

Abstract
Background
Alcohol intoxication occurs in nearly half of all trauma patients and increases the morbidity, mortality and healing complications of these patients. Prior studies in our lab and elsewhere have demonstrated impairments in re-epithelialization, angiogenesis, and inflammation in wounds following acute ethanol exposure. Clinically, acute ethanol has been shown to cause an increased breakdown of wounds. To date, the mechanisms by which acute ethanol exposure modifies wound strength have received little experimental attention.
Methods
To examine how ethanol influences functions critical to the development of wound strength, the effect of ethanol exposure on fibroblast proliferation and extracellular matrix production was examined. Normal human dermal fibroblasts (NHDF) were exposed to ethanol (100 mg/dl) and then examined for proliferative capacity and mRNA production of collagen I, collagen III, and lysyl oxidase (LOX). In in vivo studies, the wound breaking strength, LOX activity, collagen and hyaluronic acid (HA) contents of wounds of ethanol-exposed (100 mg/dl) mice were examined.
Results
At 24, 48 and 72 hours after acute ethanol exposure (8 hours duration), NHDF displayed a significant impairment in proliferative capacity (up to 50% at 24 hours p < 0.001). After ethanol exposure, NHDF produced less collagen I and LOX mRNA, but more collagen III mRNA than control fibroblasts (p < 0.05). Ethanol exposure in vivo caused a reduction in wound breaking strength of up to 40% when compared to control mice (p < 0.01). LOX activity, collagen and HA contents in the wounds of ethanol-exposed mice were significantly reduced (p < 0.01).
Conclusions
These studies reveal that a single exposure to ethanol prior to injury can cause a significant decrease in wound breaking strength. Our studies suggest that ethanol directly impairs fibroblast function, leading to decreased collagen production. The results provide a possible explanation for how acute ethanol exposure might increase in wound complications and wound failure.
Keywords: Ethanol, wound healing, fibroblast, collagen
Introduction
Almost half of all trauma patients presenting to the emergency room have an elevated blood alcohol content (BAC) at the time of admission (Rivara et al., 1993). These patients display higher morbidity and mortality than non-intoxicated patients, including wound healing impairments (Cherpitel, 1993a; Cherpitel, 1993b; Gentilello et al., 1993; Girasek et al., 2002; McGill et al., 1995; Nickelsen et al., 2005). Ethanol exposure can cause a variety of local and systemic effects in trauma patients (Choudhry and Chaudry, 2006). Changes seen with ethanol exposure include impairment of the inflammatory response and increased complications such as increased infections along with wound and anastomosis breakdown. It is important to note that nearly 11 million injuries in the United States occur in people with alcohol exposure (Barnes and Edwards, 2005; Thal et al., 1985). Roughly 25% of alcohol related trauma victims have acute alcohol exposure not chronic exposure (Rivara et al., 1993). Thus, there is clearly a need for further understanding the role that acute exposure plays in trauma and the wound healing setting.
The goal of dermal wound repair is to restore damaged tissue to a state as near normal skin as possible. This occurs through a series of repair stages including inflammation, proliferation, and remodeling, all of which are impacted by acute ethanol exposure prior to injury (Fitzgerald et al., 2007; Radek et al., 2007; Radek et al., 2008; Radek et al., 2005). While the proliferation and remodeling phases include broad activities such as re-epithelialization, angiogenesis, and reconstitution of dermal appendages, all of these activities rely on an intact extracellular matrix (ECM) for coordination. Cell adhesion and migration are critical processes for wound closure, angiogenesis, and scar formation, and are dependent upon an intact extracellular matrix (Ehrlich, 1988; Hebda et al., 1993; Raza and Cornelius, 2000; Tonnesen et al., 2000). The ECM provides the scaffold upon which cells migrate and also acts as a reservoir for growth factors.
Many studies examine the effects of alcohol on wound related areas such as immune responsiveness and infection resistance, yet there is a dearth of information on alcohol and wound strength. Several clinical studies have shown an increase in anastomotic leakage rates in patients undergoing intestinal surgery following alcohol use (Makela et al., 2003; Sorensen et al., 1999; Tonnesen et al., 1987). While there are a fair number of studies showing decreased strength in intestinal or mucosal healing, there are relatively few detailing similar effects in other tissues like long bones (Rai et al., 2008), and virtually none that do so in dermal wounds. Prior studies have evaluated the role of acute alcohol exposure on ECM components and processes including increased matrix proteolytic activity, decreased angiogenesis and decreased collagen content (Radek et al., 2007; Radek et al., 2008; Radek et al., 2005), but have not specifically looked at the role of fibroblasts, the key cell responsible for producing the majority of the ECM.
During wound repair, dermal fibroblasts exhibit a significant proliferative response, migrate across the wound, secrete growth factors, and lay down ECM components (Clark, 1995). As the fibroblasts migrate towards the center of the wound, they lyse the fibrin clot and replace it with fibronectin and hyaluronic acid, which develops into early granulation tissue. This is then replaced first by collagen type III, and then collagen type I. Immature wounds contain a higher degree of collagen III which is organized in parallel fibrils while mature wounds contain a high degree of cross-linked collagen I fibers.
The dermis is slowly remodeled through a continued balance of degradative and synthetic activity, along with the cross linking of collagen. LOX is the enzyme responsible for cross linking collagen in a specific pattern and maintains an association with the collagen fibrils preventing non-specific cross linking (Szauter et al., 2005). Enzymatic cross linking occurs in normal and wounded skin and contributes to improved collagen architecture and tissue strength. This remodeling process yields an organized matrix that reestablishes the strength and flexibility of the wounded tissue (Bernstein, 1996; Clark, 1993; Singhal et al., 1999; Soo et al., 2000; Swift et al., 1999). In normally healing wounds, approximately 90% of the original strength of the skin is restored upon completion of tissue repair (Bernstein, 1996).
