

Abstract
The nervous system is protected by blood barriers that use multiple systems to control extracellular solute composition, osmotic pressure, and fluid volume. In the human nervous system, misregulation of the extracellular volume poses serious health threats. Here we show that the glial cells that form the Drosophila blood-nerve barrier have a conserved molecular mechanism that regulates extracellular volume: the Serine/Threonine kinase Fray, which we previously showed is an ortholog of mammalian PASK/SPAK; and the Na-K-Cl cotransporter NCC69, which we show is an ortholog of human NKCC1. In mammals, PASK/SPAK binds to NKCC1 and regulates its activity. In Drosophila, larvae mutant for NCC69 develop a peripheral neuropathy, where fluid accumulates between glia and axons. The accumulation of fluid has no detectable impact on action potential conduction, suggesting that the role of NCC69 is to maintain volume or osmotic homeostasis. Drosophila NCC69 has kinetics similar to human NKCC1, and NKCC1 can rescue NCC69, suggesting that they function in a conserved physiological mechanism. We show that fray and NCC69 are coexpressed in nerve glia, interact in a yeast-two-hybrid assay, and have an essentially identical bulging nerve phenotype. We propose that normally functioning nerves generate extracellular solutes that are removed by NCC69 under the control of Fray. This mechanism may perform a similar role in humans, given that NKCC1 is expressed at the blood-brain barrier.
Keywords: Na-K-Cl transporter, osmoregulation, blood-nerve barrier, blood-brain barrier
Introduction
In animals as diverse as humans and Drosophila, the nervous system is isolated from the blood or hemolymph by cellular junctions that restrict the passage of solutes (reviewed in Carlson et al. 2000; Rubin and Staddon 1999). This paracellular barrier permits a tight regulation of the extracellular environment of the nervous system, with ion pumps, cotransporters, exchangers, and channels maintaining ionic homeostasis. In humans, improper ionic homeostasis in the nervous system has serious clinical consequences, including seizures, coma, and death (Somjen 2002). Following brain injury, elevated extracellular solutes cause water to accumulate, resulting in edema and the pathological rise of intracranial pressure (Iadecola 1999). Understanding the mechanisms controlling volume and ionic homeostasis is thus critical for developing rational treatments for brain injury.
The Na-K-Cl cotransporter, NKCC1, has been shown to mediate the early swelling response in a mouse model of focal ischemia in the brain (Chen et al. 2005; O’Donnell et al. 2004; Yan et al. 2003). NKCC1 is widely expressed in the nervous system, where it has been proposed to regulate the intracellular level of Cl in neurons and the swelling of glia in response to neuronal activity (Chen and Sun 2005; Delpire 2000; Simard and Nedergaard 2004). Knockdown of NKCC1 reduces the swelling response to ischemia, but its role in vivo under normal conditions is incompletely understood. Analysis of mouse NKCC1 mutant phenotypes is hampered by the complex anatomy of the nervous system and the masking of behavioral changes caused by the disruption of the vestibular system (Delpire et al. 1999; Dixon et al. 1999; Flagella et al. 1999).
Glial cells have an important role in regulating extracellular ionic composition and volume (Newman 2005; Somjen 2002; Wang and Bordey 2008), and may also be critical for blood-brain barrier function. Oligodendroctyes associated with myelinated axons are connected to astrocytes by gap junctions (Orthmann-Murphy et al. 2008), and may function as a syncitial unit to move K from the axons to blood vessels by a process known as “K siphoning” (Newman 1986). Despite this attractive hypothesis, the full range of glial activities at the blood-brain barrier remain incompletely understood.
Drosophila is a useful model system to elucidate the molecular, cellular and physiological mechanisms in the nervous system. The larval nervous system is far simpler than the mammalian. In larvae a single glial cell layer surrounding the nervous system forms the paracellular barrier that restricts the flow of ions (Baumgartner et al. 1996; Stork et al. 2008). This anatomy allows easy analysis of phenotypes associated with ion homeostasis and volume regulation. By contrast, in humans and mice the analogous barrier is associated with the blood microvessels that penetrate deep within the central and peripheral nervous systems (Zlokovic 2008). Despite these differences, the nervous systems of these organisms face similar physiological challenges. Many of the ion channels, exchangers, and transporters of the Drosophila nervous system are evolutionarily conserved, and have orthologs in humans (Rubin 2001). Furthermore, molecules that form paracellular barriers are conserved (Banerjee et al. 2006), suggesting that many molecular mechanisms that mediate ionic and volume homeostasis are also conserved. We show here that fly glia have a molecular mechanism to regulate extracellular volume in larval nerves that is directly related to NKCC1 function in mammals.
One of us (B. F.) has demonstrated that NKCC1 activity is regulated by rat PASK, known as SPAK in humans and mice (Dowd and Forbush 2003). We previously demonstrated that PASK is the ortholog of Drosophila Fray, and that loss of function of fray in nerve glia results in the accumulation of extracellular fluid between axons and glia (Leiserson et al. 2000). Here, we show that reducing the activity of the cation chloride cotransporter, NCC69, in larval nerve glia causes the same phenotype. NCC69 and Fray function in the subperineurial glia (SPG), the cells that provide the paracellular blood-nerve barrier. The fluid in NCC69 mutant nerves supports normal action potential activity, suggesting that NCC69 normally regulates extracellular volume by removing solutes from the extracellular space, with little effect on the extracellular ion composition. Measurement of the kinetic parameters of NCC69 in vitro and rescue experiments in vivo demonstrate that NCC69 is the ortholog of human NKCC1. These results suggest that under normal conditions, nerve glia remove excess extracellular solutes to maintain volume and osmotic homeostasis, using Na-K-Cl cotransport mediated by the highly conserved Fray-NCC69 pathway.
Materials and Methods
Genetics
The genotypes of the stocks used in this study are listed in Table S1. The two NCC69 alleles referred to in this study were made by mobilizing the P-element line EY168 (Bellen et al. 2004), in which the P-element had inserted 107 bp 5′ of the transcription start site. We screened 112 independent lines by PCR to map excision events. For the r1 and r2 alleles, a segment that spanned the breakpoints was amplified by PCR (primer pairs NCC69-U1..L1 and U8..L1, respectively), sequenced, and the location of the breakpoints determined. The r1 allele has a 537 bp deletion (3L:12,384,465..12,385,007; release r5.22) and the r2 allele, a 8,743 bp deletion (3L:12,375,902..12,384,644).
Molecular Biology
A list of primers is given in Table S2. A cDNA clone of NCC69, GH27027, was obtained from BACPAC Resources and was used for in situ hybridization, yeast-two-hybrid experiments, cell transformation, and transgenics. The human NKCC1 clone was previously reported (Payne et al. 1995). Sequencing was performed by the Keck Sequence Facility and the DNA Analysis Facility on Science Hill (Yale University). Nucleotide queries were performed on the NCBI BLAST server. Protein sequences alignments and distance relationships were performed using ClustalW (Larkin et al. 2007).
