
| 0.5 | 95.5 ± 2.5 | 96.1 ± 3.0 | 92.4 ± 1.3 | 86.7 ± 2.1 | 36.0 ± 4.5 | 42.1 ± 5.1 | 1.9 ± 0.5 | 3.1 ± 0.9 | 0.61 ± 0.03 | 0.63 ± 0.04 | 1.44 ± 0.05 | 1.56 ± 0.06 |
| 0.25 | 87.4 ± 3.6 | 93.8 ± 3.2 | 79.5 ± 2.8 | 72.3 ± 2.1 | 43.2 ± 3.7 | 47.5 ± 3.0 | 3.8 ± 0.9 | 5.6 ± 1.5 | 0.62 ± 0.04 | 0.70 ± 0.05 | 1.39 ± 0.04 | 1.51 ± 0.06 |
| 0.125 | 78.0 ± 4.8 | 83.8 ± 5.6 | 62.5 ± 2.8 | 54.3 ± 2.5* | 47.3 ± 4.4 | 50.7 ± 2.8 | 7.3 ± 1.1 | 7.5 ± 1.1 | 0.67 ± 0.05 | 0.74 ± 0.05 | 1.44 ± 0.05 | 1.51 ± 0.04 |
Data are presented as mean ± 1 SEM.
*p < 0.05 compared with WT mice at 0.125 s stimulus duration.
**p < 0.001 for an overall effect of stimulus duration. Accuracy measures are also depicted in the graph in Figure 5A.
Figure 5.

The nicotinic receptor α5 subunit is required for normal attentional performance under challenging conditions. WT mice expressing the α5 subunit and mice in which the α5 subunit had been genetically deleted (α5−/−) were trained on the five-choice serial reaction time test for sustained visual attention. A, WT and α5−/− mice responded with equal percentage accuracy at the 1 s stimulus duration used during acquisition of the task. Lowering the stimulus duration to make the task more challenging resulted in a main effect on percentage accuracy overall (two-way repeated-measures ANOVA, p < 0.0001), and WT mice responded with greater percentage accuracy compared with α5−/− mice when the stimulus duration was lowered (two-way repeated-measures ANOVA, genotype effect, p = 0.004; Bonferroni's post hoc test at 0.125 s, *p < 0.05). Values are mean ± 1 SEM. Complete results for this experiment are presented in Table 1. B, In a subsequent experiment, each mouse was run on the task with the stimulus duration set at 0.063 s 5 min after administration each of saline and 0.03 mg/kg nicotine. WT mice performed with greater percentage accuracy compared with α5−/− mice after saline treatment (two-way repeated-measures ANOVA, genotype effect, p = 0.02; Bonferroni's post hoc test with saline treatment, *p < 0.05). Although there was no overall effect of nicotine (two-way repeated-measures ANOVA, treatment effect, p = 0.2), separate analysis within each genotype group found that percentage accuracy was significantly decreased by nicotine in the WT group only (two-tailed paired t test, ‡p = 0.005). Values are mean ± 1 SEM. Complete results for this experiment are presented in Table 2.
Under the baseline training condition of 1 s stimulus duration, the mice performed at high accuracy, and there were no differences in performance between genotypes. Performance under conditions of increased attentional demand was then assessed by testing each mouse for one session each in which the stimulation duration was 1 (training control), 0.5, 0.25, and 0.125 s. The order of sessions was determined from a Latin square, with 3 d of regular training (1 s stimulus duration) between test days. During this experiment, one wild-type and one α5−/− mouse stopped performing and were therefore excluded from the subsequent analysis. As illustrated in Figure 5A and Table 1, accuracy of responding was reduced overall by shorter stimulus duration (two-way repeated-measures ANOVA, F(3,66) = 154.77; p < 0.0001), and this effect was more pronounced in α5−/− mice (main effect of genotype, F(1,66) = 10.28; p = 0.004). A Bonferroni's post hoc analysis demonstrated that wild-type mice responded with greater accuracy compared with α5−/− mice at the stimulus duration of 0.125 s (62.5 ± 2.8 vs 54.3 ± 2.5%, respectively; p < 0.05). It is interesting to note that significant differences in attention performance of similar magnitude (∼1 SD) have been reported in human studies (Sunderland et al., 1989; Foldi et al., 1992; Okonkwo et al., 2008b) and are believed to have profound consequences on the performance of complex tasks (Okonkwo et al., 2008a). As illustrated in Table 1, we found no main effect of α5 genotype on the other measures within this experiment, including the number of trials performed (F(1,66) = 1.21; p = 0.3), percentage omissions (F(1,66) = 1.47; p = 0.2), number of premature responses (F(1,66) = 0.84; p = 0.4), correct latency (F(1,66) = 1.18; p = 0.3), or reinforcer latency (F(1,66) = 2.62; p = 0.12). In addition to the effect on percentage accuracy described above, we also found significant effects of stimulus duration on the number of trials performed (F(3,66) = 14.64; p < 0.0001), percentage omissions (F(3,66) = 23.68; p < 0.0001), number of premature responses (F(3,66) = 19.20; p < 0.0001), and correct latency (F(3,66) = 6.32; p < 0.0008) but not on reinforcer latency (F(3,66) = 0.81; p = 0.5). There were no significant interactions between genotype and stimulus duration for any measure.