Our understanding of normal wound repair mechanisms has advanced considerably in recent years, but this enhanced understanding has also highlighted the lack of knowledge on the role of exogenous influences on abnormal wound healing. Pathological factors such as old age, diabetes, and other diseases, as well as exogenous factors including ethanol and its metabolites have all demonstrated distinct changes in the cellular and tissue response, resulting in abnormal wound healing (Molina et al., 2002; Swift et al., 1999; Yamaguchi and Yoshikawa, 2001). The focus of this study is to examine the effect that a single acute ethanol exposure prior to injury can have on the healing wound. Paying particular attention to the fibroblast, we demonstrate that ethanol has a direct effect on the ability of fibroblasts to proliferate and produce vital ECM components. The results demonstrate that the fibroblast impairment has demonstrable phenotypic changes including decreased collagen content. Our findings also represent the first demonstration of the negative effects of acute ethanol exposure on dermal wound breaking strength.
Materials and Methods
Materials
Normal human dermal fibroblasts (NHDF) and appropriate media were obtained from Promocell (Heidelberg, Germany). All chemicals were obtained from Sigma (St. Louis, MO) unless otherwise stated. Cell culture reagents, Trizol™, and DNase I kit were purchased from Invitrogen (Carlsbad, Ca). CellTiter 96 AQueous One Solution Cell Proliferation Assay was purchased from Promega (Madison, WI). SYBR Green, RETROscript reverse transcription kits and reagents were purchased from Applied Biosystems (Foster City, CA).
Cell culture
NHDF were cultured in fibroblast growth medium (Promocell) supplemented with penicillin/streptomycin (100 IU/ml/100μg/ml). For all experiments, cells were incubated overnight in low serum fibroblast basal medium prior to experiments. Cells were used between passages 7 and 10 for all experiments.
Ethanol proliferation assay
NHDF at 40–50% confluency were incubated for 8 h in 96 well plates with either fibroblast basal media/10% FBS or fibroblast basal media/10% FBS with 100 mg/dl of ethanol at 37°C with 5% CO2. After 8 h, the media was removed, cells were washed once with sterile PBS and ethanol free fibroblast growth media was replaced. At each time point tested (immediately after media replacement, 24, 48 and 72 hours) proliferation was assessed by adding 100 μl of media and 20 μl of CellTiter 96 AQueous One Solution Reagent to each well and incubating at 37°C for 90 minutes. Optical density (OD) was then read at 490 nm using a microplate spectrophotometer (SPECTRA PLUS, Molecular Devices, Sunnyvale, CA). Experiments were run in quadruplicate.
Semi-Quantitative real-time PCR of Collagen I, Collagen III and LOX mRNA
NHDF at 70–80% confluency were incubated for 8 h in 6 well plates with either fibroblast basal media/10% FBS or fibroblast basal media/10% FBS with 100 mg/dl of ethanol at 37°C with 5% CO2. After 8 hours of ethanol exposure, media was removed and RNA was extracted from cells using Trizol™, following the manufacturer’s directions. The samples were then treated with DNase I. One microgram of the total RNA was reverse transcribed using the RETROscript RT kit. The reactions were then subjected to real time PCR analysis. Real time primers were designed and constructed using Primer Express software (Applied Biosystems, Foster City, CA). Real time RT-PCR was performed using the StepOnePlus™ Real-Time PCR System. Gene expression was determined via the 2−ΔΔCt method normalized to GAPDH expression. The primers that were used are below in Table 1.
Table 1.
PCR primers
| Forward | Reverse | |
|---|---|---|
| GAPDH | 5′-GGCTGCTTTTAACTCTGGTAAAGT-3′ | 5′-AACCATGTAGTTGAGGTCAATGAA-3′ |
| Collagen I | 5′-GCTTCACCTACAGCGTCACTGTCG-3′ | 5′-AGAGGAGTTTACAGGAAGCAGACAG-3′ |
| Collagen III | 5′-CCGATGGGTTGCCAGGATCCATG-3′ | 5′-GAAGGGCATTGTGCTGAACTTGCG-3′ |
| Lysyl Oxidase | 5′-ATGATCACAGGGTGCTGCTCAGAT-3′ | 5′-TTCCCAGGAATATCTTGGTCGGCT-3′ |
Administration of ethanol and excisional wounds
Six to eight week-old female BALB/c mice (Harlan Sprague Dawley, Indianapolis, IN), weighing between 17 and 21 g, were administered an intraperitoneal injection of a 20% ethanol solution (1.4 g/kg) or equivalent volume of saline as a control, as previously described (Radek et al., 2005). BAC was determined using an enzymatic colorimetric assay (Pointe Scientific Inc., Canton, MI), as previously described (Faunce et al., 1997). After 30 min, the ethanol treated mice had an average circulating BAC of 100 mg/dl, a BAC that is just above the legal limit in most states (Messingham et al., 2000). The mice were subsequently anesthetized with an intraperitoneal injection of Ketamine/Xylazine (100 mg/kg/5 mg/kg), according to their body weight. When completely anesthetized, each mouse had its dorsal skin shaved, and six full-thickness excisional wounds were placed on the dorsum using a 3-mm biopsy punch (Acu Punch, Acuderm, Fort Lauderdale, FL). After 8 h, the mice had a circulating BAC of 23 mg/dl, but by 24 h the ethanol was undetectable (Radek et al., 2008). At various times after wound placement, mice were euthanized and wounds harvested for analyses. For each of the different excisional wound analyses, one of the six wounds was randomly selected from each mouse, and this single wound utilized for analysis. Animal protocols used in these studies were reviewed and approved by the University of Illinois at Chicago Office of Animal Care and Institutional Biosafety.
