

Abstract
Background
Proton leak (H+ leak) dissipates mitochondrial membrane potential (mΔΨ) through the reentry of protons into the mitochondrial matrix independent of ATP synthase. Changes in H+ leak may affect reactive oxygen species (ROS) production. We measured H+ leak and ROS production during ischemia-reperfusion and ischemic preconditioning (IPC) and examined how changing mitochondrial respiration affected mΔΨ and ROS production.
Materials/Methods
Isolated rat hearts (n=6/group) were subjected to either Control-IR or IPC. Rate pressure product (RPP) was measured. Mitochondria were isolated at end reperfusion. Respiration was measured by polarography and titrated with increasing concentrations of malonate (0.5-2mM). mΔΨ was measured using a tetraphenylphosphonium electrode. H+ leak is the respiratory rate required to maintain membrane potential at -150mV in the presence of oligomycin-A Mitochondrial complex III ROS production was measured by fluorometry using Amplex-Red.
Results
IPC improved recovery of RPP at end reperfusion (63±4% vs. 21±2% in Control-IR, p<0.05). Ischemia-reperfusion caused increased H+ leak (94±12 vs. 31±1 nanomoles O/mg protein/min in Non-Ischemic Control, p<0.05). IPC attenuates these increases (55±9 nanomoles O/mg protein/min, p< 0.05 vs. Control-IR). IPC reduced mitochondrial ROS production compared to Control-IR (31±2 vs. 40±3 nanomoles/mg protein/min, p<0.05). As mitochondrial respiration decreased, mΔΨ and mitochondrial ROS production also decreased. ROS production remained lower in IPC than in Control-IR for all mΔΨ and respiration rates.
Conclusions
Increasing H+ leak is not associated with increased ROS production. IPC decreases both the magnitude of H+ leak and ROS production after ischemia-reperfusion.
Keywords: Ischemia, reperfusion, reactive oxygen species, mitochondria, proton leak, uncoupling, heart
Introduction
Ischemia-reperfusion damages the mitochondrial electron transport chain and increases the production of reactive oxygen species (ROS).1-4 Ischemia-reperfusion injury causes electrons to leak from respiratory complex I and III, passing one electron to oxygen generating superoxide radical (O2·−), the major free radical ROS produced by the mitochondria.2, 5 The burst of ROS produced upon reperfusion of the postischemic heart is responsible for many pathologic changes in the myocardium including lipid peroxidation, which damages cellular membranes, and loss of contractile function.6 Increased ROS also causes activation of the mitochondrial permeability transition pore (MPTP) leading to possible loss of ion homeostasis and cell death.1, 7
The term ischemic preconditioning (IPC) is used to describe the process by which brief periods of ischemia-reperfusion (3 to 5 minutes) protects the myocardium from the damaging effects of subsequent longer episodes of ischemia-reperfusion.1 IPC leads to preservation of cardiac contractile function and reduction of infarct size during reperfusion.1 IPC is also known to preserve mitochondrial function after ischemia-reperfusion injury.8 Our lab has shown that IPC specifically preserves the function of mitochondrial respiratory complex I and II and decreases mitochondrial superoxide production.9-11 The exact mechanism by which IPC leads to such effects on the mitochondria has not been determined. We hypothesize that they may be related to changes in mitochondrial membrane potential (mΔΨ). mΔΨ regulates ROS production by changing the redox status of the electron transport chain. 12, 13 Previous studies from our laboratory have confirmed that the in vivo redox status of the myocardium changes dramatically throughout an episode of ischemia-reperfusion with associated changes in ROS production.14 When the mΔΨ is sufficiently high the ETC becomes reduced, the flow of electrons slows down, and electrons are leaked to oxygen generating O2·−.12, 13, 15, 16 Mild depolarization of the inner mitochondrial membrane can restore the flow of electrons along the electron transport chain and decrease O2·− production.12 H+ leak depolarizes mΔΨ through the reentry of protons into the mitochondrial matrix independent from ATP synthesis (uncoupling). The reduction of mΔΨ without the production of ATP leads to loss of mitochondrial efficiency. By depolarizing mΔΨ, H+ leak may decrease ROS production and lead to cardio-protection.12, 17-19 Previous studies have demonstrated differences in the rate and mechanism of H+ leak in IPC and non-preconditioned mitochondria,19 but the relationship between the observed H+ leak and ROS production in these two groups have yet to be determined.
The current experiments measured the magnitude of mitochondrial H+ leak in IPC and non-preconditioned rat hearts to determine how H+ leak correlates with ROS production after an episode of ischemia-reperfusion. Previous studies have shown that mild uncoupling through mechanisms such as H+ leak can decrease ROS production.12, 16 Our results indicate that preconditioning decreases H+ leak and also decreases ROS production when compared to non-preconditioned mitochondria. Therefore, IPC is shown to preserve mitochondrial efficiency by limiting H+ leak while preventing the formation of increased amounts of ROS after an episode of ischemia-reperfusion.
Materials/Methods
Isolated heart preparation
Male Sprague–Dawley rats (275–300 g) were anesthetized with sodium pentobarbital (60 mg/kg intraperitoneally, ip) and heparinized (heparin sodium, 500 U ip). Hearts were excised quickly and arrested in cold Krebs–Henseleit solution. Hearts were then perfused in a non-recirculating Langendorff apparatus at 37°C with Krebs– Henseleit buffer consisting of [in mM] NaCl [118]; KCl [4.6]; KH2PO4 [1.17]; MgSO4 [1.17]; CaCl2 [1.16]; NaHCO3 [23]; and glucose [5.3]; pH: 7.4 and equilibrated with 95% O2 and 5% CO2 gas. Left ventricular rate pressure product (RPP, peak systolic pressure minus end diastolic pressure multiplied by heart rate) was recorded using an intraventricular latex balloon connected to a pressure transducer.20 Data were continuously recorded using a PowerLab Chart v4.2 (AD Instruments Inc., Milford, MA) and a Dell GenuineIntel ×86 Family 6 Model Stepping 6 computer (Dell Computer Corp., Round Rock, TX).