Effect of nicotine on attentional performance
We then assessed how nicotine affects attentional performance in the 5-CSRTT in both genotypes. Acute nicotine has been shown to influence attentional performance in rodents and in humans (Mirza and Stolerman, 1998; Mumenthaler et al., 1998; Hahn et al., 2003; Lambe et al., 2005; Hoyle et al., 2006; Day et al., 2007; Pattij et al., 2007), and our results in mouse brain slice show that the presence of the α5 subunit allows for greater nicotine-elicited inward currents in mPFC neurons involved in attention circuits. We therefore hypothesized that the α5 subunit may be necessary for nicotine to influence mouse performance in the 5-CSRTT under challenging conditions.
This experiment was performed with the same mice used for the stimulus duration experiment described above and took place approximately 2 months later. To ensure that the mice would be adequately challenged on the task, we set the stimulus duration to 0.063 s. We tested wild-type and α5−/− mice in the 5-CSRTT starting 5 min after subcutaneous administration of either saline or 0.03 mg/kg nicotine in a randomized and balanced cross-over design. During the 5-CSRTT experiment, one wild-type mouse stopped performing the task and was therefore excluded from the analysis. Based on experimental values obtained in previous mouse studies (Petersen et al., 1984; Thumwood et al., 1988; Siu and Tyndale, 2007) (our calculations are described in Materials and Methods), the subcutaneous dose of 0.03 mg/kg likely results in peak mouse brain nicotine concentrations near 117 nm, a concentration that is sufficient to both activate and desensitize nAChRs in layer VI pyramidal neurons (Figs. 3, 4) and is relevant to the levels of nicotine observed in the human brain during cigarette smoking (Rose et al., 2010). Results from this experiment are shown in Table 2 and Figure 5B.
Table 2.
Performance on the 5-CSRTT after saline or nicotine administration
| Number of trials |
Percentage accuracy |
Percentage omissions |
Premature*** |
Correct latency (s)*** |
Reinforcer latency (s) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | a5−/− | WT | α5−/− | WT | α5−/− | WT | α5−/− | WT | α5−/− | WT | α5−/− | |
| Saline | 66.3 ± 5.4 | 71.2 ± 6.9 | 69.5 ± 2.4 | 60.4 ± 2.5* | 53.9 ± 4.7 | 53.2 ± 2.6 | 2.5 ± 1.0 | 3.2 ± 0.7 | 0.74 ± 0.05 | 0.79 ± 0.06 | 1.67 ± 0.11 | 1.59 ± 0.04 |
| Nicotine | 71.0 ± 6.2 | 78.9 ± 6.1 | 64.9 ± 2.2** | 59.7 ± 2.7 | 49.3 ± 4.2 | 50.9 ± 3.4 | 3.9 ± 0.7 | 5.1 ± 1.1 | 0.69 ± 0.03 | 0.66 ± 0.03 | 1.62 ± 0.07 | 1.70 ± 0.17 |
Data are presented as mean ± 1 SEM.
*p < 0.05 compared with WT mice after saline treatment.
**p = 0.005 compared with WT mice after saline treatment.
***p < 0.05 for an overall effect of nicotine. Accuracy measures are also depicted in the graph in Figure 5B.
The results from the saline condition of the study (Fig. 5B) replicated our previous finding that wild-type mice performed with greater accuracy on the demanding attention task compared with α5−/− mice. Two-way repeated-measures ANOVA found that there was a significant effect of genotype (F(1,23) = 5.91; p = 0.02) on accuracy; Bonferroni's post hoc test found that wild-type mice exhibited greater percentage accuracy than α5−/− mice with saline treatment only (Fig. 5B) (p < 0.05). As seen in previous work (Mirza and Stolerman, 1998; Hahn et al., 2003; Pattij et al., 2007), nicotine resulted in a small but significant increase in the number of premature responses (F(1,23) = 7.15; p = 0.01) and decreased correct latency (F(1,23) = 4.82; p = 0.04) in both genotypes, suggesting that these effects may be mediated by nicotinic receptors that do not contain the α5 subunit. It has been suggested recently that the nicotinic receptors relevant for the effects of nicotine on impulsivity are α4β2 receptors in the infralimbic prefrontal cortex (Tsutsui-Kimura et al., 2010). There were no statistically significant effects of nicotine on percentage accuracy as tested by two-way ANOVA (F(1,23) = 1.91; p = 0.2). However, we noticed that nicotine decreased accuracy in 11 of 12 wild-type mice, and a paired Student's t test analysis showed a significant effect of nicotine in wild-type mice (two-tailed paired t test, p = 0.005). No such relationship was seen in α5−/− mice (two-tailed paired t test, p = 0.8). These results suggest that nicotine is able to affect performance under demanding conditions in wild-type mice but not in α5−/− mice. As illustrated in Table 2, we found no main effect of genotype on the number of trials performed (F(1,23) = 0.66; p = 0.4), percentage omissions (F(1,23) = 0.01; p = 0.9), number of premature responses (F(1,23) = 0.66; p = 0.4), correct latency (F(1,23) = 0.02; p = 0.9), or reinforcer latency (F(1,23) = 0.01; p = 0.9). Beyond the effects of nicotine on premature responding and correct latency described above, there were no significant effects of nicotine on the number of trials performed (F(1,23) = 2.38; p = 0.14), percentage omissions (F(1,23) = 1.57; p = 0.2), or reinforcer latency (F(1,23) = 0.09; p = 0.8). There were no significant interactions between genotype and nicotine for any measure.