Wound breaking strength
Six to eight week-old female BALB/c mice (n = 6 per group, per time point) were anesthetized and prepped as above. A 2-cm longitudinal skin incision was made through the dermis and panniculus carnosus in a paraspinal location and closed with surgical clips. The surgical clips were removed on postoperative day 5. At 5, 7, 10, 14, 21, 28 and 35 days post injury, the animals were sacrificed and the pelts harvested for analysis.
Wound disruption strength was measured as previously described (Greenhalgh and Gamelli, 1987). Skin strips for testing were cut at right angles to the long axis of the wound with specially designed cutters. The cutters are dumbbell shaped with the narrower portion, containing the incision, measuring 4 mm across. The wider portions of the skin strips were placed in the jaws of a motorized tensiometer (Mark-10, Copiague, NY) and held in place with thumbscrews. Skin strips were stretched at a constant rate (3 cm/min) until disruption occurred. Wound breaking strength was determined by continuous recording on a digital readout. Two skin strips per wound were subjected to analysis and the average was recorded as the wound breaking strength for an individual animal.
LOX assay
One 3 mm excisional wounds (n=6) at 5, 7, 10, 14, and 21 days post injury were collected and homogenized in CelLytic tissue lysis buffer (Sigma). Supernatants were harvested after centrifugation at 13,000rpm for 15 min and stored at −80°C until use. Activity of LOX in the skin homogenates was determined using an Amplite Fluorimetric LOX assay kit (AAT Bioquest, Inc, Sunnyvale, CA) according to the manufacturer’s directions. The relative activity of LOX was recorded as OD570.
Analysis of wound collagen content
The hydroxyproline content of single individual excisional wounds (n = 4–6 mice per group, one wound per mouse) was determined according to a standard protocol (Woessner, 1961). Each wound was harvested from the pelt with the use of a uniform 3-mm biopsy punch. The initial punch wounds from all animals were retained and used as normal skin samples (n = 59). Briefly, frozen tissue was hydrolyzed in 2 ml of 6N HCl overnight at 90°C, neutralized with 2.5N NaOH and diluted 40-fold with MilliQ water. A 2 ml aliquot of the sample was incubated with 1 milliliter of a 0.05 M chloramine-T solution for 20 min at room temperature, followed by 1 milliliter of 3.15 M perchloric acid for 5 min at room temperature and 1 milliliter of 20% p-dimethylaminobenzaldehyde for 20 min at 60°C in a water bath. The samples were then cooled by immersion in cold tap water for 5 minutes. The amount of hydroxyproline was determined by comparison to a standard curve measured spectrophotometrically at an absorbance of 557 nm. Samples were analyzed in duplicate.
Analysis of wound HA
The concentration of HA in wound tissue homogenates of ethanol treated and control mice at different time points were determined using an HA ELISA kit (R&D systems, Minneapolis, MN) according to manufacturer’s instructions. The quantity of HA is expressed per milligram of wet weight tissue.
Statistical analysis
Data were analyzed using GraphPad Prism (version 3.02, GraphPad Software, San Diego, CA). The values were calculated as means ± SEM for each data set. Data were analyzed by a two-way ANOVA and either Student’s t-test or Bonferroni’s posttest. p values < 0.05 were considered significant.
Results
Acute ethanol exposure impairs in vitro fibroblast proliferation
Our previous studies demonstrated an impairment of the proliferative phase of wound healing and the extracellular matrix, but did not specifically look at the role of the key cell responsible for the ECM (Radek et al., 2007; Radek et al., 2005). In this study, we began by examining if acute ethanol exposure would have a direct effect on fibroblasts, the primary cell responsible for the proliferative phase of wound healing. In vitro acute ethanol exposure (100 mg/dl for 8 hours) had no effect on fibroblast viability but did cause a significant decrease in fibroblast proliferation when compared to fibroblasts grown in medium alone (Figure 1). At the end of the 8 hour treatment (time 0), there was no statistically significant difference in fibroblast number between control or ethanol treated fibroblasts. At 24 hours after replacement with fresh growth media, control fibroblasts were nearly twice as numerous as ethanol treated fibroblasts (absorbance values of 0.41 ± 0.02 vs. 0.21 ± 0.02). By 72 hours, the control fibroblasts were still nearly 1.6 times as numerous as the ethanol treated fibroblasts (0.81 ± 0.13 vs. 0.49 ± 0.09). These data suggest that acute ethanol exposure has a direct effect on fibroblast proliferation, and this effect was persistent even after ethanol removal.
Figure 1.