Rats were acclimated in a quiet environment and fed a standard diet. They were treated in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources of the National Research Council, 1996.
Ischemic preconditioning protocol
Hearts were assigned to Control-IR and Ischemic preconditioning (IPC) group. The Control-IR group (n=6) was subjected to 30 minutes of equilibration, 30 minutes of global normothermic ischemia, and 30 minutes of reperfusion. The IPC group (n=6) was subjected to 10 minutes of equilibration, then ischemic preconditioning was induced by two 5-minute episodes of ischemia each followed by 5 minutes of re-equilibration, followed by 30 minutes of global normothermic ischemia, and 30 minutes of reperfusion. Ischemia was achieved by a stopcock located directly above the aorta. Hearts were immersed at all times in a water jacketed non-gassed perfusate bath to maintain normothermia (37°C). Both control and IPC hearts were homogenized for mitochondria isolation described below at end equilibration and end reperfusion.
Isolation of subsarcolemmal (SS) mitochondria
SS mitochondria were isolated by differential centrifugation according to Palmer et al.22 Briefly, hearts were minced in the isolation buffer (MSH): [in mmol/L] mannitol [230]; sucrose [70]; Hepes [3]; pH: 7.4 at 4°C, with the addition of 0.2% bovine serum albumin (BSA) and 1mM EGTA, and homogenized with a polytron tissue processor (Brinkman Instruments, Westbury, NY) for 3 seconds at a rheostat setting of 6.5. Further homogenization was performed with a glass homogenizer (Kontes Glass Co., Vineland, NJ). The polytron homogenate was centrifuged at 500 × g for 10 minutes. The supernatant was centrifuged at 5000 × g for 10 minutes for isolation of SS mitochondria. Subsequent pellets were washed twice at 5000 × g for 10 minutes each with MSH buffer and lastly, suspended in 0.5 ml of MSH buffer with 0.2% BSA and EGTA 1mM. Protein concentration was determined by the Lowry method.23
Measurement of mitochondrial oxygen consumption
Mitochondrial respiration was measured by the polarographic method of Chance and Williams using a Clark oxygen electrode.24 The incubation media contained [in mmol/L]: KCl [100]; KH2PO4 [1]; HEPES [50], pH: 7.4 at 37°C. Succinate (5mM) was used as substrate for complex II. Rotenone (4.15 μM) was used to inhibit backflow to complex I. ATP synthase was inhibited with oligomycin-A [1.67μM] to measure proton leak. Mitochondria (approximately 0.5mg of protein) were added last to initiate state 2 respiration. Increasing concentrations of malonate [0.5-2mM] were used to titrate respiration, and its effect on H2O2 production and membrane potential were measured.
Measurement of hydrogen peroxide production by fluorometry
Complex III H2O2 production was measured using Amplex-red. In a polystyrene cuvette containing 3mL of buffer solution [in mM] KCl [100]; KH2PO4 [1] and HEPES [50], horseradish peroxidase [0.01mM]; succinate [5 mM]; rotenone [4.15 μM]; KPi [3μM] Amplex-Red [50μM] and oligomycin-A [1.67μM] were added followed lastly by mitochondria (approximately 0.05 mg of protein). Fluorescence was recorded for 5 minutes at excitation 560nm and emission 583nm at 37°C. The slope of the fluorescence curve of each sample was used to measure the rate of H2O2 formation by inputting the measured slope into the formula for a standard curve. The standard curve was obtained by adding known concentrations of H2O2 to buffer solution containing horseradish peroxidase and Amplex-Red. To measure the effect of respiration on ROS production, increasing concentrations of malonate [0.5-2mM] were added to the cuvette.
Membrane potential
Membrane potential was measured using a tetraphenylphosphonium (TPP) electrode.25-28 A calibration curve was obtained prior to the start of each experiment using a total of 2.8μM TPP; membrane potential was calculated from the Nernst equation using the TPP binding correction factor and temperature corrected to 37°C. The incubation media contained [in mmol/L]: KCl [100]; KH2PO4 [1]; HEPES [50], pH: 7.4, at 37°C. Succinate (5mM) was used as substrate for complex II. Rotenone (4.15 μM) was used to inhibit backflow of electrons to complex I.
Proton leak
H+ leak was measured as the state 2 (oxygen consumption in the absence of ATP phosphorylation) respiration in the presence of oligomycin-A (1.67μM). Malonate was added to the reaction chamber in concentrations ranging from 0.5mM to 2mM to titrate respiration. Respiration and membrane potential were measured simultaneously as previously described. H+ leak was defined as the state 2 respiratory rate necessary to maintain mΔΨ of -150mV.29
Statistical analysis
Data are expressed as means ± SEM. Students t-test was used to compare pairs; for multiple group comparisons, Two-way ANOVA with the Holm-Sidak post-hoc test was used to determine significance. p< 0.05 was considered to be statistically significant.
Materials
Amplex-Red was obtained from Invitrogen Corp., Carlsbad, California. All other chemicals were obtained from Sigma-Aldrich Inc., St. Louis, Missouri. Rats were obtained from Harlan Laboratories, Indianapolis, Indiana.
Results
Mechanical Function
Figure 1A is a representative pressure tracing which graphically demonstrates contractile function throughout the experimental protocol. After two episodes of preconditioning (End EQ), the rate pressure product (RPP) for IPC hearts was 87±3% compared to the first 10 minutes of equilibration. This indicates that the brief episodes of preconditioning did lead to mild loss of mechanical function of approximately 13±3%. The protective effects of IPC are evident at end reperfusion where there is greater preservation of mechanical function compare to Control-IR. At end reperfusion, IPC hearts recovered 63± 4% of their end equilibration RPP compared to 21±2% in the Control-IR group (p<0.05, Figure 1B).