Control experiment to assess effects of nicotine on locomotor activity
Because systemic nicotine administered at 0.5 mg/kg (more than an order of magnitude above the dose used in our 5-CSRTT) has been shown previously to inhibit locomotor activity in wild-type mice (Salas et al., 2003; Jackson et al., 2010), we later assessed the effect of nicotine on locomotor activity in both genotypes. The same mice used for the 5-CSRTT experiments were subcutaneously administered either 0.03 or 0.3 mg/kg nicotine in a random balanced order, and locomotor activity was measured over a 40 min span as described in Materials and Methods. We measured general motor activity (Fig. 6A, Total beam breaks) in addition to the number of times that the mouse crossed the full length of monitoring cage (Fig. 6B, Beam lengths). For both measures, two-way repeated-measures ANOVA found significant effects of nicotine (p < 0.02), genotype (p < 0.03), and the interaction between nicotine and genotype (p < 0.004). Bonferroni's post hoc test found a significant effect of genotype at 0.3 mg/kg nicotine only (p < 0.05). One-way ANOVAs performed for each genotype found a significant effect of nicotine for both measures in WT mice (p < 0.0002) but not in α5−/− mice (p = 0.7). For WT mice, Bonferroni's post hoc tests found that locomotor activity after administration of 0.3 mg/kg nicotine was different from that after administration of either saline or 0.03 mg/kg nicotine (p < 0.05), which were not different from each other. Together with the data in Table 2, the locomotor results suggest that the 0.03 mg/kg concentration of nicotine used in our 5-CSRTT experiment may have relatively selective effects on aspects of attentional performance; however, administration of higher acute concentrations of nicotine risks locomotor sequelae in WT mice.
Figure 6.

Acute nicotine administration inhibits locomotor activity in WT mice expressing the α5 subunit mice at 0.3 mg/kg but not at 0.03 mg/kg. General locomotor activity (A, Total beam breaks) and the number of times that mice crossed the full length of the monitoring cage (B, Beam lengths) were measured for 40 min after nicotine administration. For both measures, there were significant effects of nicotine (two-way repeated-measures ANOVA, p < 0.02), genotype (p < 0.03), and the interaction between nicotine and genotype (p < 0.004). The genotype difference was evident at 0.3 mg/kg nicotine only (Bonferroni's post hoc test, p < 0.05). There was a significant effect of nicotine for both measures within the WT group (one-way ANOVA, p < 0.0002) but not in the α5−/− group (p = NS). For WT mice, locomotor activity after administration of 0.3 mg/kg nicotine was significantly different from that after administration of either saline or 0.03 mg/kg nicotine (Bonferroni's post hoc test, *p < 0.05), which were not different from each other (p = NS). Values are shown as mean ± 1 SEM.
Discussion
This study represents a novel demonstration that attention circuitry and accuracy are significantly influenced by the α5 nicotinic subunit. This conclusion is based on a series of functional and pharmacological experiments performed using slice electrophysiology and cognitive performance in wild-type and α5−/− mice. We found that presence of the α5 nicotinic subunit greatly enhances the ability of ACh to excite layer VI pyramidal neurons of the mPFC by increasing its efficacy and by conferring a monophasic high-affinity response to cholinergic stimulation. Next, we examined neuronal responses to nicotine and found that the presence of the α5 subunit enhances the inward current elicited by nicotine and allows it to persist under conditions of prolonged stimulation. Consistent with the ability of α5 subunits to enhance nicotinic excitation at the neuronal level, we then demonstrated that the presence of the α5 nicotinic subunit enhances attention in mice under challenging conditions that would require optimal performance of attention pathways. Interestingly, presence of the α5 nicotinic subunit makes the accuracy of wild-type mice vulnerable to nicotine exposure, suggesting that the fully enhanced, phasic, and nondesensitized excitatory response to ACh within layer VI of the mPFC is important for normal attentional performance under challenging conditions. Our combined electrophysiological and behavioral results thus demonstrate a previously unknown role for the nAChR α5 subunit to influence signaling within attention pathways and suggest this relatively restricted receptor isoform as a potential therapeutic target for attention-related disorders.