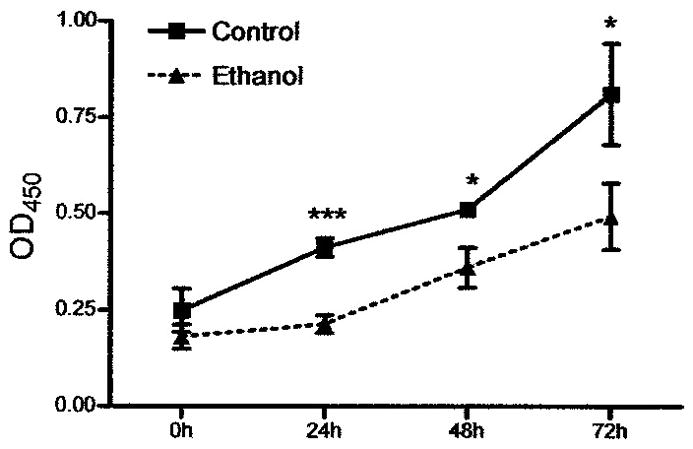
Acute ethanol exposure impairs in vitro fibroblast proliferation. Fibroblasts were exposed to ethanol (100mg/dl) or control medium for 8 hours, and then fresh medium was replaced. Cell proliferation was assayed immediately after exposure and at 24, 48, and 72 hours after exposure. Data presented are absorbance (OD 450) means ± SEM for each time point; n = 4 for each treatment group and time point. * p < 0.05, *** p < 0.001
Acute ethanol exposure alters in vitro fibroblast mRNA expression of collagen and lysyl oxidase
The above experiment established that acute ethanol exposure has a direct impairment on the regulatory function of fibroblasts. Next we wanted to see if it would also have a direct effect on the genetic expression of collagen I, collagen III and lysyl oxidase, all of which are vital ECM components produced by fibroblasts during wound healing. For collagen I and lysyl oxidase, 8 hours of exposure to ethanol led to a significant reduction in the mRNA levels compared to control fibroblasts (60% ± 7 and 70% ± 6, respectively). For collagen III, 8 hours of exposure led to double the mRNA level compared to control fibroblasts (Figure 2).
Figure 2.

Acute ethanol exposure alters in vitro fibroblast mRNA expression of collagen and lysyl oxidase. Fibroblasts were exposed to ethanol (100mg/dl) or control medium for 8 hours, and then RNA was extracted, subjected to real time RT-PCR, normalized to GAPDH expression and compared to control fibroblasts (n = 7 – 10). Data are expressed as mean fold change in target expression level ± SEM. Ethanol exposure reduced collagen I mRNA expression, and LOX expression, but increased collagen III mRNA expression. * p < 0.05
Acute ethanol exposure reduces wound breaking strength and wound lysyl oxidase activity
The previous experiments established that acute ethanol exposure can directly impair fibroblast function in vitro. To determine if this direct in vitro fibroblast impairment would manifest any detectable in vivo phenotypic changes in wound healing, wound breaking strength was analyzed in a murine incisional wound model. The major component contributing to wound breaking strength changes over time with early strength determined by the epithelial layer, later breaking strength correlates with the relative amount of collagen present in the wound and at prolonged time frames the architecture of the collagen in the wound and the remodeling of the ECM are the greatest contributors to wound breaking strength. Prior to day 10 post wounding, there was no significant difference between ethanol treated and control mice (Figure 3A). At day 14 post wounding, the ethanol treated mice had wound breaking strengths only 60% as great as control mice. By day 28 post wounding the ethanol treated mice still showed a 20% reduction in wound breaking strength compared to the control mice. Day 21 presented what might be considered an anomaly as the breaking strength actually decreased in each experimental replicate for the control wounds. While this result is puzzling, the finding was consistent and reproducible in duplicate experiments. This might possibly reflect the time point at which the wound switches from active collagen synthesis to remodeling, a period known to involve proteolytic activity.
Figure 3.

Acute ethanol exposure reduces wound breaking strength and LOX enzymatic activity. 30 minutes prior to incisional wounding, mice were injected with an ethanol solution resulting in a BAC of 100 mg/dl. (A) At time points indicated, groups of mice were harvested and two test strips were prepared for each mouse and the breaking strength results (in kg of force) were averaged for a single data point (n = 13 for normal skin, n = 9 – 10 for all others). Breaking strength was then normalized as a percentage of normal skin strength and is presented as % relative strength ± SEM. At 14 days after acute exposure and wounding, ethanol treated mice had a breaking strength only 60% as great as control mice. At 28 days after acute exposure and wounding, ethanol treated mice still showed a 20% reduction in wound breaking strength as compared to control mice. (B) At the time points indicated, a single wound from each mouse was harvested and tissue homogenates were prepared. LOX activity was then determined as described in Materials and Methods. Acute ethanol exposure significantly reduces LOX activity in wounds compared to non-treated group. * p < 0.05, ** p < 0.01
We further investigated if ethanol treatment affected the LOX activity in the wounds. A LOX assay was used to detect LOX enzymatic activity in homogenates from normal skin and skin wounds from ethanol treated or untreated mice. The results demonstrated that LOX activity in non-ethanol treated group was significantly increased after wounding at day 5 to day 21 compared to normal skin. In control wounds, LOX activity reached its peak at day 7 at a level that was more than a 2 fold increase over normal tissue (p<0.05, Figure 3B). This result was consistent with our previous genearray study which showed that LOX gene expression in mouse skin wounds was indeed significantly elevated from day 3 after wounding and peaked at day 5 (data not shown). In contrast to wounds of control mice, LOX activity in the wounds of ethanol treated mice was significantly decreased from normal skin levels at all time points (p<0.05, Figure 3B). Moreover, LOX activity was significantly lower in wounds of ethanol treated mice than control at all time points from day 5 to day 21 (p<0.01, Figure 3B).
Acute ethanol exposure delays and reduces peak in vivo collagen content
Based on the time points that breaking strength were significantly reduced it seemed likely that an impairment in the ECM may be responsible for the decreased breaking strength. To assess whether collagen content may be responsible for the decreases in breaking strength observed, hydroxyproline testing was performed on 3mm excisional punch wounds at the given time points (Figure 4A). Hydroxyproline, used as an index for total collagen content, was significantly reduced at day 10 post wounding in wounds from ethanol-treated mice compared with saline controls (46.6 ± 2.2 vs. 37.8 ± 0.5 μg per wound). Day 10 represents the time of maximal collagen content in control wounds. The decrease in total collagen that was seen at this time point in ethanol treated mice suggests that ethanol prevents maximal collagen synthesis. In other words, an ethanol induced deficit in collagen production is most pronounced when collagen synthetic demands peak. The time to peak collagen content was also delayed in wounds of ethanol treated mice, moving from day 10 in control wounds to day 14 in wounds of ethanol treated mice. When peak HP content was compared irrespective of time point (day 10 control, day 14 ethanol), the result was a statistically significant reduction in collagen content (46.6 ± 2.2 vs. 40.2 ± 0.6, p < 0.05).