Expression Constructs
A list of expression constructs is given in Table S3. We used a three way ligation strategy to clone HA:NCC69 into pIZT/V5-His (Invitrogen) for expression in cell culture (pIHANCC69) and pP{UAST} (Brand and Perrimon 1993) for expression in flies (pUHANCC69). First, the insert from GH27027 was amplified with primers HANCC69-EcoRI and 4357RA-703, then digested with EcoRI and XhoI. This fragment was ligated to the appropriate vector, which had been digested with EcoRI and XbaI, along with a 3.3kb XhoI-XbaI fragment from GH27027. For the human NKCC1 rescue construct, we used the Gateway cloning system (Invitrogen) following the manufacturer’s instructions, to clone the human NKCC1 cDNA into the pTHW vector (Drosophila Gateway Collection, http://www.ciwemb.edu/labs/murphy/Gateway%20vectors.html). Human NKCC1 was amplified using primers TEF-TOPO1 and TEF-L1, then cloned into pENTR/D to make an entry clone, which was used in the topoisomerase reaction to make pTH-TEF, a plasmid that contains P{UAS-HA:NKCC1}.
Yeast-two-hybrid
A list of bait and prey constructs is shown in Tables S4 and S5. PCR-amplified fragments of Fray (p4624R6, Leiserson et al. 2000) and NCC69 were cloned into pGBT9 (Bartel et al. 1993) and pGADGH (Clontech). The PJ69-4A yeast strain (James et al. 1996) was transformed using the LiAc method as described by (Gietz and Woods 2001), with minor modifications (see Supplemental Material). Clones that received both bait and prey plasmids were streaked onto plates lacking Trp, Leu, and His. Growth was assessed after two days at 30C.
P-element-mediated transformation and rescue
For the NCC69 construct (pUHANCC69), we made transgenic lines as described (Leiserson et al. 2000) and obtained six independent insertions. Eight transgenic lines with the human NKCC1 construct (pUHANKCC1) were obtained from BestGene (Chino Hills, CA).
Histology
Immunostaining was performed as described (Johansen et al. 1989) and analyzed by digital optical microscopy (Halpern et al. 1991) or confocal microscopy (Bio-Rad 1024 and Zeiss LSM510). Image processing was performed with Adobe Photoshop, Zeiss Zen, Bitplane Imaris, and NIH ImageJ software. Primary antibodies used were: rabbit-anti-HRP (1:250; Cappel), goat-anti-HRP (1:200; Cappel), 8D12 mouse-anti-Repo (1:10; Alfonso and Jones 2002) and 16B12 mouse-anti-HA.11 (1:2,000; Covance). The 8D12 monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
Secondary antibodies used were HRP-donkey-anti-goat (1:200, Jackson ImmunoResearch Laboratories); and the following Alexa antibodies (1:1,000; Molecular Probes): 488-goat-anti-mouse, 568-goat anti-mouse, 488-goat-anti-rabbit (1:500 for the in situ double-labeling), 568-goat-anti-rabbit, 568-donkey anti-goat, and 488-donkey-anti-mouse.
Transmission electron microscopy
Larval fillets were fixed at RT for 1 hour in 4% glutaraldehyde in 0.1M phosphate buffer, pH7.4. The fillets were washed in PBS, stained for 1 hour in 1% osmium tetroxide, dehydrated and embedded in Epon, then silver sections were cut, stained 10 min in 3% uranyl acetate followed by 3 min in 0.3% lead citrate.
In situ hybridization
This was performed with digoxigenin-labeled probes as described in Tautz and Pfeifle (1989), with modifications described in Doe et al. (1991) and the Supplemental Material. Single stranded probes were synthesized using a single primer in a PCR reaction. Prior to hybridization, the probes were boiled for 30 min. Following hybridization, the embryos were stained for at least one hour with alkaline-phosphatase-conjugated sheep-anti-DIG antibodies (Roche), which had been preabsorbed with embryos, at a final dilution of 1:2000. The staining was visualized with NBT/BCIP or Fast Red (Biomeda).
Cell culture and flux assays
The flux experiments were done with Drosophila SL2 cells (kindly provided by the Artavanis-Tsakonas lab, Harvard Medical School), an adherent derivative of S2. They were grown at RT in SFM (Gibco) supplemented with 2% fetal bovine serum (FBS, heat-inactivated, Gibco) or Schneider’s medium (Gibco) supplemented with 10% FBS (SS). In a 12-well plate, 2.5×10^5 cells were transfected with 1 μg plasmid (pIHANCC69 or pIZT, the vector control) using Cellfectin (Invitrogen) according to the manufacturer’s guidelines. The cells were expanded for ca. 2 weeks, then selected in SFM containing 100 to 800 μg/ml Zeocin (Invitrogen). After three months, the cells were adapted to SSZ400 (SS supplemented with 400 μg/ml Zeocin).
For the flux experiments, cells were grown in shaking cultures, then plated in 96 well plates at 10^5 cells/well, and grown overnight. The 86Rb flux was assayed robotically as described (Darman and Forbush 2002), with the modifications described in the Supplemental Material. The Km and Ki values were determined for each experiment by fitting the data to single- or double-site binding using a non-linear least-squares approximation (Solver plug-in of Excel).
Electrophysiology
Third instar larvae were filleted and mounted on slides in physiological saline (150 mM Na, 5 mM K, 4 mM HCO3, 6 mM Mg, 1 mM Ca; 5mM TES; 50mM sucrose, pH 7.2) as previously described (Halpern et al. 1991). Peripheral nerves were stimulated with suction electrodes (6–8 μm wide opening) placed at least 500 micrometers from the bodywall musculature. Evoked excitatory junctional currents (EJCs) were recorded using two electrode voltage clamp (Dagan 8500) with pClamp 9.0 software, as previously described (White et al. 2001). Spontaneous neuromuscular activity was monitored both by two electrode voltage clamp and by single electrode current clamp.
Quantitation of nerve phenotype
Five nerve widths were taken from ca. 220 μm nerve segments of each nerve overlying the middle of bodywall segment A2 to divide the nerve segment into four equal lengths. For the analysis shown in Figures 6A and 6B, the nerve measurements were binned in 0.5 μm intervals, and a normalized cumulative distribution determined. Statistics of the Kolmogorov-Smirnov (K-S) type were used to test whether the difference between the distributions was statistically significant. The critical values were calculated using the large sample approximation for quantiles of the Smirnov test statistic for two samples of different size (Conover 1999). To test for significant differences between the quantiles shown in Figure 6B, we used the “quantile test,” a variation of the median test (Conover 1999). For this analysis, the quantiles were calculated without binning.
Figure 6.

Quantitation of the nerve phenotype. A: Cumulative frequency graphs of nerve widths. Curves from animals with a higher proportion of large nerve widths are shifted to the right; those with a lower proportion are shifted to the left. The rightmost curve is for NCC69. The fly rescue (expressing HA:NCC69 in the nerve glia in NCC69 mutants) shifts the curve leftward to overlap the wild type curve. The human rescue (expressing HA:NKCC1 in the nerve glia of NCC69 mutants) shifts the curve leftward as well, but for the most part does not overlap the wild type curve. The NCC69 distribution is significantly different from the others (p<0.005; see Results and Table S6). B: Percentiles from the distributions shown in A. The 50th, 75th, 80th, 85th and 90th percentiles are shown. In each case, NCC69 has a value that shows a statistically significant difference from the values of the other genotypes (p<10−4; see Results and Table S7). C: Average cross-section area of the nerves from the genotypes shown in A. The average cross-section area from NCC69 is significantly larger than the other genotypes (one-tail t test p<0.005; Table 3).
For Figure 6C, the width measurements from each nerve segment were treated as an ordered quintuplet (d1, d2, d3, d4, d5), that was used to estimate the average cross-sectional area:
| (Eq 1) |
The value A gives the cross-sectional area of a cylinder that has the same volume as the estimated volume of the nerve segment (See Supplemental Material). To arrive at the mean area values shown in Figure 6C and Table 3, we averaged all the A values for each animal, then took the mean of those averages. Standard Student’s t-tests were used to determine the p-values.