Figure 1.
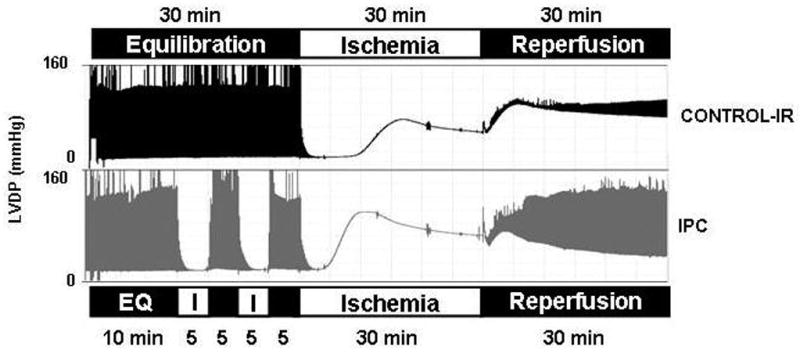

1A Representative left ventricular developed pressure (LVDP) tracings during Control-IR and IPC experiments. 1B) Recovery of left ventricular function at end-reperfusion expressed as percent of end-equilibration rate pressure product (RPP) in IPC and Control-IR. IPC hearts recovered 63±4% of their RPP vs. 21±2% in Control-IR (p<0.05, n=6/group).
Proton leak
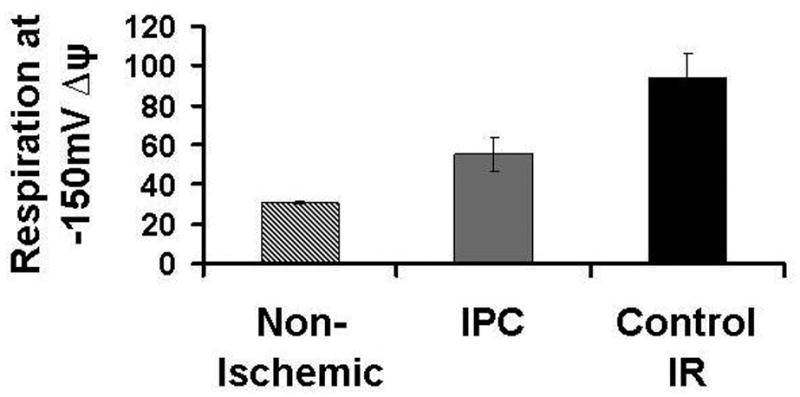
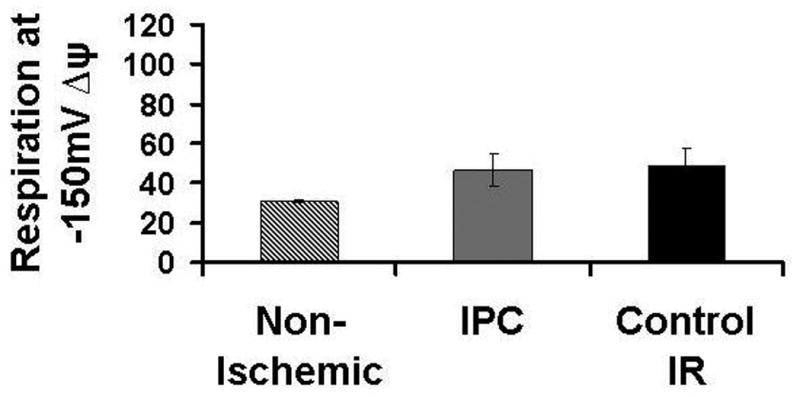
H+ leak was measured as the state 2 respiratory rate required to maintain mΔΨ of -150mV. At end equilibration, there is an increase in H+ leak in both Control-IR and IPC mitochondria when compared to Non-Ischemic, however, this difference is marginally significant. H+ leak was 49 ±9 nanomoles O/mg protein/min for Control-IR, 47±8 nanomoles O/mg protein/min for IPC and 31±1 nanomoles O/mg protein/min for Non-Ischemic (p=o.o5 for both IPC and Control-IR vs. Non-Ischemic; figure 2C). At end reperfusion, H+ leak was 94±12 nmoles O/minute/mg protein for Control-IR vs. 55±9 nmoles O/minute/mg protein for IPC and 31±1 nmoles O/minute/mg protein for Non-Ischemic mitochondria (p<0.05, Figure 2A, B). This indicates that IPC attenuates the increase in H+ leak caused by ischemia-reperfusion injury. Increasing malonate concentrations led to decreasing mΔΨ. Malonate inhibits respiration at complex II; inhibiting the respiratory activity of the electron transport chain decreases the transport of H+ from the matrix into the intermembrane space which normally occurs during respiration. Continued H+ leak into the matrix along with decreased flow of H+ back to the intermembrane space leads to dissipation of mΔΨ (Figure 2A).
Figure 2. Mitochondrial H+ leak.