Figure 4.

Acute ethanol exposure delays and reduces peak in vivo wound collagen and hyaluronic acid contents. 30 minutes prior to a 3mm excisional punch biopsy wound, mice were injected with an ethanol solution resulting in a BAC of 100 mg/dl. (A) At time points indicated, groups of mice were harvested and wounds were hydrolyzed. Hydroxyproline content, used as an index for the presence of collagen, was determined using a standard biochemical assay and represented as micrograms of HP per wound. HP content data are means ± SEM for each day; n = 59 for normal skin, n = 4–6 for all others. At day 10 post wounding, ethanol treated mice displayed a 20% reduction in HP content compared to control mice. Peak HP content irrespective of time (day 10 for control, day 14 for ethanol, represented by the bracket) was reduced in ethanol treated mice by nearly 15%. Time to peak HP content was delayed in ethanol treated mice. (B) At the time points indicated, the quantity of HA per mg wound tissue homogenates was determined by an HA ELISA as described in Materials and Methods. The results show that wounds contain less HA than normal skin. Importantly, when compared to control, ethanol exposure causes a significant reduction in wound HA content. * p < 0.05, ** p < 0.01
Acute ethanol exposure further reduces hyaluronic acid level in wounds
To examine if ethanol treatment could affect HA in the wounds, HA contents were determined after ethanol exposure at day 5 to day 21 after wounding. Results demonstrated that normal skin had HA of 26.6±5.6 ng/mg tissue. After wounding, HA levels fell sharply to 10.8–12.54ng/mg between day 5 and d 21 in non-ethanol treated group (p<0.01 compared to normal skin, Figure 5). However, ethanol treatment further significantly decreased the levels of HA in the wounds to levels between 5.5–7.2ng/mg, which were significantly lower not only than normal skin, but also non-ethanol treated group (p<0.01, Figure 4B).
Discussion
The reconstitution of the ECM is a dynamic process with synthetic and proteolytic activity both occurring concomitantly. When these processes are appropriately balanced, the result is a wound that approximates unwounded tissue, with well organized matrix and minimal scarring. As the balance of these two opposing processes shifts, the final outcome of the healing wound is changed. Derangements to this process can occur as a result of changes in signaling events, pathologic conditions or exogenous factors (Branton and Kopp, 1999; Kovacs and DiPietro, 1994; Leask and Abraham, 2004; Radek et al., 2007; Steffensen et al., 2001; Uchiyama et al., 2000; Yamaguchi and Yoshikawa, 2001). Shifts can favor enhanced ECM synthesis and/or decreased proteolytic activity resulting in hypertrophic scars or keloids; alternatively they can favor depressed ECM synthesis and/or enhanced proteolytic activity resulting in fragile wounds and dehiscence (Beare et al., 2003; Casini et al., 1999; Lois et al., 1999; Molina et al., 2002).
Interestingly, the fibroblast sits at the crossroads of the ECM as it is responsible for the majority of the ECM synthesis and is also involved in the proteolytic activity directly through secretion of MMPs and indirectly through cytokine production. The results of this study demonstrate that acute ethanol exposure directly inhibits fibroblast function during wound healing. Our results demonstrate that ethanol exposure can inhibit fibroblast proliferation. Acute ethanol exposure also reduces the production of mRNA for the major structural component, collagen I by fibroblasts. This data supports our previous finding that collagen I mRNA was reduced in vivo in wounds from ethanol treated mice (Radek et al., 2007). The mechanisms that explain the ability of ethanol exposure to reduce collagen I gene expression are currently unknown. One mechanism that might explain this reduction in collagen I mRNA in vivo is an indirect effect resulting from decreased transforming growth factor (TGF)-β1. In vivo, wounds from ethanol exposed mice contain significantly less TGF-β1 at the later time points. Since TGF-β1 is known to promote collagen synthesis in fibroblasts (Branton and Kopp, 1999; Kovacs and DiPietro, 1994), an ethanol mediated reduction in TGF-β1 might contribute to decreased collagen I production. Other studies corroborate this notion and have shown that acute ethanol exposure reduces TGF-β1 induced collagen synthesis in fibroblasts using an in vitro model (Stephens et al., 1996).
Taken together, our data suggests that at least two levels of inhibition are at work regarding collagen production. A baseline decline in type I collagen mRNA production as a direct result of acute ethanol exposure is likely, given our current findings. A decline in TGF-β1 levels in wounds at later time points, as demonstrated in prior studies, would serve to limit pro-fibrotic responses of fibroblasts by removing their primary stimulus for collagen I mRNA production.
The current studies demonstrate that ethanol exposure can directly impair fibroblast proliferation. In the context of the healing wound, as reduction of the number of fibroblasts within the wound, along with reduced production of collagen I, would be expected to lead to reduced collagen content. In the current study ethanol exposure resulted in a highly significant decrease in wound collagen deposition at day 10, which is the time point of maximal collagen content in this wound model As discussed above, the ability of ethanol to significantly decrease the total amount of collagen in wounds seems to involve direct effects on fibroblasts. Other aspects of healing that have been described to be affected by ethanol may also contribute to an impairment of ECM production. These include increased collagen degradation, and decreased nutrients in the form of hypoxia and decreased angiogenesis (Grenett et al., 1998; Lois et al., 1999; Radek et al., 2005). Our previous studies have shown that the inflammatory response in wounds is perturbed following ethanol exposure (Fitzgerald et al., 2007). Because inflammatory cells can influence protease production, this effect of ethanol might lead to altered protease activity. The enzymes that are most responsible for the balance of collagen degradation and synthesis are the matrix metalloproteinases (MMPs) and their naturally occurring inhibitors, tissue inhibitors of metalloproteinases. The MMPs are secreted by various cells including keratinocytes, endothelial cells and fibroblasts and collectively are known to degrade collagen, gelatin, and other ECM components (Salo et al., 1994). Previous studies in our lab have investigated the effect of ethanol on MMPs. These studies demonstrated that levels of matrix metalloproteinase-8, a collagen type I proteinase, are increased in the wounds of ethanol treated mice, contributing to an overall inhibition of appropriate ECM formation (Radek et al., 2007).