Table 3.
Mean cross-sectional areas of NCC69 and rescues
| Genotype | mean area A | std err | n | width |
|---|---|---|---|---|
| CS (wild type) | 52.4 | 3.7 | 10 | 8.2 |
| NCC69r2 | 106.4 | 8.5 | 5 | 11.6 |
| Fly rescue | 50.8 | 1.7 | 4 | 8.0 |
| Human rescue | 66.9 | 3.8 | 4 | 9.2 |
For each animal, the average area, A (μm2), was calculated from 10 to 12 nerve segments as described in Experimental Procedures. These values of A were then averaged for each animal. The mean of those averages is what is presented in column 2, with the corresponding standard error in column 3. The number of animals (n) is shown in column 4. Column 5 gives the corresponding nerve width (μm), derived from the value in column 2.
Results
Cation chloride cotransporters (CCCs) mediate the electroneutral cotransport across the plasma membrane of Cl, along with K (KCCs), Na (NCCs) or Na and K (NKCCs). The CCC family (also known as SLC12) has four major branches (Fig. 1A; reviewed in Hebert et al. 2004; Pullikuth et al. 2003). Two branches contain proteins that transport Na, K, and Cl: the sodium-dependent potassium chloride cotransporter group, N(K)CC; and the sodium-independent potassium chloride cotransporter group, KCC. Humans have four KCC genes with multiple alternatively spliced isoforms, while flies have a single KCC gene (Hekmat-Scafe et al. 2006).
Figure 1.

NCC69 is a member of the Drosophila N(K)CC family. A: A cladogram showing the relationship between the Drosophila (red) and human (black) cation chloride cotransporters (CCCs). Humans have three Na-(K)-Cl cotransporters: one Na-Cl and two Na-K-Cl cotransporters, denoted NCC and NKCC, respectively. Drosophila has two related cotransporters, denoted NCC69 and NCC83, but the phylogenetic analysis fails to reveal to which class (NCC or NKCC) these proteins belong. B: A model of human NKCC1 showing homology to NCC69. The color-coding shows the similarity to NCC69. The residues annotated with P’s are conserved phosphorylation sites that are important for NKCC1 activation in the shark (Darman and Forbush 2002; Vitari et al. 2006). C: Map of the NCC69 genomic region. Two transcripts are predicted, which encode the same protein of 1,171 amino acid residues. The deleted regions are shown of r1 and r2, two alleles that were used in this study, which span 537 and 8,743 bp, respectively.
In humans, the N(K)CC group comprises two NKCCs, NKCC1 and NKCC2, and one Na-Cl cotransporter, NCC. Both NKCC2 and NCC are associated with the kidney. NKCC1 has widespread expression, including the brain and peripheral nerves (Hebert et al. 2004). Flies have two members of the N(K)CC group, NCC69 and NCC83 (Fig. 1A), that diverged too early in the N(K)CC lineage to indicate whether they are K-dependent transporters. Here we focus on NCC69, which has extensive primary and secondary amino acid sequence homology to human NKCC1 (Fig. 1B), and which we show below is expressed in nerve glia.
NCC69 is expressed and required in nerve glia
To look for a possible role of NCC69 in larval nerves, we examined whether the gene is expressed in subperineurial glia, which form pleated septate junctions that provide a paracellular barrier (Stork et al. 2008). These cells are evident in the embryo by their expression of the glial transcription factor Repo, and by their close association with nerves (Fig. 2A, B, and C). Tissue in situ hybridization experiments revealed that these glia express NCC69 transcripts (Fig 2D and E). The only other prominent expression of NCC69 was in the dorsal median cells of the ventral ganglion (not shown).
Figure 2.

NCC69 is expressed and required in subperineurial glia. A: Drosophila whole embryo, stage 16, labeled with anti-HRP to reveal neurons and neuronal projections (green) and anti-Repo to label glial nuclei (magenta). At this magnification, the nerves (arrowheads) can be easily identified linking the CNS to the periphery in a segmentally repeated and bilaterally symmetric pattern. Bar: 50 microns. B, C: Higher magnification of region boxed in A showing the double label B and the anti-Repo (magenta) channel C. At this magnification, the labeling reveals individual nuclei (arrows) of glial cells that are closely associated with the axons. These glial cells have been shown to express fray and Gliotactin, and will give rise to the subperineurial glia in larval nerves. Bar: 20 microns. D, E: Drosophila whole embryo, stage 16, labeled with anti-HRP (green) and probed with NCC69 anti-sense DNA (magenta). The nerve glia express the NCC69 mRNA (arrowheads), visible in the double-labeled image D and the NCC69 (magenta) channel E. Bar: 20 microns. F, G: Fillet preps from 3rd instar larvae, stained with DAB anti-HRP to label the nervous system. Wild type F nerves appear uniform in width (arrowheads). By contrast, NCC69 G mutant nerves have localized bulges and swellings (arrowheads). Fillet preps from 3rd instar larvae expressing an RNAi construct targeted against NCC69 in neurons H or subperineurial glia I. When the RNAi construct is expressed in neurons, the nerves appear wild type (compare with F). Expression of the RNAi construct in nerve glia causes bulges in the nerves G. Genotypes: F, CS. G, NCC69[r1]. H, w, C155-GAL4/+; UAS-RNAi(NCC69)/+. I, w; Gli-GAL4/+; UAS-RNAi(NCC69). F–I are to the same scale (Bar = 20 microns).
We generated two mutant alleles of NCC69 and examined the larval abdominal nerves in homozygotes. The alleles have different deletions, but both span the 5′ exon of the NCC69-RA transcript (Fig. 1C). The abdominal nerves of these mutants have localized swellings or bulges (Fig. 2G). The phenotype is completely penetrant, with bulges appearing along the lengths of the nerves in all the animals examined (n>50), and which could occur in any of the abdominal nerves. NCC69 mutants survive to adulthood with no other apparent abnormality. Placing either allele in combination with a deficiency that spans NCC69 does not make the phenotype more severe, suggesting that these alleles are genetic nulls, at least with respect to the nerve phenotype.
Both the expression pattern and the mutant phenotype of NCC69 suggest that it is required in subperineurial glia. To test this we expressed two independent NCC69-RNAi constructs in separate experiments in subperineurial glia. Knockdown of NCC69 in these glia caused bulges similar to those seen in the NCC69 mutants (Fig. 2I). By contrast, knockdown of NCC69 in adjacent neurons had no effect (Fig. 2H). Expressing the NCC69 cDNA in subperineurial cells of NCC69 mutants rescues the bulging phenotype (see below and Fig. 5B), while expression in neurons has no effect (data not shown). These experiments demonstrate that the action of NCC69 is required in subperineurial nerve glia—the same cells in which we observe NCC69 expression.
Figure 5.