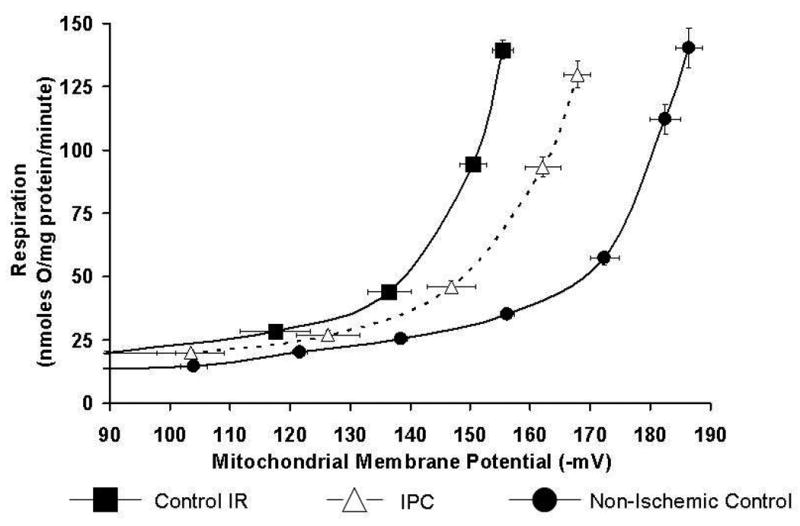
2A: H+ leak curves in Control-IR and IPC at end-reperfusion and in Non-Ischemic Control mitochondria. H+ leak is defined as the respiratory rate required to maintain mΔΨ of -150mV. Respiration and mΔΨ was titrated with malonate (see methods section). Increasing malonate concentrations led to decreasing mΔΨ and state 2 respiration. N=6 for each data points; 6 separate experiments.
2B: H+ leak in Control-IR and IPC at end-perfusion and in Non-Ischemic Control extrapolated from 2A curves. IPC had a higher H+ leak compared to Non-Ischemic Control, while Control-IR had a higher H+ leak compared to both Non-Ischemic and IPC. *p< 0.05 vs. Non-Ischemic, § p<0.05 vs. IPC and Non-Ischemic.
2C: H+ leak in Control-IR and IPC at End EQ and in Non-Ischemic Control. There was an increase in H+ leak in both Control-IR and IPC mitochondria when compared to Non-Ischemic; however this difference was only marginally significant. H+ leak was 49±9 in Control-IR, 47±8 for IPC and 31±1 nanomoles O/mg protein/min for Non-Ischemic (p=0.05 for both IPC and Control-IR vs. Non-Ischemic).
Mitochondrial respiration
There was no difference in baseline rate of mitochondrial state 2 respiration between IPC and Control-IR at end reperfusion. Increasing additions of malonate decreased the respiratory activity equally in both groups of mitochondria (Figure 3). While specific complex activities were not measured in this experiment, we previously demonstrated that IPC protects the respiratory activity of complex I and II following an episode of ischemia-reperfusion.9, 10
Figure 3.

Effect of increasing malonate concentrations on mitochondrial respiration in both IPC and Control-IR. Baseline respiration is approximately the same in both groups (n=6/group) and declines at an equal rate with increasing concentrations of malonate.
Hydrogen Peroxide Production
Baseline hydrogen peroxide (H2O2) production at end reperfusion for IPC mitochondria was 31±2 nmoles/mg protein/minute compared to 40±3 nmoles/mg protein/minute for Control-IR (p<0.05). As malonate concentration increases, H2O2 production in IPC remains lower than Control-IR for all rates of respiration (Figure 4A). Previously recorded values of ROS production in heart mitochondria are somewhat lower than the values recorded in the current experiment.2, 30; however, those experiments did not assess the effect of reperfusion injury, and one looked at ROS production in pigeon hearts. It is now known that reperfusion injury is responsible for a large increase in free radical formation observed in heart mitochondria.4, 6, 31, 32 It is also known that different organ tissues produce ROS at different rates,13 and variation can be expected among different species. Figure 4B is a representative tracing of the fluorometry studies measuring H2O2 production. Catalase was used as a negative control. In the presence of catalase, H2O2 is converted to O2 and H2O. The addition of catalase to our reaction chamber, with succinate as a substrate, attenuated the production of H2O2 to baseline levels. This indicates that the increased fluorescence observed in our experiments was indeed due to H2O2 formation.
Figure 4.
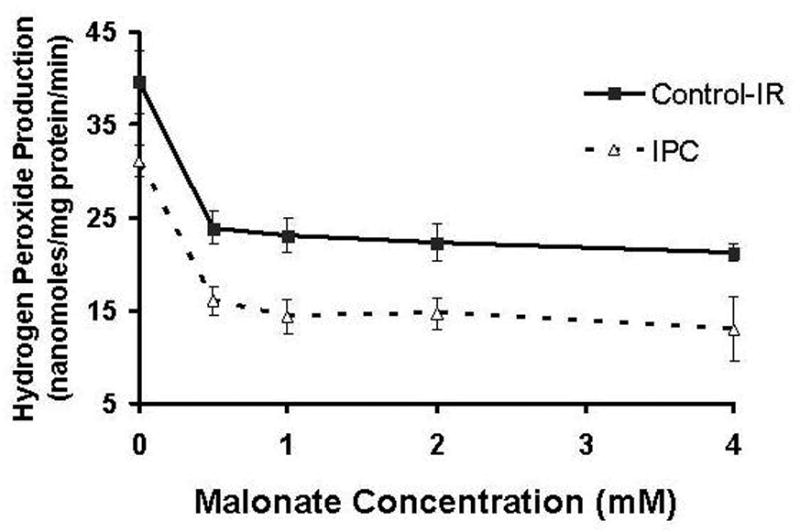

4A. Effect of increasing concentration of malonate on mitochondrial hydrogen peroxide production. At different respiratory rates, IPC mitochondria produced less H2O2. IPC had a lower basal rate of H2O2 production (omM malonate point). Inhibition of respiration decreased ROS production in both groups, with IPC producing less at all points. *p<0.05 vs. Control IR, n=6/group.
4B. H2O2 production as measured by fluorometry. In the presence of horseradish peroxidase (catalyst), Amplex-red reacts with H2O2 to produce the highly fluorescent compound resorufin. Catalase reduces H2O2 to H2O and was used as a negative control. The addition of catalase to the reaction chamber abolishes any increase in fluorometric signal.