As previously stated, mature wounds contain a high degree of cross-linked collagen I fibers, while immature wounds contain high degrees of parallel collagen III fibers. The ratio of collagen I to collagen III fibers, and their degree of architecture strongly influence the rapidity that the collagen can be remodeled and the resulting strength of the wound (Bornstein and Sage, 1980; Clark, 1995; Ehrlich, 1988). The excisional mouse wounds for the early time points in this study showed no difference in HP content, while the in vitro study showed a decrease in collagen I mRNA with an increase in collagen III mRNA. In the presence of acute alcohol exposure, early wound fibroblasts are directed to produce fewer collagen I fibers and more collagen III fibers. These collagen III fibers provide less strength for the wound than collagen I fibers, and place an additional burden on the wound as they need to first be degraded and replaced with collagen I prior to remodeling. This combination of decreased overall collagen content and shifting the ratio of collagen away from collagen I should impair the strength of the wound, and this is indeed borne out by our study. Furthermore, we found that injury increased LOX activity in the skin wounds at least from day 5 to day 21 and peaked at day 7. The result was consistent with a previous report in a rat skin wound model (Fushida-Takemura et al., 1996). However, ethanol exposure significantly reduced LOX activity, both compared to normal skin and control wounds. Given the critical roles that LOX plays in the cross linking of collagen (Szauter et al., 2005) the reduced wound breaking strength in ethanol treated mice may be partially due to the decreased activity of LOX.
In addition to collagen, HA is a major component of the ECM. HA is a nonsulfated, high-molecular-weight glycosaminoglycan with a simple, repeated disaccharide linear copolymer structure (Chen and Abatangelo, 1999; Jiang et al., 2007). Recently studies suggested that HA multifaceted roles in biology, but the main function seems to be in tissue repair. HA is involved in activation and moderation of the inflammatory response, promoting cell migration and proliferation as well as angiogenesis (Chen and Abatangelo, 1999; Jiang et al., 2007). Topical use of HA for acute or chronic wounds has been shown to improve wound healing (Gao et al., 2010; Nolan et al., 2006; Voinchet et al., 2006). In the current study, we found that the HA content of wounds was significantly decreased by acute ethanol exposure. Alterations in both collagen and HA might therefore contribute to the reduced wound breaking strength seen in ethanol treated mice. One possibility is that the decrease of HA levels in the wounds of ethanol treated mice involves an inhibition of HA synthetases; this will need to be examined in future studies.
Acute ethanol exposure has been previously shown to increase the risk of intestinal anastomosis leakage (Makela et al., 2003; Nickelsen et al., 2005; Sorensen et al., 1999), but an effect on wound breaking strength has not been demonstrated in dermal wounds until now. Our finding may explain many of the clinical observations that demonstrate increased incidence of wound failure with alcohol exposure. Our finding that even a low BAC can cause such impairment raises the significance of ethanol as a risk factor for trauma patients as the BAC of such patients are typically 2 to 3 times the ethanol level in our study. Ultimately, therapeutics aimed at reversing the ethanol impairment that might be administered to intoxicated trauma patients prophylactically. The development of such therapeutics will be needed to counteract the effects of ethanol on the repair process.
Acknowledgments
We thank Anna Turabelidze, Mateusz Wietecha, Shujuan Guo, and the Alcohol Research Program at Loyola University Medical Center for helpful advice and assistance.
This publication was supported by NIH Grants RO1-GM50875 (LAD), P20-GM078426 (LAD), and 5T32AA013527 (MJR). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS, NIAAA, or NIH.