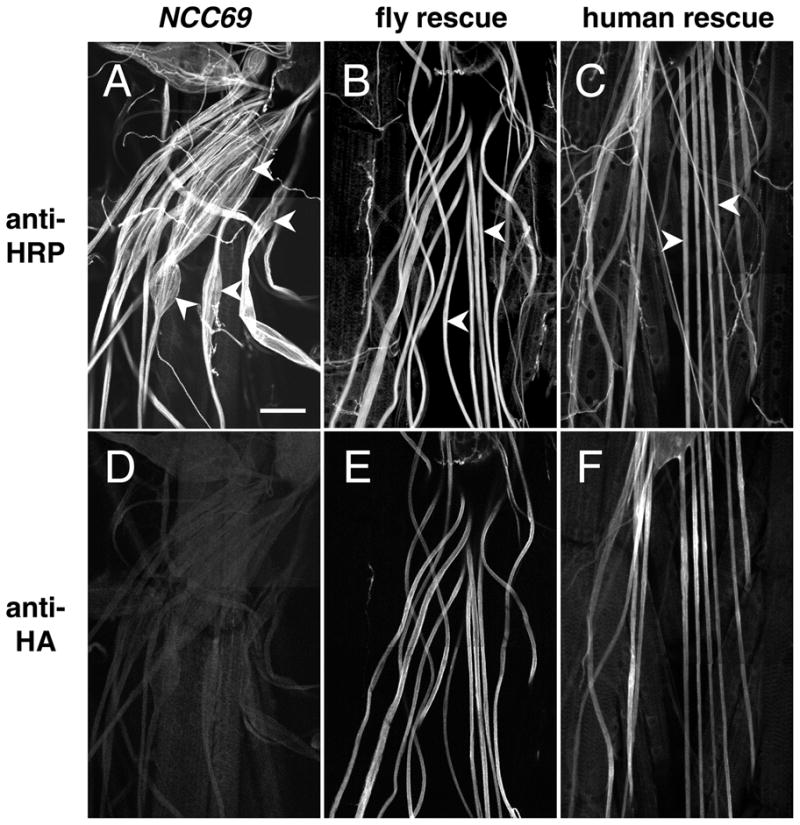
Human NKCC1 can substitute for NCC69 in vivo. 3rd instar fillets stained with anti-HRP, A–C or anti-HA D–F. A, D: NCC69. Many nerve bulges are visible (arrowheads in A), corresponding to the accumulation of extracellular fluid between glia and axons. Staining of this prep with anti-HA D serves as a negative control for the rescue experiments. Genotype: w; NCC69r2. B, E: Fly rescue. Expressing the NCC69 cDNA in subperineurial glia in NCC69 mutants reduces the nerve swelling considerably (arrowheads in B). Staining with anti-HA E reveals ample expression of the HA:NCC69 transgene in the nerve. Genotype: w; Gli-GAL4; NCC69r2, UAS-HA:NCC69. C, F: Human rescue. Expressing the human NKCC1 cDNA in subperineurial glia in NCC69 mutants also reduces nerve swelling (arrowheads in C), demonstrating that these two genes encode functional orthologs. Genotype: w; Gli-GAL4; UAS-HA:NKCC1 NCC69r1. Bar: 50 microns; A–F are all to the same scale.
We next examined whether the peripheral nerve edema of NCC69 null mutants affects action potential activity and nerve conduction. In NCC69 mutant larvae, we did not observe phenotypes associated with mutations that affect neuronal activity, such as lack of coordination when crawling, loss of nociception, or temperature-sensitive paralysis. Mutant larvae developed at a normal pace. Thus, although axons in NCC69 mutants traversed abnormal fluid-filled extracellular environments, the effect on nerve conduction appeared to be minor.
It remained possible, however, that a phenotype might be revealed under conditions of elevated action potential activity. This was examined both electrophysiologically and through genetic tests. Both mutant and wild type nerves of 3rd instar larvae were subjected to trains of tetanic stimuli of increasing frequency (50 pulses at 10, 20, 50, and 100 Hz), and the ability of the motoneuron axons to conduct the pulses was monitored by recording evoked excitatory junction currents (EJCs) from the innervated muscle fibers (Fig. 3 and Table 1). The peripheral nerves were stimulated at least 500 microns from where they branch in the periphery. This approach ensured that action potentials evoked by the stimulus would encounter nerve bulges of NCC69 mutants as they traveled toward the nerve terminal. Both the wild type and mutant nerves showed little evidence of action potential failure, even at the highest stimulation frequencies (at 100 Hz, p≈0.99; Table 1).
Figure 3. Evoked action potential activity is unaffected in NCC69 mutants.

Excitatory junctional currents (EJCs) were recorded from specific ventral longitudinal muscle fibers of a wild type (CS) and an NCC69r2 larva, in response to trains of stimuli at 10 and 50 Hz evoked by shocking the nerve at a distance of at least 500 micrometers from the innervated segment. This ensured that in NCC69 mutants, the resulting action potentials would encounter bulges as they traveled toward the nerve terminal. The failure of an action potential to arrive at the nerve terminal results in a missing EJC (asterisks above the wild type trace at 50 Hz). The all-or-nothing response indicates that the EJCs are a reliable indicator of action potential propagation. The shock artifacts, which appeared as upward deflections on the traces, have been clipped for easier viewing.
Table 1.
Failure rates of EJCs in wild type and NCC69 mutants
| frequency (Hz) | ||||
|---|---|---|---|---|
| Genotype | 10 | 20 | 50 | 100 |
| CS (wild type) | 0 (0) | 1.1 (0.9) | 9.5 (3.2) | 23.1 (7.2) |
| NCC69r2 | 0 (0) | 0 (0) | 0 (0) | 2.9 (3.2) |
| p-value (t-test) | NA | 0.886 | 0.995 | 0.989 |
Nerves were subjected to trains of 50 shocks at the frequencies shown, and currents recorded from various muscles (MFs 6, 7, 12, or 13) under voltage clamp. Failures of the muscle to respond were tallied. Mean numbers of failures (and standard error in parentheses) are given for the two genotypes. For CS, n=12 muscles except at 100 Hz where n=8. For NCC69, n=7 muscles, except at 100 Hz where n=6. The p-values result from testing the hypothesis that NCC69 nerves have more failures than wild type.
Because an NCC69 nerve conduction phenotype might not be evident with the acute excitation described above, we used the eag Sh (ether-a-go-go Shaker) mutation combination to examine the effects of chronic hyperexcitation. In eag Sh mutants, multiple voltage-gated K channels are affected, resulting in elevated action potential activity throughout larval development (Budnik et al. 1990; Ganetzky and Wu 1983; Zhong et al. 1992; Zhong and Wu 2004). As measured by the frequency of spontaneous EJCs, the endogenous activity of eag Sh; NCC69 larvae (4.2 to 12.2 Hz, n=5 muscles) was comparable to eag Sh; + (0.3 to 12.1 Hz, n=22). We found no evidence that NCC69 nerve bulges have any effect on action potential function and nerve conduction, when challenged by either acute or chronic hyperstimulation.
Finally, we used a sensitive genetic test to detect reduced action potential function. NCC69 mutants crawl normally at 37° C. By contrast, mutations that reduce voltage-gated Na channels typically have a temperature-sensitive paralysis phenotype. For example, the parats1 mutation, that affects the alpha subunit of the Na(v)1 voltage gated Na channel, results in a characteristic 37° C paralysis in larvae (Ganetzky 1984). The mutation provides a sensitized genetic background useful for identifying mutations that affect action potential function. When combined with a second mutation that reduces membrane excitability (such as mlenap-ts1), the double mutant paralyzes at either a lower temperature, or dies (Ganetzky 1984). We found that the NCC69 mutation did not enhance the severity of the parats1 phenotype: parats1; NCC69r2 double mutant larvae paralyzed at the same temperature as parats1 alone (37° C, n>10 animals). At 29° C, a temperature at which parats1 larvae are somewhat uncoordinated, larvae mutant for both parats1 and NCC69r2 appear similar to larvae mutant for parats1 alone (n=7 animals, each). Thus, the NCC69 bulges did not further reduce neural activity. In all these assays of nerve function— electrophysiology, genetic and behavioral—NCC69 bulges had no detectable impact.