Implications of α5 nicotinic subunits for cholinergic stimulation of prefrontal neurons
Here, we show that the α5 accessory subunit has a major influence on α4β2* nAChRs in whole neurons of prefrontal attention circuits. Our research allows us to describe not only the functional properties of α5-containing layer VI nicotinic currents but also the nicotinic currents that are present after genetic deletion of this subunit. We find that the α5 subunit in layer VI neurons confers a monophasic, high-affinity response to ACh and that its deletion results in a biphasic response to ACh with a small high-affinity component and a large low-affinity component [likely with β2 and α4 accessory subunits, respectively (Gotti et al., 2009)]. These α5−/− results are relevant to understanding α4β2* nicotinic receptors in human attention circuitry in the presence of the α5 D398N polymorphism, which results in reduction of its characteristic function (Bierut et al., 2008). Our concentration–response findings show that the activation of α4β2α5 nicotinic receptors in intact neurons can be accurately modeled in reduced preparations (Moroni et al., 2006; Kuryatov et al., 2008; McClure-Begley et al., 2009). The monophasic activation curve in the wild-type mice suggests that recorded neurons in our preparation contain α5 accessory subunits in a high percentage, if not in all α4β2* nAChRs. This contrasts greatly with immunoprecipitation analyses suggesting that only 14–16% of α4β2* nAChRs in the whole cerebral cortex contain the α5 subunit (Brown et al., 2007; Mao et al., 2008), suggesting that layer VI neurons contain a disproportionate level of the unusual α4β2α5 subtype of nicotinic receptor.
Nicotinic α5 subunits appear necessary to allow a low concentration of nicotine to activate nicotinic receptors in prefrontal attention circuitry with prolonged and repeated exposures. This finding differs sharply from that predicted by previous work in reduced preparations in which the presence of the α5 subunit appeared to enhance desensitization of α4β2* nAChRs (Ramirez-Latorre et al., 1996; Kuryatov et al., 2008). Nicotine-induced desensitization is a complex process involving alterations in receptor conformation and phosphorylation (Quick and Lester, 2002) that is likely differentially regulated in neurons compared with non-neuronal expression systems.
Implications of α5 nicotinic subunits for behavior
What are the behavioral sequelae of the enhanced, excitatory nicotinic currents in layer VI pyramidal neurons of the mPFC? Cholinergic excitation in mPFC is important for attention (Passetti et al., 2000; Parikh et al., 2007). By using mice genetically manipulated for a rare but seemingly important nicotinic subunit, we suggest that the endogenous ACh responsivity of layer VI neurons in mice expressing the α5 subunit (Figs. 1, 2) is required for normal performance in the 5-CSRTT under challenging conditions (Fig. 5A). We are mindful that mRNA for the α5 subunit is expressed in several additional discrete regions of the adult brain (Wada et al., 1990; Salas et al., 2003; Winzer-Serhan and Leslie, 2005). Of these brain regions, however, the mPFC is most likely directly responsible for attention in the 5-CSRTT (Muir et al., 1996; Chudasama et al., 2003). There is a dense band of α5 subunit expression in layer VI (Wada et al., 1990; Marks et al., 1992; Salas et al., 2003; Winzer-Serhan and Leslie, 2005), and this main corticothalamic projection layer gives feedback to the thalamus that is important in attention (Gabbott et al., 2005; Zikopoulos and Barbas, 2006). Although there is also expression of the α5 subunit in a small subset of inhibitory cells in prefrontal cortex (Porter et al., 1999; Christophe et al., 2002), recent work in mice suggests that manipulations of GABAergic transmission alter impulsivity instead of accuracy (Oliver et al., 2009). In our 5-CSRTT experiments, measures of impulsivity (premature responses), reaction time (correct latency), and motivation (reinforcer latency) were not altered by genetic deletion of the α5 subunit, suggesting that reduced accuracy of responding was not secondary to alterations in these other behavioral processes. A potential confounding factor after genetic manipulation is the compensatory upregulation of related genes. Analysis of the α5−/− mouse used in our study found that they express normal mRNA levels for nAChR α4, α6, α7, β2, and β4 subunits, in addition to having normal binding levels for [125I]epibatidine (labeling non-α7 nAChRs) and for [125I]α-bungarotoxin (labeling α7 nAChRs), in every brain region examined (Salas et al., 2003). However, future work is necessary to appreciate the developmental consequences of α5 subunit deletion on attention circuitry and performance (King et al., 2003; Kassam et al., 2008; Heath and Picciotto, 2009).
In human studies, nicotine benefits attentional performance in subjects dependent on nicotine (Parrott and Roberts, 1991; Warburton and Arnall, 1994; Dawkins et al., 2007) and in patient populations having impaired attention (Newhouse et al., 1988; Zabala et al., 2009). However, the effects of nicotine on attention in healthy nonsmoking subjects are less clear (Newhouse et al., 2004). Nicotine generally increases processing speed across studies; however, although some studies found improvements in attentional performance accuracy (Wesnes and Warburton, 1984; Mumenthaler et al., 1998; Levin et al., 1998), other studies found no change (Le Houezec et al., 1994; Kleykamp et al., 2005) or impairments (Heishman et al., 1993; Foulds et al., 1996; Heishman and Henningfield, 2000) in performance accuracy. It is interesting that the reduced latency to respond to the light stimuli observed in our mice when treated with nicotine is consistent with the faster processing speed observed in many human studies. However, our results and that of Pattij et al. (2007) suggest that acute nicotine does not improve attention in mice, whereas nicotine can improve attentional performance in rats under certain conditions (Mirza and Stolerman, 1998; Hahn et al., 2003; Day et al., 2007), suggesting possible species differences. Indeed, the complexity of the effects of nicotine on rodent neuropharmacology and behavior are widely accepted (Picciotto, 2003).