Discussion
The main findings of this study are that IPC significantly decreases the rate of both H+ leak and ROS production after ischemia-reperfusion injury. We also demonstrated that decreasing the respiratory activity of the mitochondrial electron transport chain decreases mΔΨ and ROS production in both preconditioned and Control-IR heart mitochondria after ischemia-reperfusion injury.
Proton leak
It has previously been shown that increased ROS production increases mitochondrial H+ leak. 17, 18, 33, 34 It is also known that increased H+ leak decreases ROS production.17, 33, 35 This led to the theory of a protective feedback loop, where increased ROS production activates mechanisms that lead to increased H+ leak; the increased H+ leak then leads to decreased production of ROS, limiting further damage to mitochondrial function.17, 18, 33, 34 It is unknown whether H+ leak is a part of the protective mechanism of IPC.
The current study demonstrated that ischemia-reperfusion injury increased H+ leak in isolated mitochondria. IPC decreased H+ leak after ischemia-reperfusion when compared to non-preconditioned mitochondria (Figure 2C). Our findings are in agreement with those of Nadtochiy et al.19 This group found that mitochondria that underwent ischemia-reperfusion injury had a higher rate of H+ leak compared to IPC. The findings of our study and that of Nadtochiy et al may initially seem to be in contrast to the theory that increased H+ leak is a protective mechanism and would seem to indicate that the protective effects of IPC are not due to an increased H+ leak. To explain this apparent discrepancy, we must discuss two important processes that affect both H+ leak and ROS production: 1) The different mechanisms responsible for H+ leak in IPC and Control-IR mitochondria; and 2) The change in redox status of the mitochondria induced by ischemia-reperfusion injury.
Other groups have shown that mitochondrial uncoupling with agents such as carbonyl cyanide p-(tri-fluromethoxy)phenyl-hydrazone (FCCP) or 2,4-Dinitrophenol (DNP) protects mitochondrial and myocardial function from ischemia-reperfusion injury in a manner similar to IPC.33, 36 Several mechanisms are involved in uncoupling. Among them are activation of uncoupling proteins (UCPs), the adenine nucleotide translocase (ANT), and the MPTP.19, 34, 37-39 Nadtochiy et al. demonstrated that the mechanisms responsible for increased H+ leak were different in the IPC and control groups. The small increase in H+ leak in IPC was mediated by uncoupling proteins UCPs, while the larger increase seen in the control group was mediated by the ANT.18, 19 The ANT is believed to be one of the components the MPTP.39 While mild cardio-protective increases in H+ leak may be mediated by UCPs, large increases may be mediated by the MPTP.1, 7, 18, 37, 38-40 More severe episodes of ischemia lead to more severe disruption of mitochondrial membrane and greater H+ leak.41, 42 It is possible to postulate that high rates of H+ leak as seen in the Control-IR group, is a marker of inner mitochondrial membrane damage/disruption. It is still possible that a small degree of H+ leak may in fact be beneficial in protecting against the effects of ischemia-reperfusion injury.19 Figure 2B shows the rate of H+ leak for Control-IR, IPC and Non-Ischemic mitochondria at End RP. IPC has a lower H+ leak when compared to Control-IR; however, the rate of H+ leak in IPC is also significantly higher than Non-Ischemic mitochondria. We postulate that the small increase in H+ leak seen in IPC is indeed protective. The moderate increase in H+ leak in IPC is due to activation of UCPs and is due to the increased ROS caused by ischemia-reperfusion injury.18, 19 The larger H+ leak seen in ischemia-reperfusion injury is the result of activation of the ANT19 which has been shown to be a part of the MPTP (for review see 40). This increased H+ leak mediated by the MPTP is detrimental and can lead to cell death.7, 40 This indicates that the degree of H+ leak and the mechanism involved determine whether there is a protective effect.
Aon et al. recently reported on the effects of increased uncoupling on both isolated mitochondria and cardiac myocytes.46 In their experiment, increasing the rate of H+ leak to achieve uncoupling led to decreasing ROS production; however, this decrease in ROS was dependent on the redox status of the mitochondria.46 In isolated, energized mitochondria, the redox potential is high, leading to increased ROS production as previously described. Mild uncoupling with FCCP led to a decrease in ROS production in mitochondria that were in a highly reduced state (high redox potential). When isolated mitochondria were oxidatively stressed and pushed towards the other extreme of the redox potential (i.e. highly oxidized), there was an increase in ROS production with mild uncoupling.46 In intact myocytes, uncoupling increased ROS production.46 Our group has previously demonstrated that ischemia-reperfusion changes the redox status of the in vivo postischemic myocardium.14 Ischemia caused a shift in tissue redox status to a more reduced state, while the induction of reperfusion caused a change in redox status to a more oxidized state.14 We could postulate that IPC preserves the redox balance of mitochondria, preventing the large changes in redox status and ROS production. Further studies are needed to definitively determine how IPC affects the redox status of mitochondria throughout an episode of ischemia-reperfusion.
Hydrogen Peroxide Production
Measuring H2O2 production is one method of examining ROS production by mitochondria. O2·− is rapidly dismutated to H2O2 by the enzyme manganese superoxide dismutase (SOD2) located in the inner mitochondrial membrane. H2O2 then diffuses across the mitochondrial membrane. In the presence of horseradish peroxidase, H2O2 reacts with Amplex-red to form the highly fluorescent product resorufin, which can be detected by fluorometry.