NCC69 is a Na-K-Cl cotransporter
We used in vitro cotransporter flux assays to address whether NCC69 functions as a Na-K-Cl cotransporter analogous to mammalian NKCC1. Our assay was modeled from studies of human and shark cotransporters expressed in a human embryonic kidney cell line (Darman et al. 2001; Payne et al. 1995; Xu et al. 1994). We used a Drosophila cell line, SL2, expressing an HA-tagged NCC69 construct to assay ion transport (Fig. 4). A stable line transfected with HA-tagged NCC69 shows membrane expression (Fig. 4A).
Figure 4.

In vitro analysis of NCC69 cotransporter function. A: Anti-HA staining of 1B4, a stable SL2 cell line transfected with a construct to express HA:NCC69. A high proportion (ca. 85%) of the cells express HA:NCC69 at varying levels. The construct is detected at the cell membrane (inset), as well as in intracellular compartments. Bar: 10 microns (inset, 5 microns).
B: Assays of 86Rb flux in SL2 transfected cell lines expressing HA:NCC69 versus IZT (vector alone). Cell lines were subjected to four different “activation” conditions: isotonic (control), hypertonic, hypotonic, and 0 Cl isotonic media. In the three activation conditions, the HA:NCC69-transfected cell line responded with a greater 86Rb flux than the control. The flux was inhibited by bumetanide, a compound that inhibits cotransporters from humans and other species. C–F: Dependence of 86Rb flux in HA:NCC69 cells on bumetanide C, Na D, Rb E, and Cl F concentrations. The results are shown for a representative experiment with individual data points and a curve fit using a non-linear least squares algorithm. For Na and Rb, the data best fit using a model that assumes a single ligand-binding site; for Cl, the best fit is consistent with two ligand-binding sites. The data points shown as black diamonds with error bars represent the average of four replicates with the standard error about the mean. Where the error bars are not visible, the standard error is smaller than the symbols.
SL2 cotransporter assays indicate that NCC69 is a Na-K-Cl cotransporter. To assay for functional NCC69, we treated cells with a variety of “activation” conditions, then returned the cells to normal isotonic saline containing 86Rb, and assayed how much 86Rb entered the cell (86Rb is a convenient radioisotope congener of K). Transport is activated when SL2 cells experience a change in osmolarity or a change in Cl concentration. In all conditions tested, the cells transfected with HA-NCC69 yielded a higher 86Rb flux than vector controls (Fig. 4B). The flux is largely abolished by the loop diuretic, bumetanide (Fig. 4B, rightmost category), an inhibitor of mammalian and shark CCCs (Haas and Forbush 2000). The flux varies inversely with increasing concentrations of bumetanide (Fig 4C), consistent with bumetanide acting as a non-competitive inhibitor.
A key feature of cotransporters is the dependence of the transport (flux) of one ion species on the concentrations of other ions. For NCC69, the transport of Rb depends on the concentrations of Na (Fig. 4D), Rb (Fig. 4E), and Cl (Fig. 4F). NCC69 displays kinetics consistent with the cotransport of one Na, one K, and two Cl ions. From the dependence relationship between the flux and the ion concentrations, we calculated the apparent affinity constants (Km) of the cotransporter for the different ion species. Similarly, from the varying bumetanide concentrations, we calculated its inhibition affinity Ki. These values are presented in Table 2, along with published values for human NKCC1. The Km values for the three ions are remarkably similar to the corresponding Km values of human NKCC1. The Ki values show that NCC69 has a ten-fold greater sensitivity to bumetanide over NKCC1.
Table 2.
Kinetic parameters of NCC69
| Km (mM) | Ki (μM) |
|||
|---|---|---|---|---|
| Na | Rb | Cl | Bu | |
| Dros NCC69 | 7.44 (0.66) | 1.48 (0.16) | 30.32 (1.85) | 0.049 (0.007) |
| Human NKCC1 | 14.57 (1.24) | 1.95 (0.09) | 31.25 (0.79) | 0.69 (0.13) |
The kinetic parameters for NCC69 were calculated from data represented in Figure 4. The parameters were calculated using a non-linear, least squares fit algorithm (see Materials and Methods). The mean from a minimum of six experiments is shown with the standard error about the mean shown in parentheses. The parameters for human NKCC1 are from published data (Isenring et al. 1998), and are presented here for comparison.
Human NKCC1 can substitute for Drosophila NCC69 in vivo
Expression of either Drosophila NCC69 or human NKCC1 in subperineurial glia rescued the NCC69 bulging nerve phenotype (Fig. 5A–C). The expression of the HA-tagged constructs of either fly NCC69 (Fig. 5E) or human NKCC1 (Fig. 5F) was detected with anti-HA antibody. The human construct expressed less well than the fly. This difference in expression may account for the fact that HA:NCC69 produced a greater degree of rescue than HA:NKCC1 (see below).
A quantitative examination of the degree of nerve bulging confirms the rescue of the NCC69 mutant phenotype. The nerve width distribution for NCC69 is shifted toward larger values compared to the wild type control (Fig. 6A). The graph of the fly NCC69 rescue coincides with the wild type control, indicating full rescue, while the human NKCC1 rescue is in the middle, indicating partial rescue. Comparisons of selected percentiles (Fig. 6B) and average cross-sectional areas (Fig. 6C) for the four genotypes show a similar trend. The average cross-section area of NCC69 nerves measured over twice normal (p<0.005 to reject the hypothesis H0 that NCC69 is the same as wild type or either rescue), while the human rescue gave a value about one quarter above normal (Table 3; p=0.02 to reject the hypothesis H0 that the NKCC1 rescue and wild type are the same). Kolmogorov-Smirnov tests on the distributions shown in Fig. 6A (see Table S6 for p-values) and quantile tests for the data shown in Fig. 6B (see Table S7 for p-values) gave similar results. While the NKCC1 rescue deviates significantly from wild type, it nevertheless shows a statistically significant rescue of the NCC69 mutant phenotype, demonstrating that the two molecules perform homologous functions in vivo.
NCC69 and Fray function to regulate extracellular volume in larval nerves
Loss of function of the Ser/Thr kinase Fray causes bulging nerves, similar to the NCC69 phenotype. Like NCC69, we previously showed that fray is expressed in subperineurial glia, and is required in these cells for normal nerve morphology. We further showed that rat PASK (known in mouse and humans as SPAK) can rescue the fray mutant phenotype, demonstrating that Fray and PASK/SPAK are orthologs (Leiserson et al. 2000). There is evidence that the mammalian orthologs of NCC69 and Fray, NKCC1 and PASK/SPAK, bind to one another, and that NKCC1 cotransport is activated by PASK/SPAK. (Dowd and Forbush 2003; Piechotta et al. 2003; Piechotta et al. 2002). Given the strong molecular and functional homologies of these mammalian molecules in flies, we suspected analagous regulation could be occurring in Drosophila. To look for an interaction between NCC69 and Fray, we used the yeast-two-hybrid assay. Portions of the Fray protein were assayed against the N-termini of the three putative Drosophila CCCs: KCC, NCC69, and NCC83. Of the three, NCC69 showed an interaction (Fig. 7A). This was specific to the C-terminal PF2 domain of Fray, a conserved region among Fray orthologs that was found to interact with NKCC1 in mice (Piechotta et al. 2002). Neither the N-terminal catalytic domain nor the PF1 domains of Fray displayed any interactions with NCC69.