In combination with our electrophysiological data, we interpret our finding that acute nicotine decreased accuracy in 11 of 12 wild-type mice to suggest that, to be detected above tonic background “noise,” there may need to be a minimum threshold of phasic ACh “signal” to excite layer VI mPFC neurons for normal attention performance under challenging conditions. Acute nicotine exposure may interfere with optimal performance in the wild-type mice by (1) increasing baseline noise through tonic activation of nicotinic receptors, (2) decreasing the phasic signal through partially desensitizing the response to ACh, or (3) a combination of these effects. In line with this hypothesis, the lower optimal performance of the α5−/− mice with or without nicotine may result from phasic ACh excitation of layer VI neurons that is always below an important threshold.
The clinical implications of our findings are significant. Human imaging studies show that nicotine from one cigarette can remain bound to prefrontal α4β2* nAChRs for hours (Brody et al., 2006), which has been widely interpreted to suggest that all receptors in this class would be desensitized to nicotine and ACh during this time. However, our findings show that prefrontal layer VI α4β2* nAChRs containing the α5 subunit may be persistently activated by bound nicotine and remain partially responsive to ACh neurotransmission. A number of very recent genetic studies in human have found that the risk for, and strength of, nicotine addiction are statistically linked with single nucleotide polymorphisms in the α5 subunit gene CHRNA5 (Bierut et al., 2008; Schlaepfer et al., 2008; Stevens et al., 2008; Weiss et al., 2008; Saccone et al., 2009; Caporaso et al., 2009), including a nonsynonymous coding polymorphism that reduces the function of α5 subunit-containing nicotinic receptors (Bierut et al., 2008). The strength of the link between polymorphisms in the CHRNA5 gene and human addiction has been suggested to arise from a cognitive behavioral profile, including deficits in sustained and selective attention, that promotes early experimentation with drugs and alcohol (Rigbi et al., 2008; Schlaepfer et al., 2008; Weiss et al., 2008) that may be explained mechanistically by our finding that the α5 subunit is required for normal attentional control. The α4β2* nAChR is currently under consideration as a therapeutic target for a number of indications, including attention deficit hyperactivity disorder, depression, Alzheimer's disease, and Parkinson's disease (Taly et al., 2009), all of which are associated with impaired attention (Newhouse et al., 2004; Wisco, 2009). Therapeutic intervention to enhance activity of α5 subunit-containing α4β2* nAChRs may be an efficient manner in which to treat attentional dysfunction across a number of psychiatric and neurological disorders.
Footnotes
This work was supported by Canadian Institutes of Health Research Grant MOP 89825, the Canada Research Chairs Program, and the Canadian Foundation for Innovation (all to E.K.L.). We sincerely thank Judy Sinyard for assistance in performing the five-choice serial reaction time test.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitto HJ, Usrey WM. Corticothalamic feedback and sensory processing. Curr Opin Neurobiol. 2003;13:440–445. doi: 10.1016/s0959-4388(03)00096-5. [DOI] [PubMed] [Google Scholar]
- Alves NC, Bailey CD, Nashmi R, Lambe EK. Developmental sex differences in nicotinic currents of prefrontal layer VI neurons in mice and rats. PLoS One. 2010;5:e9261. doi: 10.1371/journal.pone.0009261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165:1163–1171. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG. Cigarette smoking saturates brain α4β2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63:907–915. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RW, Collins AC, Lindstrom JM, Whiteaker P. Nicotinic alpha5 subunit deletion locally reduces high-affinity agonist activation without altering nicotinic receptor numbers. J Neurochem. 2007;103:204–215. doi: 10.1111/j.1471-4159.2007.04700.x. [DOI] [PubMed] [Google Scholar]
- Caporaso N, Gu F, Chatterjee N, Sheng-Chih J, Yu K, Yeager M, Chen C, Jacobs K, Wheeler W, Landi MT, Ziegler RG, Hunter DJ, Chanock S, Hankinson S, Kraft P, Bergen AW. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One. 2009;4:e4653. doi: 10.1371/journal.pone.0004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophe E, Roebuck A, Staiger JF, Lavery DJ, Charpak S, Audinat E. Two types of nicotinic receptors mediate an excitation of neocortical layer I interneurons. J Neurophysiol. 2002;88:1318–1327. doi: 10.1152/jn.2002.88.3.1318. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Passetti F, Rhodes SE, Lopian D, Desai A, Robbins TW. Dissociable aspects of performance on the 5-choice serial reaction time task following lesions of the dorsal anterior cingulate, infralimbic and orbitofrontal cortex in the rat: differential effects on selectivity, impulsivity and compulsivity. Behav Brain Res. 2003;146:105–119. doi: 10.1016/j.bbr.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Bouger P, Chudasama Y, Cardinal RN, Robbins TW. Cortical cholinergic function and deficits in visual attentional performance in rats following 192 IgG-saporin-induced lesions of the medial prefrontal cortex. Cereb Cortex. 2004;14:922–932. doi: 10.1093/cercor/bhh052. [DOI] [PubMed] [Google Scholar]