Previous studies have shown that reperfusion after an episode of ischemia causes myocardial hyperoxygenation and a subsequent burst of ROS.6 This increase in ROS is responsible for pathologic changes such as membrane lipid peroxidation and activation of the MPTP leading to cell death.6, 7, 11 Our lab has shown that IPC prevents myocardial hyperoxygenation after reperfusion and preserves myocardial O2 metabolism.11 The results of the current experiment indicate that IPC mitochondria produce less ROS at end reperfusion compared to Control-IR. This is consistent with our previous studies using electron paramagnetic resonance and spin trapping,9 which is a specific method for measuring O2·− production.43 Our group has also previously demonstrated that IPC decreases ROS formation in vivo and attenuates the negative effects mediated by ischemia-reperfusion injury.11 Understanding what mechanisms attenuate ROS formation in IPC after ischemia-reperfusion injury is important; ROS generated by mitochondria during ischemia-reperfusion play a key role in myocardial cell dysfunction and death.16
It is known that mitochondrial production of ROS is directly affected by mΔΨ12, 15, 16 At high levels of mΔΨ (greater than -160mV), the flow of electrons through the mitochondrial ETC slows down and electrons leak from complex I and complex III.12, 13, 42 As shown in figure 5, these electrons react with oxygen to produce O2·− the principal free radical ROS produced by mitochondria, with complex III being the major source of ROS production.5 Mitochondrial uncoupling restores the flow of electrons through the electron transport chain, therefore fewer electrons leak from the complexes to form O2·−.13, 33, 37, 39 mΔΨ also affects the redox state of the mitochondrial NAD(P)H system which in turn affects the rate of ROS formation.14, 16 At high mΔΨ, in isolated mitochondria, the mitochondrial NAD(P)H system is in a highly reduced state with an associated increase in the rate of ROS production; 14 this is also associated with a deceleration of state 3 respiration toward a resting state 4 respiration.12, 16 Uncoupling of mΔΨ is associated with greater oxidation of NAD(P)H and decreased ROS production in isolated mitochondria.16 Chen et al have also shown that ischemia-reperfusion injury leads to reduction of state 3 respiration with an increase in state 4 respiration and an associated increase in ROS production at complex I and III.2 These studies indicate that ischemia-reperfusion injury causes a highly reduced redox state with increased state 4 respiration and associated increased ROS production. Our results show that IPC decreases ROS production under these conditions. In the current experiment, isolated mitochondria subjected to ischemia-reperfusion injury and undergoing state 4 respiration demonstrated lower rates of ROS production in the IPC group compared to Control-IR (Figure 4, o mM Malonate point). Reducing ROS production after ischemia-reperfusion injury may be a part of the mechanism of IPC.
Figure 5.
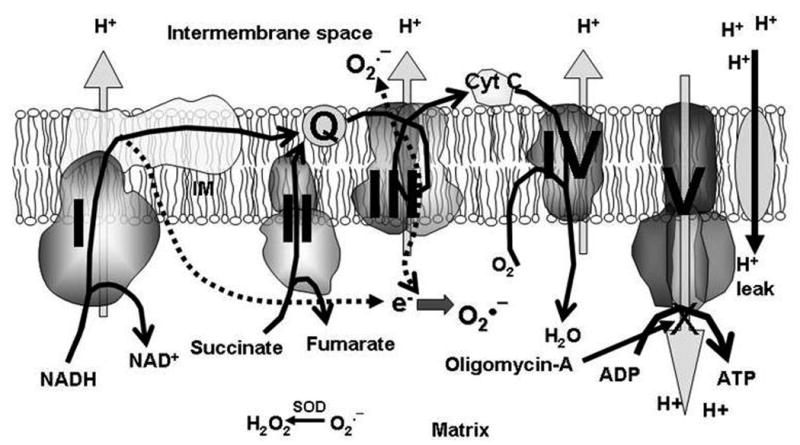
Physiology of the respiratory electron transport chain. Heavy curved arrows indicate the normal flow of electrons through the ETC. Substrates, such as NADH and succinate, normally enter at complex I and II where they are oxidized. The electrons are then transported to complex III by ubiquinol. Cytochrome c transfers electrons from complex III to complex IV which reduces O2 to form H2O. In the process, electrons are pumped from the matrix into the intermembrane space at complex I, III and IV generating an electrochemical gradient, the mΔΨ. This gradient is then used by complex V, (ATP synthase), to power the production of ATP from ADP. H+ leak is the reentry of protons into the mitochondrial matrix independent of complex V. Complex V is normally the point of reentry for H+ back into the matrix, with only minimal H+ leak. In the setting of ischemia-reperfusion injury, the respiratory complexes are damaged, and electrons leak from complex I and III (indicated by broken arrows). These electrons bind to O2 to produce O2·−. O2·− is dismutated to form H2O2 by the enzyme superoxide dismutase (SOD). H2O2 inside the matrix crosses the inner mitochondrial membrane to the intermembrane space. In the setting of ischemia-reperfusion injury H+ leak increases. Oligomycin-A is a complex V inhibitor; when complex V is inhibited, all reentry of H+ into matrix is due to H+ leak.
Several sites have been shown to be responsible for ROS production in the mitochondria including complex I, complex III, and ά-ketoglutarate dehydrogenase; there is also a significant dependence on the redox status of coenzyme Q.13, 15 Complex III has been identified as the dominant site of ROS production in heart mitochondria and in the setting of ischemia-reperfusion injury.5, 13, 42 In the current study, rotenone was used to inhibit backflow of electrons into complex I, eliminating production of ROS at complex I.42 Using succinate as a complex II substrate and in the presence of rotenone, ROS production measured in the current protocol emanated from complex III.5, 42 In the presence of rotenone (an inhibited system), uncoupling leads to increased ROS production compared to baseline.1, 2, 5, 17, 42 In uninhibited mitochondria, uncoupling decreases ROS production.1 The higher H+ leak and increased ROS production seen in the Control-IR group of the current study shows that mitochondria undergoing ischemia-reperfusion injury behave more like inhibited mitochondria, with increased depolarization (H+ leak) being associated with increased ROS production. IPC attenuates H+ leak after ischemia-reperfusion injury and decreases ROS production (Figure 2C, Figure 4).