Figure 7.

NCC69 and Fray interact by yeast-two-hybrid assay and have strikingly similar mutant nerve phenotypes. A: Yeast-two-hybrid interactions between NCC69 and Fray. B–D: Transmission electron micrographs of nerves from 3rd instar larvae. In wild type B, axons (“A”) and glial cell processes (“G” and arrowheads) are closely associated, with little extracellular space. By contrast, in NCC69 C and fray D mutant nerve bulges, axons and glia may be separated by a large amount of extracellular space (“E”). Bar in D: 1 micron; B–D are to the same scale. Genotypes: B, CS. C, w; NCC69r2. D, ry frayr1.
As a test of whether Fray positively regulates NCC69 activity in vivo, we made a fray NCC69 double mutant chromosome to test for any additive effects in the nerve phenotype. If Fray is the only kinase that upregulates NCC69, double mutants would have a nerve phenotype that is comparable to that of either mutant alone. Double mutant larvae die before the 3rd instar precluding quantitation of the nerve phenotype comparable to that of the single mutants, presented above. However, the shift in lethal phase shows an additive effect of the lethal phenotype. This effect likely results from a function outside the nerves, as fray has vital functions, and bulging nerves in and of themselves have little effect on viability (this work; and Leiserson et al. 2000).
Another approach to test whether Fray positively regulates NCC69 activity in vivo is to compare the mutant nerve phenotypes of the two genes—they would be expected to be very similar. Indeed, by light microscopy, the nerve phenotype of fray and NCC69 look very similar. Bulging nerves, however, can result from the accumulation of fluid in different compartments. In fray mutants, the accumulation occurs in the extracellular space, between axons and glia (Leiserson et al. 2000). There are other mutants, such as inebriated, in which perineurial glia become abnormally thick (Yager et al. 2001). To see which compartment is abnormally large in NCC69 larval nerves, we examined thin sections by TEM (Fig. 7C). From this analysis, it is clear that fluid accumulates in the extracellular space between glia and axons. The ultrastructure of this phenotype is strikingly similar to fray (compare Fig. 7C with D), suggesting these two genes function in the same cellular process.
Discussion
NCC69 mutant larvae have a peripheral neuropathy similar to fray, with swollen nerves in which fluid accumulates between axons and surrounding glia. Our data show that the nerve bulges in NCC69 mutants arise from the loss of ion cotransport. The morphological and ultrastructural similarity of the fray and NCC69 phenotypes suggests that these genes share similar cellular functions. Both genes are expressed in subperineurial glia, and expression in these glia is necessary and sufficient to rescue the mutant phenotype (Figs. 2 and 5; Leiserson et al. 2000). In flux assays, NCC69 functions as a Na-K-Cl cotransporter, with biochemical characteristics remarkably similar to human NKCC1. Despite several million years in which to diverge, human NKCC1 has kinetic behavior similar to NCC69 and can substitute for NCC69 function in larval glia, showing they perform the same molecular function and are orthologs.
We previously demonstrated that rat PASK and Fray are orthologs. In mammals, it has been shown that PASK can bind and regulate NKCC1 (Dowd and Forbush 2003; Piechotta et al. 2003). Our data shows that the C-terminus of Fray can physically interact with the N-terminus of NCC69 in a yeast-two-hybrid assay, similar to the interaction seen between mammalian homologs of Fray and CCCs (Piechotta et al. 2002). We extend the findings from mammals by showing that in flies, the two molecules function in the same cellular process in vivo: they are both required in subperineurial glia, and loss of function of either molecule in these glia causes the accumulation of fluid in the extracellular compartment of the nerves.
In NCC69 mutants, we did not observe any of the changes associated with altered neural activity, suggesting that the extracellular fluid that accumulates in NCC69 mutants nevertheless supports normal action potential activity. In Drosophila, mutations that reduce electrical activity commonly causes temperature-sensitive paralysis, and mutation combinations often display an enhanced phenotype, such as a lowered temperature of paralysis or lethality (Budnik et al. 1990; Ganetzky and Wu 1986). The NCC69 mutation did not show these properties. We found that NCC69 motoneurons functioned normally in electrophysiology assays and the muscle was able to respond faithfully to trains of stimuli of up to 100 Hz. Even when NCC69 mutant nerves experienced hyperactivity throughout larval life, as occurs in the mutant combination eag Sh; NCC69, there was no evidence of any failure of motoneurons to reliably transmit action potentials. Similarly, fray mutant larvae failed to show any evidence of reduced neural activity either in their behavior or in recordings from eag Sh; fray mutants (our unpublished observations).
Recently, Sun et al. (2010) reported a survey of the expression and function of Drosophila SLC12 gene family members, including NCC69 (referred to as CG4357). Although they were unable to ascribe a cellular function to NCC69 from their expression and mutation analysis, they did show that it functioned as a Na-K-Cl cotransporter in a heterologous cell line.
A model for NCC69 function in Drosophila nerve glia
Figure 8 shows a working model of ion fluxes in Drosophila nerves that accounts for the bulging nerve phenotype seen in NCC69 mutants. Action potentials leave increased K in the extracellular compartment. The Na/K ATPase moves three Na ions out of the cell for every two K ions it moves into the cell. It is thus both electrogenic (creating net negative charge inside) and osmogenic (increasing the osmotic strength outside). The septate junctions prevent free diffusion of ions to relieve the osmotic stress. We propose that the role of NCC69 is to move ions (and water) into the SPG, which then secretes ions toward the hemolymph. The paracellular barrier in these cells is consistent with a secretory function. The model predicts a stoichiometry of one cycle of Na-K-Cl cotransport for every two cycles of the Na/K ATPase to maintain osmotic equilibrium, resulting in the movement of 5 K into, and 5 Na out of the glial cell.
Figure 8.

A model of NCC69 function in larval nerves, showing how the Na/K ATPase could create the need for solute removal from the extracellular space in larval nerves. The diagram shows a simplified view of the nerve, consisting of an axon and the subperineurial glia, whose septate junctions (SJ) restrict paracellular flow. The model leaves out two classes of glia, the wrapping glia, which are located inside the nerve, and the perineurial cells which are located on the outside, because they do not form septate junctions, and would not pose much of a barrier to paracellular ion flow. The ion flows in the diagram are represented by arrows. Several known ion flows (e.g., Ih and INa) have been omitted for the sake of simplicity. 1: The flow of ions is initiated by the action potential which leaves increased extracellular K in its wake through voltage-gated K channels. 2: In each cycle, the Na/K ATPase removes 2 K ions from the extracellular space, and replaces them with 3 Na ions. 3: Cl ions move to balance the gain in positive charge. These Cl ions could flow from other parts of the extracellular space and/or from intracellular sources, e.g., through Cl channels or transporters. The net effect of the Na/K ATPase is to accumulate NaCl in the extracellular space. If left unchecked (as in NCC69 mutants), the accumulation of NaCl draws water into the extracellular space through osmosis, causing swelling. 4: NCC69 relieves the pressure by transporting solutes into the subperineurial glia, causing it to swell. 5: The subperineurial cell exports solutes, presumably into the hemolymph, to maintain volume homeostasis. 6: K flows down the axoplasm to replace the K that is lost.