- Dawkins L, Powell JH, West R, Powell J, Pickering A. A double-blind placebo-controlled experimental study of nicotine. II. Effects on response inhibition and executive functioning. Psychopharmacology (Berl) 2007;190:457–467. doi: 10.1007/s00213-006-0634-6. [DOI] [PubMed] [Google Scholar]
- Day M, Pan JB, Buckley MJ, Cronin E, Hollingsworth PR, Hirst WD, Navarra R, Sullivan JP, Decker MW, Fox GB. Differential effects of ciproxifan and nicotine on impulsivity and attention measures in the 5-choice serial reaction time test. Biochem Pharmacol. 2007;73:1123–1134. doi: 10.1016/j.bcp.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Foldi NS, Jutagir R, Davidoff D, Gould T. Selective attention skills in Alzheimer's disease: performance on graded cancellation tests varying in density and complexity. J Gerontol. 1992;47:P146–P153. doi: 10.1093/geronj/47.3.p146. [DOI] [PubMed] [Google Scholar]
- Foulds J, Stapleton J, Swettenham J, Bell N, McSorley K, Russell MA. Cognitive performance effects of subcutaneous nicotine in smokers and never-smokers. Psychopharmacology (Berl) 1996;127:31–38. doi: 10.1007/BF02805972. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Schwarz A, Reese T, Bertani S, Crestan V, Bifone A. Region-specific effects of nicotine on brain activity: a pharmacological MRI study in the drug-naive rat. Neuropsychopharmacology. 2006;31:1690–1703. doi: 10.1038/sj.npp.1300955. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, McIntosh JM, Marks MJ, Collins AC. Mouse striatal dopamine nerve terminals express α4α5β2 and two stoichiometric forms of α4β2*-nicotinic acetylcholine receptors. J Mol Neurosci. 2010;40:91–95. doi: 10.1007/s12031-009-9263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn B, Shoaib M, Stolerman IP. Involvement of the prefrontal cortex but not the dorsal hippocampus in the attention-enhancing effects of nicotine in rats. Psychopharmacology (Berl) 2003;168:271–279. doi: 10.1007/s00213-003-1438-6. [DOI] [PubMed] [Google Scholar]
- Heath CJ, Picciotto MR. Nicotine-induced plasticity during development: modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacology. 2009;56(Suppl 1):S254–S262. doi: 10.1016/j.neuropharm.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heishman SJ, Henningfield JE. Tolerance to repeated nicotine administration on performance, subjective, and physiological responses in nonsmokers. Psychopharmacology (Berl) 2000;152:321–333. doi: 10.1007/s002130000541. [DOI] [PubMed] [Google Scholar]
- Heishman SJ, Snyder FR, Henningfield JE. Performance, subjective, and physiological effects of nicotine in non-smokers. Drug Alcohol Depend. 1993;34:11–18. doi: 10.1016/0376-8716(93)90041-n. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- Hoyle E, Genn RF, Fernandes C, Stolerman IP. Impaired performance of alpha7 nicotinic receptor knockout mice in the five-choice serial reaction time task. Psychopharmacology (Berl) 2006;189:211–223. doi: 10.1007/s00213-006-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA, Damaj MI. Role of α5 nicotinic acetylcholine receptors in the pharmacological and behavioral effects of nicotine in mice. J Pharmacol Exp Ther. 2010;334:137–146. doi: 10.1124/jpet.110.165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK. Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci. 2008;28:8756–8764. doi: 10.1523/JNEUROSCI.2645-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SL, Marks MJ, Grady SR, Caldarone BJ, Koren AO, Mukhin AG, Collins AC, Picciotto MR. Conditional expression in corticothalamic efferents reveals a developmental role for nicotinic acetylcholine receptors in modulation of passive avoidance behavior. J Neurosci. 2003;23:3837–3843. doi: 10.1523/JNEUROSCI.23-09-03837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleykamp BA, Jennings JM, Blank MD, Eissenberg T. The effects of nicotine on attention and working memory in never-smokers. Psychol Addict Behav. 2005;19:433–438. doi: 10.1037/0893-164X.19.4.433. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J. Roles of accessory subunits in α4β2* nicotinic receptors. Mol Pharmacol. 2008;74:132–143. doi: 10.1124/mol.108.046789. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. J Neurosci. 2005;25:5225–5229. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Houezec J, Halliday R, Benowitz NL, Callaway E, Naylor H, Herzig K. A low dose of subcutaneous nicotine improves information processing in non-smokers. Psychopharmacology (Berl) 1994;114:628–634. doi: 10.1007/BF02244994. [DOI] [PubMed] [Google Scholar]
- Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J, Rose JE. Transdermal nicotine effects on attention. Psychopharmacology (Berl) 1998;140:135–141. doi: 10.1007/s002130050750. [DOI] [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The α4β2α5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem. 2008;104:446–456. doi: 10.1111/j.1471-4159.2007.05011.x. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure-Begley TD, King NM, Collins AC, Stitzel JA, Wehner JM, Butt CM. Acetylcholine-stimulated [3H]GABA release from mouse brain synaptosomes is modulated by α4β2 and α4α5β2 nicotinic receptor subtypes. Mol Pharmacol. 2009;75:918–926. doi: 10.1124/mol.108.052274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza NR, Stolerman IP. Nicotine enhances sustained attention in the rat under specific task conditions. Psychopharmacology (Berl) 1998;138:266–274. doi: 10.1007/s002130050671. [DOI] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. α4β2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–768. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- Muir JL, Everitt BJ, Robbins TW. The cerebral cortex of the rat and visual attentional function: dissociable effects of mediofrontal, cingulate, anterior dorsolateral, and parietal cortex lesions on a five-choice serial reaction time task. Cereb Cortex. 1996;6:470–481. doi: 10.1093/cercor/6.3.470. [DOI] [PubMed] [Google Scholar]
- Mumenthaler MS, Taylor JL, O'Hara R, Yesavage JA. Influence of nicotine on simulator flight performance in non-smokers. Psychopharmacology (Berl) 1998;140:38–41. doi: 10.1007/s002130050736. [DOI] [PubMed] [Google Scholar]
- Newhouse PA, Sunderland T, Tariot PN, Blumhardt CL, Weingartner H, Mellow A, Murphy DL. Intravenous nicotine in Alzheimer's disease: a pilot study. Psychopharmacology (Berl) 1988;95:171–175. doi: 10.1007/BF00174504. [DOI] [PubMed] [Google Scholar]
- Newhouse PA, Potter A, Singh A. Effects of nicotinic stimulation on cognitive performance. Curr Opin Pharmacol. 2004;4:36–46. doi: 10.1016/j.coph.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Okonkwo OC, Crowe M, Wadley VG, Ball K. Visual attention and self-regulation of driving among older adults. Int Psychogeriatr. 2008a;20:162–173. doi: 10.1017/S104161020700539X. [DOI] [PubMed] [Google Scholar]
- Okonkwo OC, Wadley VG, Ball K, Vance DE, Crowe M. Dissociations in visual attention deficits among persons with mild cognitive impairment. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2008b;15:492–505. doi: 10.1080/13825580701844414. [DOI] [PubMed] [Google Scholar]
- Oliver YP, Ripley TL, Stephens DN. Ethanol effects on impulsivity in two mouse strains: similarities to diazepam and ketamine. Psychopharmacology (Berl) 2009;204:679–692. doi: 10.1007/s00213-009-1500-0. [DOI] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–154. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott AC, Roberts G. Smoking deprivation and cigarette reinstatement: effects upon visual attention. J Psychopharmacol. 1991;5:404–409. doi: 10.1177/026988119100500435. [DOI] [PubMed] [Google Scholar]
- Passetti F, Dalley JW, O'Connell MT, Everitt BJ, Robbins TW. Increased acetylcholine release in the rat medial prefrontal cortex during performance of a visual attentional task. Eur J Neurosci. 2000;12:3051–3058. doi: 10.1046/j.1460-9568.2000.00183.x. [DOI] [PubMed] [Google Scholar]
- Pattij T, Janssen MC, Loos M, Smit AB, Schoffelmeer AN, van Gaalen MM. Strain specificity and cholinergic modulation of visuospatial attention in three inbred mouse strains. Genes Brain Behav. 2007;6:579–587. doi: 10.1111/j.1601-183X.2006.00284.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Petersen DR, Norris KJ, Thompson JA. A comparative study of the disposition of nicotine and its metabolites in three inbred strains of mice. Drug Metab Dispos. 1984;12:725–731. [PubMed] [Google Scholar]
- Picciotto MR. Nicotine as a modulator of behavior: beyond the inverted U. Trends Pharmacol Sci. 2003;24:493–499. doi: 10.1016/S0165-6147(03)00230-X. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci. 1999;19:5228–5235. doi: 10.1523/JNEUROSCI.19-13-05228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L. Functional contributions of α5 subunit to neuronal acetylcholine receptor channels. Nature. 1996;380:347–351. doi: 10.1038/380347a0. [DOI] [PubMed] [Google Scholar]
- Rigbi A, Kanyas K, Yakir A, Greenbaum L, Pollak Y, Ben-Asher E, Lancet D, Kertzman S, Lerer B. Why do young women smoke? V. Role of direct and interactive effects of nicotinic cholinergic receptor gene variation on neurocognitive function. Genes Brain Behav. 2008;7:164–172. doi: 10.1111/j.1601-183X.2007.00329.x. [DOI] [PubMed] [Google Scholar]