References
- Barnes EV, Edwards NL. Treatment of osteoarthritis. South Med J. 2005;98:205–209. doi: 10.1097/01.SMJ.0000153116.71823.24. [DOI] [PubMed] [Google Scholar]
- Beare AH, O’Kane S, Krane SM, Ferguson MW. Severely impaired wound healing in the collagenase-resistant mouse. J Invest Dermatol. 2003;120:153–163. doi: 10.1046/j.1523-1747.2003.12019.x. [DOI] [PubMed] [Google Scholar]
- Bernstein E. Principles of Cutaneous Surgery. McGraw Hill; New York: 1996. [Google Scholar]
- Bornstein P, Sage H. Structurally distinct collagen types. Annu Rev Biochem. 1980;49:957–1003. doi: 10.1146/annurev.bi.49.070180.004521. [DOI] [PubMed] [Google Scholar]
- Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- Casini A, Galli A, Calabro A, Di Lollo S, Orsini B, Arganini L, Jezequel AM, Surrenti C. Ethanol-induced alterations of matrix network in the duodenal mucosa of chronic alcohol abusers. Virchows Arch. 1999;434:127–135. doi: 10.1007/s004280050316. [DOI] [PubMed] [Google Scholar]
- Chen WY, Abatangelo G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999;7:79–89. doi: 10.1046/j.1524-475x.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- Cherpitel CJ. Alcohol and injuries: a review of international emergency room studies. Addiction. 1993a;88:923–937. doi: 10.1111/j.1360-0443.1993.tb02110.x. [DOI] [PubMed] [Google Scholar]
- Cherpitel CJ. Alcohol and violence-related injuries: an emergency room study. Addiction. 1993b;88:79–88. doi: 10.1111/j.1360-0443.1993.tb02765.x. [DOI] [PubMed] [Google Scholar]
- Choudhry MA, Chaudry IH. Alcohol intoxication and post-burn complications. Front Biosci. 2006;11:998–1005. doi: 10.2741/1857. [DOI] [PubMed] [Google Scholar]
- Clark R. Wound repair. In: Clark R, editor. The Molecular and Cellular Biology of Wound Repair. Plenum Press; New York: 1995. pp. 3–21. [Google Scholar]
- Clark RA. Biology of dermal wound repair. Dermatol Clin. 1993;11:647–666. [PubMed] [Google Scholar]
- Ehrlich HP. The role of connective tissue matrix in wound healing. Prog Clin Biol Res. 1988;266:243–258. [PubMed] [Google Scholar]
- Faunce DE, Gregory MS, Kovacs EJ. Effects of acute ethanol exposure on cellular immune responses in a murine model of thermal injury. J Leukoc Biol. 1997;62:733–740. doi: 10.1002/jlb.62.6.733. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DJ, Radek KA, Chaar M, Faunce DE, DiPietro LA, Kovacs EJ. Effects of acute ethanol exposure on the early inflammatory response after excisional injury. Alcohol Clin Exp Res. 2007;31:317–323. doi: 10.1111/j.1530-0277.2006.00307.x. [DOI] [PubMed] [Google Scholar]
- Fushida-Takemura H, Fukuda M, Maekawa N, Chanoki M, Kobayashi H, Yashiro N, Ishii M, Hamada T, Otani S, Ooshima A. Detection of lysyl oxidase gene expression in rat skin during wound healing. Arch Dermatol Res. 1996;288:7–10. doi: 10.1007/BF02505035. [DOI] [PubMed] [Google Scholar]
- Gao F, Liu Y, He Y, Yang C, Wang Y, Shi X, Wei G. Hyaluronan oligosaccharides promote excisional wound healing through enhanced angiogenesis. Matrix Biol. 2010;29:107–116. doi: 10.1016/j.matbio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Gentilello LM, Cobean RA, Walker AP, Moore EE, Wertz MJ, Dellinger EP. Acute ethanol intoxication increases the risk of infection following penetrating abdominal trauma. J Trauma. 1993;34:669–674. doi: 10.1097/00005373-199305000-00009. discussion 674–675. [DOI] [PubMed] [Google Scholar]
- Girasek DC, Gielen AC, Smith GS. Alcohol’s contribution to fatal injuries: a report on public perceptions. Ann Emerg Med. 2002;39:622–630. doi: 10.1067/mem.2002.122864. [DOI] [PubMed] [Google Scholar]
- Greenhalgh D, Gamelli RL. Immunomodulators and wound healing. J Trauma. 1987;27:510–514. doi: 10.1097/00005373-198705000-00009. [DOI] [PubMed] [Google Scholar]
- Grenett HE, Aikens ML, Torres JA, Demissie S, Tabengwa EM, Davis GC, Booyse FM. Ethanol transcriptionally upregulates t-PA and u-PA gene expression in cultured human endothelial cells. Alcohol Clin Exp Res. 1998;22:849–853. [PubMed] [Google Scholar]
- Hebda PA, Collins MA, Tharp MD. Mast cell and myofibroblast in wound healing. Dermatol Clin. 1993;11:685–696. [PubMed] [Google Scholar]
- Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- Kovacs EJ, DiPietro LA. Fibrogenic cytokines and connective tissue production. Faseb J. 1994;8:854–861. doi: 10.1096/fasebj.8.11.7520879. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. Faseb J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Lois M, Brown LA, Moss IM, Roman J, Guidot DM. Ethanol ingestion increases activation of matrix metalloproteinases in rat lungs during acute endotoxemia. Am J Respir Crit Care Med. 1999;160:1354–1360. doi: 10.1164/ajrccm.160.4.9811060. [DOI] [PubMed] [Google Scholar]
- Makela JT, Kiviniemi H, Laitinen S. Risk factors for anastomotic leakage after left-sided colorectal resection with rectal anastomosis. Dis Colon Rectum. 2003;46:653–660. doi: 10.1007/s10350-004-6627-9. [DOI] [PubMed] [Google Scholar]
- McGill V, Kowal-Vern A, Fisher SG, Kahn S, Gamelli RL. The impact of substance use on mortality and morbidity from thermal injury. J Trauma. 1995;38:931–934. doi: 10.1097/00005373-199506000-00019. [DOI] [PubMed] [Google Scholar]
- Messingham KA, Fontanilla CV, Colantoni A, Duffner LA, Kovacs EJ. Cellular immunity after ethanol exposure and burn injury: dose and time dependence. Alcohol. 2000;22:35–44. doi: 10.1016/s0741-8329(00)00100-2. [DOI] [PubMed] [Google Scholar]