The normal nerve electrophysiology in NCC69 mutants is consistent with this model, because it strongly suggests that the bulging phenotype neither results from altered neuronal excitability, nor significantly impacts neuromuscular function. Rather, NCC69 functions to regulate extracellular volume in larval nerves by removing solutes from the extracellular space without dramatically affecting ion composition. With the movement of ions, water naturally follows by osmotic forces. Failure to transport these ions out of the extracellular space causes a build-up of ions in the extracellular space, which in turn draws water into the space by osmosis causing the nerves to bulge.
The model is consistent with data on glia functioning to remove K and restore the normal extracellular ionic environment in the wake of action potentials in axons (Menichella et al. 2006; Newman et al. 1984; Orkand et al. 1966; Walz 2000). In support of the idea that K efflux promotes nerve swelling in Drosophila, we find that nerves in hyperactive mutants (eag Sh) have larger widths, relative to controls (our unpublished observations). Consequently, the only place for the ions to go is into glia or back into the axons. In this view, neuronal activity causes an accumulation of extracellular solutes which are normally removed by the Fray-NCC69 pathway.
Drosophila nerves as a model for volume regulation of the human nervous system
This mechanism of volume regulation may well function in other systems including humans. Drosophila nerves share common traits with human nerves at the level of morphology, phenotypes, and molecules. Drosophila nerves resemble non-myelinated fibers of mammals, in which a single Schwann cell can associate with multiple axons. Disruption of ion flow in mammalian systems gives rise to the accumulation of extracellular fluid, with phenotypes that have striking similarities to fray and NCC69 bulging nerves. For example, loss of function of Cx32 (Scherer et al. 1998), Kir4.1 (Neusch et al. 2001), Cx47 (Menichella et al. 2003; Menichella et al. 2006; Odermatt et al. 2003), or KCC3 (Byun and Delpire 2007) all lead to glial misregulation of ion homeostasis, resulting in the accumulation of extracellular fluid in nerves, nerve tracts, or white matter. Similarly, loss of function of NKCC1 (Arnold et al. 1981; Santi and Lakhani 1983), the isk K channel (Vetter et al. 1996), and Cx30 (Cohen-Salmon et al. 2007) all result in the accumulation of extracellular fluid in the inner ear. All these mutations result in the misregulation of ion homeostasis, and at the ultrastructural level look remarkably similar to NCC69 and fray bulging nerves.
Finally, this work demonstrates a conserved molecular mechanism, the Fray-NCC69 pathway, that functions in Drosophila nerve glia, the cells that form the blood-nerve barrier. In humans, NKCC1 is also expressed in glia and in the cells that form the blood-brain barrier (Alvarez-Leefmans et al. 2001; O’Donnell et al. 2004; Su et al. 2002; Wang et al. 2003). While a full characterization of its function in these cell types has yet to be done, there is mounting evidence that NKCC1 plays an important role in mediating the swelling response to focal ischemia in glia and the blood-brain barrier (Chen and Sun 2005; O’Donnell et al. 2004; Yan et al. 2003). Our study suggests that in flies, the NKCC1 and SPAK orthologs normally function in the nervous system to regulate extracellular volume, presumably in response to neuronal activity. Given the advantages of Drosophila, a proven system for uncovering molecular mechanisms, the relative simplicity of the blood-nerve barrier, and the multiple levels of homology between the fly and human nervous systems, we are confident further work in flies will provide further insights into extracellular volume regulation of the human nervous system.
Supplementary Material
Acknowledgments
We thank Judy Cole and Ester Bashi for technical assistance; Barry Piekos for help with the EM; Steven Ma for help with statistical analysis; Daria Hekmat-Scafe, Jessica Tanis, and Louise Nicholson for critical review of the manuscript; and members of the Keshishian, Wyman, and Carlson laboratories for discussions. We are grateful for the support of Flybase, the Berkeley Drosophila Genome Project, the Bloomington and Vienna Stock Centers, and the many members of the Fly community who shared protocols, stocks, reagents and ideas. The research was supported by grants to HK from the NIH (5R01NS031651 and 1R21NS053807), and the NSF (IBN 0641915); and to BF from the NIH (R01GM083340).
References
- Alfonso TB, Jones BW. gcm2 promotes glial cell differentiation and is required with glial cells missing for macrophage development in Drosophila. Dev Biol. 2002;248(2):369–83. doi: 10.1006/dbio.2002.0740. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Leon-Olea M, Mendoza-Sotelo J, Alvarez FJ, Anton B, Garduno R. Immunolocalization of the Na(+)-K(+)-2Cl(−) cotransporter in peripheral nervous tissue of vertebrates. Neuroscience. 2001;104(2):569–82. doi: 10.1016/s0306-4522(01)00091-4. [DOI] [PubMed] [Google Scholar]
- Arnold W, Nadol JB, Jr, Weidauer H. Ultrastructural histopathology in a case of human ototoxicity due to loop diuretics. Acta Otolaryngol. 1981;91(5–6):399–414. doi: 10.3109/00016488109138521. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Sousa AD, Bhat MA. Organization and function of septate junctions: an evolutionary perspective. Cell Biochem Biophys. 2006;46(1):65–77. doi: 10.1385/CBB:46:1:65. [DOI] [PubMed] [Google Scholar]
- Bartel PL, Chien C-T, Sternglanz R, Fields S. Using the two-hybrid system to detect protein-protein interactions. In: Hartley DA, editor. Cellular interactions in development : a practical approach. New York: IRL Press at Oxford University Press; 1993. [Google Scholar]
- Baumgartner S, Littleton JT, Broadie K, Bhat MA, Harbecke R, Lengyel JA, Chiquet-Ehrismann R, Prokop A, Bellen HJ. A Drosophila neurexin is required for septate junction and blood-nerve barrier formation and function. Cell. 1996;87(6):1059–68. doi: 10.1016/s0092-8674(00)81800-0. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167(2):761–81. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci. 1990;10(11):3754–68. doi: 10.1523/JNEUROSCI.10-11-03754.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis. 2007;28(1):39–51. doi: 10.1016/j.nbd.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SD, Juang JL, Hilgers SL, Garment MB. Blood barriers of the insect. Annu Rev Entomol. 2000;45:151–74. doi: 10.1146/annurev.ento.45.1.151. [DOI] [PubMed] [Google Scholar]
- Chen H, Luo J, Kintner DB, Shull GE, Sun D. Na(+)-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25(1):54–66. doi: 10.1038/sj.jcbfm.9600006. [DOI] [PubMed] [Google Scholar]
- Chen H, Sun D. The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol Res. 2005;27(3):280–6. doi: 10.1179/016164105X25243. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon M, Regnault B, Cayet N, Caille D, Demuth K, Hardelin JP, Janel N, Meda P, Petit C. Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc Natl Acad Sci U S A. 2007;104(15):6229–34. doi: 10.1073/pnas.0605108104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover WJ. Practical Nonparametric Statistics. New York: John Wiley & Sons, Inc; 1999. p. 584. [Google Scholar]
- Darman RB, Flemmer A, Forbush B. Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J Biol Chem. 2001;276(37):34359–62. doi: 10.1074/jbc.C100368200. [DOI] [PubMed] [Google Scholar]
- Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277(40):37542–50. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- Delpire E. Cation-Chloride Cotransporters in Neuronal Communication. News Physiol Sci. 2000;15:309–312. doi: 10.1152/physiologyonline.2000.15.6.309. [DOI] [PubMed] [Google Scholar]
- Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999;22(2):192–5. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet. 1999;8(8):1579–84. doi: 10.1093/hmg/8.8.1579. [DOI] [PubMed] [Google Scholar]