- Robbins TW. The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology (Berl) 2002;163:362–380. doi: 10.1007/s00213-002-1154-7. [DOI] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Coleman RE. Arterial nicotine kinetics during cigarette smoking and intravenous nicotine administration: implications for addiction. Drug Alcohol Depend. 1999;56:99–107. doi: 10.1016/s0376-8716(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci U S A. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone NL, Saccone SF, Hinrichs AL, Stitzel JA, Duan W, Pergadia ML, Agrawal A, Breslau N, Grucza RA, Hatsukami D, Johnson EO, Madden PA, Swan GE, Wang JC, Goate AM, Rice JP, Bierut LJ. Multiple distinct risk loci for nicotine dependence identified by dense coverage of the complete family of nicotinic receptor subunit (CHRN) genes. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:453–466. doi: 10.1002/ajmg.b.30828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M. The nicotinic acetylcholine receptor subunit α5 mediates short-term effects of nicotine in vivo. Mol Pharmacol. 2003;63:1059–1066. doi: 10.1124/mol.63.5.1059. [DOI] [PubMed] [Google Scholar]
- Schlaepfer IR, Hoft NR, Collins AC, Corley RP, Hewitt JK, Hopfer CJ, Lessem JM, McQueen MB, Rhee SH, Ehringer MA. The CHRNA5/A3/B4 gene cluster variability as an important determinant of early alcohol and tobacco initiation in young adults. Biol Psychiatry. 2008;63:1039–1046. doi: 10.1016/j.biopsych.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu EC, Tyndale RF. Characterization and comparison of nicotine and cotinine metabolism in vitro and in vivo in DBA/2 and C57BL/6 mice. Mol Pharmacol. 2007;71:826–834. doi: 10.1124/mol.106.032086. [DOI] [PubMed] [Google Scholar]
- Stevens VL, Bierut LJ, Talbot JT, Wang JC, Sun J, Hinrichs AL, Thun MJ, Goate A, Calle EE. Nicotinic receptor gene variants influence susceptibility to heavy smoking. Cancer Epidemiol Biomarkers Prev. 2008;17:3517–3525. doi: 10.1158/1055-9965.EPI-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland T, Weingartner H, Cohen RM, Tariot PN, Newhouse PA, Thompson KE, Lawlor BA, Mueller EA. Low-dose oral lorazepam administration in Alzheimer subjects and age-matched controls. Psychopharmacology (Berl) 1989;99:129–133. doi: 10.1007/BF00634466. [DOI] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (α4)3 (β2)2 stoichiometry greatly exceeds that of (α4)2 (β2)3 human acetylcholine receptors. Mol Pharmacol. 2007;71:769–776. doi: 10.1124/mol.106.030445. [DOI] [PubMed] [Google Scholar]
- Thumwood CM, Hunt NH, Clark IA, Cowden WB. Breakdown of the blood-brain barrier in murine cerebral malaria. Parasitology. 1988;96:579–589. doi: 10.1017/s0031182000080203. [DOI] [PubMed] [Google Scholar]
- Tsutsui-Kimura I, Ohmura Y, Izumi T, Yamaguchi T, Yoshida T, Yoshioka M. Nicotine provokes impulsive-like action by stimulating α4β2 nicotinic acetylcholine receptors in the infralimbic, but not in the prelimbic cortex. Psychopharmacology (Berl) 2010;209:351–359. doi: 10.1007/s00213-010-1804-0. [DOI] [PubMed] [Google Scholar]
- Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW. The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (α5) in the rat central nervous system. Brain Res. 1990;526:45–53. doi: 10.1016/0006-8993(90)90248-a. [DOI] [PubMed] [Google Scholar]
- Warburton DM, Arnall C. Improvements in performance without nicotine withdrawal. Psychopharmacology. 1994;115:539–542. doi: 10.1007/BF02245578. [DOI] [PubMed] [Google Scholar]
- Weiss RB, Baker TB, Cannon DS, von Niederhausern A, Dunn DM, Matsunami N, Singh NA, Baird L, Coon H, McMahon WM, Piper ME, Fiore MC, Scholand MB, Connett JE, Kanner RE, Gahring LC, Rogers SW, Hoidal JR, Leppert MF. A candidate gene approach identifies the CHRNA5–A3-B4 region as a risk factor for age-dependent nicotine addiction. PLoS Genet. 2008;4:e1000125. doi: 10.1371/journal.pgen.1000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesnes K, Warburton DM. Effects of scopolamine and nicotine on human rapid information processing performance. Psychopharmacology (Berl) 1984;82:147–150. doi: 10.1007/BF00427761. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Expression of α5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol. 2005;481:19–30. doi: 10.1002/cne.20357. [DOI] [PubMed] [Google Scholar]
- Wisco BE. Depressive cognition: self-reference and depth of processing. Clin Psychol Rev. 2009;29:382–392. doi: 10.1016/j.cpr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Cholinergic switching within neocortical inhibitory networks. Science. 1998;281:985–988. doi: 10.1126/science.281.5379.985. [DOI] [PubMed] [Google Scholar]
- Zabala A, Eguiluz JI, Segarra R, Enjuto S, Ezcurra J, Pinto AG, Gutiérrez M. Cognitive performance and cigarette smoking in first-episode psychosis. Eur Arch Psychiatry Clin Neurosci. 2009;259:65–71. doi: 10.1007/s00406-008-0835-6. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci. 2006;26:7348–7361. doi: 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]