Effect of malonate concentration
Malonate is a competitive inhibitor of complex II.44 Increasing malonate concentration inhibits respiration at complex II, slowing the flow of electrons through the electron transport chain and decreasing ROS production.44 Figure 3 demonstrates that increasing malonate concentration leads to decreasing mitochondrial respiration in both Control-IR and IPC mitochondria. The results of Figure 4 indicate that increasing malonate concentration also decreases ROS production. IPC has a lower baseline rate of ROS production (Figure 4, 0 malonate point) compared to Control-IR. As malonate concentration increases, IPC continues to produce less ROS compared to Control-IR with both groups showing a plateau at a malonate concentration of approximately 1 mM. The results of figures 3 and 4 together, indicate that decreasing mitochondrial respiration decreases ROS production and that at every respiratory rate, IPC produces less ROS compared to Control-IR.
Malonate may act as an endogenous protector against the effects of ischemia-reperfusion by acting as an inhibitor of respiration and leading to decreased ROS production.44 Complex I inhibitors such as rotenone and Amobarbital (Amytal) decrease the production of ROS by inhibiting electron flow through the ETC.6, 41 The results of this experiment as well as other investigators such as Brookes and colleagues, demonstrate that inhibition of respiration at complex II can also decrease ROS production.44 The finding of decreased ROS production with inhibition of mitochondrial respiration also supports the findings of Chen et al. who showed that inhibiting mitochondrial respiration during early reperfusion decreases myocardial injury.42 Taken together, these results confirm that ROS production in isolated mitochondria is partly dependent of mitochondrial respiration.45 Allowing oxidative phosphorylation to continue in the setting of ischemia-reperfusion injury leads to an increased production of ROS, mitochondrial calcium overload and eventual apoptosis.42
Clinical significance
Coronary artery disease (CAD) is one of the leading causes of morbidity and mortality in the US. Plaque rupture in the setting of CAD leads to acute coronary syndrome and myocardial infarction. Early reperfusion is the primary mode of treatment after acute myocardial infarction and is aimed at salvaging myocardium at risk of infarction. The restoration of blood flow leads to ischemia-reperfusion injury which involves a burst of ROS production. ROS are key mediators of ischemia-reperfusion injury and inhibiting ROS production is believed to be an important part of preserving myocardial function after an episode of ischemia-reperfusion. Understanding the mechanism of how IPC decreases ROS production could lead to better pharmacologic treatments for patients with CAD and acute coronary syndrome. Previous clinical studies that aimed to reduce the formation of ROS during reperfusion through the use of scavengers have been unsuccessful (for review see 1). More precise delivery of agents that prevent the formation of ROS at the source may lead to better clinical outcomes. Since mitochondria have been identified as a major source of ROS production after ischemia-reperfusion, understanding how to attenuate ROS formation by mitochondria after ischemia-reperfusion is important in the development of improved treatment options in the setting of acute coronary syndrome.
Conclusions
We conclude that IPC attenuates both the increase in H+ leak and ROS production seen after ischemia-reperfusion injury. While previous studies showed that increased H+ leak was associated with cardio-protection, we demonstrate that a decreased rate of H+ leak in IPC mitochondria is associated with cardio-protection when compared to unconditioned mitochondria. This indicates that the mechanism involved in limiting ROS production in IPC does not involve an increased rate of H+ leak as previously suspected. Our results indicate that a moderate amount of H+ leak is associated with decreased ROS, while higher rates of H+ leak are associated with increased ROS formation. The high rate of H+ leak seen in Control-IR mitochondria may be an indicator of the severity of mitochondrial damage.
Acknowledgments
This work was supported by NIH grants: HL38324, HL63744, HL65608 and by an American College of Surgeons Faculty Research Fellowship and the Thoracic Surgery Foundation for Research and Education Research Grant to JAC.
Footnotes
Presented at the 5th annual Academic Surgical Congress, San Antonio, Texas, February 5th 2010
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiology Review. 2008;88(2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 3.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 4.Zweier JL. Measurement of superoxide-derived free radicals in the reperfused heart: evidence for a free radical mechanism of reperfusion injury. J Biol Chem. 1988;263:1353–1357. [PubMed] [Google Scholar]
- 5.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: Central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 6.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, Flaherty JY. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 7.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 8.Crestanello JA, Doliba NM, Babsky AM, Niibori K, Osbakken MD, Whitman GJ. Mitochondrial function during ischemic preconditioning. Surgery. 2002;131:172–178. doi: 10.1067/msy.2002.119490. [DOI] [PubMed] [Google Scholar]
- 9.Lee D, Steinbaugh G, Zweier J, Crestanello JA. Mitochondrial respiratory complex I and complex II activities are preserved by ischemic preconditioning [abstract] J Surg Res. 2009;151:230. [Google Scholar]
- 10.Lee DS, Steinbaugh GE, Quarrie R, Cramer BM, Pfeiffer DR, Zweier JL, Crestanello JA. Ischemic preconditioning preserves respiratory complex activity and decreases reactive oxygen species production in rat heart mitochondria [abstract] J Surg Res. 2010;158:357. [Google Scholar]
- 11.Zhu X, Liu B, Zhou S, Chen YR, Deng Y, Zweier JL, He G. Ischemic preconditioning prevents in vivo hyperoxygenation in postischemic myocardium with preservation of mitochondrial oxygen consumption. Am J Physiol Heart Circ Physiol. 2007;293:H1442–H1450. doi: 10.1152/ajpheart.00256.2007. [DOI] [PubMed] [Google Scholar]