- Doe CQ, Chu-LaGraff Q, Wright DM, Scott MP. The prospero gene specifies cell fates in the Drosophila central nervous system. Cell. 1991;65(3):451–64. doi: 10.1016/0092-8674(91)90463-9. [DOI] [PubMed] [Google Scholar]
- Dowd BF, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2003;278(30):27347–53. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, et al. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem. 1999;274(38):26946–55. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- Ganetzky B. Genetic studies of membrane excitability in Drosophila: lethal interaction between two temperature-sensitive paralytic mutations. Genetics. 1984;108(4):897–911. doi: 10.1093/genetics/108.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B, Wu CF. Neurogenetic analysis of potassium currents in Drosophila: synergistic effects on neuromuscular transmission in double mutants. J Neurogenet. 1983;1(1):17–28. doi: 10.3109/01677068309107069. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Wu CF. Neurogenetics of membrane excitability in Drosophila. Annu Rev Genet. 1986;20:13–44. doi: 10.1146/annurev.ge.20.120186.000305. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. Genetic transformation of yeast. Biotechniques. 2001;30(4):816–20. 822–6. doi: 10.2144/01304rv02. 828 passim. [DOI] [PubMed] [Google Scholar]
- Haas M, Forbush B., 3rd The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol. 2000;62:515–34. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- Halpern ME, Chiba A, Johansen J, Keshishian H. Growth cone behavior underlying the development of stereotypic synaptic connections in Drosophila embryos. J Neurosci. 1991;11(10):3227–38. doi: 10.1523/JNEUROSCI.11-10-03227.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SC, Mount DB, Gamba G. Molecular physiology of cation-coupled Cl− cotransport: the SLC12 family. Pflugers Arch. 2004;447(5):580–93. doi: 10.1007/s00424-003-1066-3. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl− cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26(35):8943–54. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Mechanisms of Cerebral Ischemic Damage. In: Walz W, editor. Cerebral Ischemia: Molecular and Cellular Pathophysiology. Totowa, New Jersey: Humana Press; 1999. pp. 1–32. [Google Scholar]
- Isenring P, Jacoby SC, Payne JA, Forbush B., 3rd Comparison of Na-K-Cl cotransporters. NKCC1, NKCC2, and the HEK cell Na-K-Cl cotransporter. J Biol Chem. 1998;273(18):11295–301. doi: 10.1074/jbc.273.18.11295. [DOI] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144(4):1425–36. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen J, Halpern ME, Keshishian H. Axonal guidance and the development of muscle fiber-specific innervation in Drosophila embryos. J Neurosci. 1989;9(12):4318–32. doi: 10.1523/JNEUROSCI.09-12-04318.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Leiserson WM, Harkins EW, Keshishian H. Fray a Drosophila serine/threonine kinase homologous to mammalian PASK, is required for axonal ensheathment. Neuron. 2000;28:793–806. doi: 10.1016/s0896-6273(00)00154-9. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23(13):5963–73. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26(43):10984–91. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21(15):5429–38. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233(4762):453–4. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Glial cell regulation of extracellular potassium. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford; New York: Oxford University Press; 2005. pp. 717–731. [Google Scholar]
- Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225(4667):1174–5. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24(9):1046–56. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Bussow H, Schilling K, Steinhauser C, et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23(11):4549–59. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29(4):788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Abrams CK, Scherer SS. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci. 2008;35(1):101–16. doi: 10.1007/s12031-007-9027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Xu JC, Haas M, Lytle CY, Ward D, Forbush B., 3rd Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem. 1995;270(30):17977–85. doi: 10.1074/jbc.270.30.17977. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Garbarini N, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem. 2003;278(52):52848–56. doi: 10.1074/jbc.M309436200. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277(52):50812–9. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- Pullikuth AK, Filippov V, Gill SS. Phylogeny and cloning of ion transporters in mosquitoes. J Exp Biol. 2003;206(Pt 21):3857–68. doi: 10.1242/jeb.00641. [DOI] [PubMed] [Google Scholar]
- Rubin GM. The draft sequences. Comparing species. Nature. 2001;409(6822):820–1. doi: 10.1038/35057277. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- Santi PA, Lakhani BN. The effect of bumetanide on the stria vascularis: a stereological analysis of cell volume density. Hear Res. 1983;12(2):151–65. doi: 10.1016/0378-5955(83)90103-x. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia. 1998;24(1):8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129(4):877–96. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8(3):254–67. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- Stork T, Engelen D, Krudewig A, Silies M, Bainton RJ, Klambt C. Organization and function of the blood-brain barrier in Drosophila. J Neurosci. 2008;28(3):587–97. doi: 10.1523/JNEUROSCI.4367-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su G, Kintner DB, Sun D. Contribution of Na+-K+-Cl− cotransporter to high-[K+]o-induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol. 2002;282(5):C1136–1146. doi: 10.1152/ajpcell.00478.2001. [DOI] [PubMed] [Google Scholar]
- Sun Q, Tian E, Turner RJ, Ten Hagen KG. Developmental and functional studies of the SLC12 gene family members from Drosophila melanogaster. Am J Physiol Cell Physiol. 2010;298(1):C26–37. doi: 10.1152/ajpcell.00376.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98(2):81–5. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron. 1996;17(6):1251–64. doi: 10.1016/s0896-6273(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397(1):223–31. doi: 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36(4–5):291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. 2008;86(4):342–67. doi: 10.1016/j.pneurobio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan Y, Kintner DB, Lytle C, Sun D. GABA-mediated trophic effect on oligodendrocytes requires Na-K-2Cl cotransport activity. J Neurophysiol. 2003;90(2):1257–65. doi: 10.1152/jn.01174.2002. [DOI] [PubMed] [Google Scholar]
- White B, Osterwalder T, Keshishian H. Molecular genetic approaches to the targeted suppression of neuronal activity. Curr Biol. 2001;11(24):R1041–53. doi: 10.1016/s0960-9822(01)00621-2. [DOI] [PubMed] [Google Scholar]
- Xu JC, Lytle C, Zhu TT, Payne JA, Benz E, Jr, Forbush B., 3rd Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci U S A. 1994;91(6):2201–5. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager J, Richards S, Hekmat-Scafe DS, Hurd DD, Sundaresan V, Caprette DR, Saxton WM, Carlson JR, Stern M. Control of Drosophila perineurial glial growth by interacting neurotransmitter-mediated signaling pathways. Proc Natl Acad Sci U S A. 2001;98(18):10445–50. doi: 10.1073/pnas.191107698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D. Inhibition of Na(+)-K(+)-Cl(−) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003;961(1):22–31. doi: 10.1016/s0006-8993(02)03832-5. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Budnik V, Wu CF. Synaptic plasticity in Drosophila memory and hyperexcitable mutants: role of cAMP cascade. J Neurosci. 1992;12(2):644–51. doi: 10.1523/JNEUROSCI.12-02-00644.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wu CF. Neuronal activity and adenylyl cyclase in environment-dependent plasticity of axonal outgrowth in Drosophila. J Neurosci. 2004;24(6):1439–45. doi: 10.1523/JNEUROSCI.0740-02.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.