- Molina PE, McClain C, Valla D, Guidot D, Diehl AM, Lang CH, Neuman M. Molecular pathology and clinical aspects of alcohol-induced tissue injury. Alcohol Clin Exp Res. 2002;26:120–128. [PubMed] [Google Scholar]
- Nickelsen TN, Jorgensen T, Kronborg O. Lifestyle and 30-day complications to surgery for colorectal cancer. Acta Oncol. 2005;44:218–223. doi: 10.1080/02841860510029707. [DOI] [PubMed] [Google Scholar]
- Nolan A, Baillie C, Badminton J, Rudralingham M, Seymour RA. The efficacy of topical hyaluronic acid in the management of recurrent aphthous ulceration. J Oral Pathol Med. 2006;35:461–465. doi: 10.1111/j.1600-0714.2006.00433.x. [DOI] [PubMed] [Google Scholar]
- Radek KA, Kovacs EJ, DiPietro LA. Matrix proteolytic activity during wound healing: modulation by acute ethanol exposure. Alcohol Clin Exp Res. 2007;31:1045–1052. doi: 10.1111/j.1530-0277.2007.00386.x. [DOI] [PubMed] [Google Scholar]
- Radek KA, Kovacs EJ, Gallo RL, DiPietro LA. Acute ethanol exposure disrupts VEGF receptor cell signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H174–184. doi: 10.1152/ajpheart.00699.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radek KA, Matthies AM, Burns AL, Heinrich SA, Kovacs EJ, Dipietro LA. Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am J Physiol Heart Circ Physiol. 2005;289:H1084–1090. doi: 10.1152/ajpheart.00080.2005. [DOI] [PubMed] [Google Scholar]
- Rai DV, Kumar G, Tewari P, Saxena DC. Acute and chronic dose of alcohol affect the load carrying capacity of long bone in rats. J Biomech. 2008;41:20–24. doi: 10.1016/j.jbiomech.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Raza SL, Cornelius LA. Matrix metalloproteinases: pro- and anti-angiogenic activities. J Investig Dermatol Symp Proc. 2000;5:47–54. doi: 10.1046/j.1087-0024.2000.00004.x. [DOI] [PubMed] [Google Scholar]
- Rivara FP, Jurkovich GJ, Gurney JG, Seguin D, Fligner CL, Ries R, Raisys VA, Copass M. The magnitude of acute and chronic alcohol abuse in trauma patients. Arch Surg. 1993;128:907–912. doi: 10.1001/archsurg.1993.01420200081015. discussion 912–913. [DOI] [PubMed] [Google Scholar]
- Salo T, Makela M, Kylmaniemi M, Autio-Harmainen H, Larjava H. Expression of matrix metalloproteinase-2 and -9 during early human wound healing. Lab Invest. 1994;70:176–182. [PubMed] [Google Scholar]
- Singhal PC, Reddy K, Ding G, Kapasi A, Franki N, Ranjan R, Nwakoby IE, Gibbons N. Ethanol-induced macrophage apoptosis: the role of TGF-beta. J Immunol. 1999;162:3031–3036. [PubMed] [Google Scholar]
- Soo C, Shaw WW, Zhang X, Longaker MT, Howard EW, Ting K. Differential expression of matrix metalloproteinases and their tissue-derived inhibitors in cutaneous wound repair. Plast Reconstr Surg. 2000;105:638–647. doi: 10.1097/00006534-200002000-00024. [DOI] [PubMed] [Google Scholar]
- Sorensen LT, Jorgensen T, Kirkeby LT, Skovdal J, Vennits B, Wille-Jorgensen P. Smoking and alcohol abuse are major risk factors for anastomotic leakage in colorectal surgery. Br J Surg. 1999;86:927–931. doi: 10.1046/j.1365-2168.1999.01165.x. [DOI] [PubMed] [Google Scholar]
- Steffensen B, Hakkinen L, Larjava H. Proteolytic events of wound-healing--coordinated interactions among matrix metalloproteinases (MMPs), integrins, and extracellular matrix molecules. Crit Rev Oral Biol Med. 2001;12:373–398. doi: 10.1177/10454411010120050201. [DOI] [PubMed] [Google Scholar]
- Stephens P, al-Khateeb T, Davies KJ, Shepherd JP, Thomas DW. An investigation of the interaction between alcohol and fibroblasts in wound healing. Int J Oral Maxillofac Surg. 1996;25:161–164. doi: 10.1016/s0901-5027(96)80065-8. [DOI] [PubMed] [Google Scholar]
- Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Lab Invest. 1999;79:1479–1487. [PubMed] [Google Scholar]
- Szauter KM, Cao T, Boyd CD, Csiszar K. Lysyl oxidase in development, aging and pathologies of the skin. Pathol Biol (Paris) 2005;53:448–456. doi: 10.1016/j.patbio.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Thal ER, Bost RO, Anderson RJ. Effects of alcohol and other drugs on traumatized patients. Arch Surg. 1985;120:708–712. doi: 10.1001/archsurg.1985.01390300058010. [DOI] [PubMed] [Google Scholar]
- Tonnesen H, Schutten BT, Jorgensen BB. Influence of alcohol on morbidity after colonic surgery. Dis Colon Rectum. 1987;30:549–551. doi: 10.1007/BF02554788. [DOI] [PubMed] [Google Scholar]
- Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5:40–46. doi: 10.1046/j.1087-0024.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Kurabayashi M, Ohyama Y, Utsugi T, Akuzawa N, Sato M, Tomono S, Kawazu S, Nagai R. Hypoxia induces transcription of the plasminogen activator inhibitor-1 gene through genistein-sensitive tyrosine kinase pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:1155–1161. doi: 10.1161/01.atv.20.4.1155. [DOI] [PubMed] [Google Scholar]
- Voinchet V, Vasseur P, Kern J. Efficacy and safety of hyaluronic acid in the management of acute wounds. Am J Clin Dermatol. 2006;7:353–357. doi: 10.2165/00128071-200607060-00003. [DOI] [PubMed] [Google Scholar]
- Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Yoshikawa K. Cutaneous wound healing: an update. J Dermatol. 2001;28:521–534. doi: 10.1111/j.1346-8138.2001.tb00025.x. [DOI] [PubMed] [Google Scholar]