- 12.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBC Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 13.Turrens JF. Mitochondrial formation of reactive oxygen species. Journal of physiology. 2003;552(Pt 2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X, Zuo L, Cardounel AJ, Zweier JL, He G. Characterization of in vivo tissue redox statuts, oxygenation, and formation of reactive oxygen species in postischemic myocardium. Antiox Redox Signal. 2007;9:447–455. doi: 10.1089/ars.2006.1389. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 17.Brookes PS. Mitochondrial H+ leak and ROS generation: an odd couple. Free Radical Biology & Medicine. 2005;38(1):12–23. doi: 10.1016/j.freeradbiomed.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Echtay KS, Roussel D, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham J, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;15:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 19.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395(3):611–618. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crestanello JA, MD, Doliba NM, PhD, Babsky AM, PhD, Doliba NM, MD, PhD, Niibori K, MD, Whitman JRG, MD, Osbakken MD., MD, PhD Ischemic Preconditioning Improves Mitochondrial Tolerance to Experimental Calcium Overload. J Surg Res. 2002:103–244. doi: 10.1006/jsre.2001.6361. [DOI] [PubMed] [Google Scholar]
- 21.Guide for the Care and Use of Laboratory Animals. Washington, DC: Natl. Acad. Press; 1996. [Google Scholar]
- 22.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 23.Peterson GL. Determination of total protein. Methods Enzymol. 1983;91:95. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- 24.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol. 1956;17:65. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 25.Barger JL, Brand MD, Barnes BM, Boyer BB. Tissue-specific depression of mitochondrial proton leak and substrate oxidation in hibernating arctic ground squirrels. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1306–R1313. doi: 10.1152/ajpregu.00579.2002. [DOI] [PubMed] [Google Scholar]
- 26.Kamo N, Muratsugu M, Hongoh R, Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J Membr Biol. 1979;49:105–121. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- 27.Marcinkeviciute A, Mildaziene V, Crumm S, Demin O, Hoek JB, Kholodenko B. Kinetics and control of oxidative phosphorylation in rat liver mitochondria after chronic ethanol feeding. Biochem J. 2000;349:519–526. doi: 10.1042/0264-6021:3490519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whipps DE, Halestrap AP. Rat liver mitochondria prepared in mannitol media demonstrate increased mitochondrial volumes compared with mitochondria prepared in sucrose media. Relationship to the effect of glucagon on mitochondrial function. Biochem J. 1984;221:147–152. doi: 10.1042/bj2210147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter RK, Joyce OJP, Farmer MK, Heneghan R, Tipton KF, Andrews JF, McBennett SM, Lund MD, Jensen CH, Melia HP. Indirect measurement of mitochondrial proton leak and its application. Int J Obes Relat Metab Disord. 1999;23:S12–S18. doi: 10.1038/sj.ijo.0800937. [DOI] [PubMed] [Google Scholar]
- 30.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide: general properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zweier JL, Kuppusamy P, Williams R, Rayburn BK, Smith D, Weisfeldt ML, Flaherty JT. Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J Biol Chem. 1989;264:18890–18895. [PubMed] [Google Scholar]
- 33.Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovas Res. 2006;72(2):313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Brookes PS, Land JM, Clark JB, Heales SJR. Peroxynitrite and brain mitochondria: evidence for increased proton leak. J Neurochem. 1998;70:2195–2202. doi: 10.1046/j.1471-4159.1998.70052195.x. [DOI] [PubMed] [Google Scholar]
- 35.Miwa S, Brand MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soci Trans. 2003;31(6):1300–1301. doi: 10.1042/bst0311300. [DOI] [PubMed] [Google Scholar]
- 36.Minners J, van den Bos EJ, Yellon DM, Schwalb H, Opie LH, Sack MN. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovasc Res. 2000;47(1):68–73. doi: 10.1016/s0008-6363(00)00069-9. [DOI] [PubMed] [Google Scholar]
- 37.Carreira RS, Miyamoto S, Di Mascio P, Gonçalves LM, Monteiro P, Providência LA, Kowaltowski AJ. Ischemic preconditioning enhances fatty acid-dependent mitochondrial uncoupling. Bioenerg Biomembr. 2007;39(4):313–320. doi: 10.1007/s10863-007-9093-y. Epub 2007 Oct 5. [DOI] [PubMed] [Google Scholar]
- 38.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109(14):1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 39.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res. 2006;72(2):210–219. doi: 10.1016/j.cardiores.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 41.Borutaite V, Morkuniene R, Budriunaite A, Krasauskaite D, Ryselis S, Toleikis A, Brown GC. Kinetic analysis of changes in activity of heart mitochondrial oxidative phosphorylation system induced by ischemia. J Molec Cell Card. 1996;28(10):2195–2201. doi: 10.1006/jmcc.1996.0211. [DOI] [PubMed] [Google Scholar]
- 42.Chen Q, Camara AKS, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292:C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 43.Roubaud V, Sankarapandi S, Kuppusamy P, Tordo P, Zweire JL. Quantitative measurement of superoxide generation using the spin trap (Diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide. Analy Biochem. 1997;247:404–411. doi: 10.1006/abio.1997.2067. [DOI] [PubMed] [Google Scholar]
- 44.Wojtovich AP, Brookes PS. The Endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta. 2008;1777:882–889. doi: 10.1016/j.bbabio.2008.03.025. Epub 2008 Apr 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liem DA, Manintveld OC, Schoonderwoerd K, McFalls EO, Heinen A, Verdouw PD, Sluiter W, Duncker DJ. Ischemic preconditioning modulates mitochondrial respiration, irrespective of the employed signal transduction pathway. Translational Res. 2008;151(1):17–26. doi: 10.1016/j.trsl.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Aon MA, Cortassa S, O'Rourke B. Redox-optomized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbabio.2010.02.016. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]