

Abstract
Protectins are newly identified natural chemical mediators that counter leukocyte activation to promote resolution of inflammation. In this study, we provide the first evidence for protectin D1 (PD1, 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid) formation from docosahexaenoic acid in human asthma in vivo and PD1 counterregulatory actions in allergic airway inflammation. PD1 and 17S-hydroxy-docosahexaenoic acid were present in exhaled breath condensates from healthy subjects. Of interest, levels of PD1 were significantly lower in exhaled breath condensates from subjects with asthma exacerbations. PD1 was also present in extracts of murine lungs from both control animals and those sensitized and aerosol challenged with allergen. When PD1 was administered before aeroallergen challenge, airway eosinophil and T lymphocyte recruitment were decreased, as were airway mucus, levels of specific proinflammatory mediators, including IL-13, cysteinyl leukotrienes, and PGD2, and airway hyperresponsiveness to inhaled methacholine. Of interest, PD1 treatment after aeroallergen challenge markedly accelerated the resolution of airway inflammation. Together, these findings provide evidence for endogenous PD1 as a pivotal counterregulatory signal in allergic airway inflammation and point to new therapeutic strategies for modulating inflammation in asthmatic lung.
Chronic airway inflammation with large numbers of eosinophils (EOS)3 and T lymphocytes (Lymphs) infiltrating respiratory tissues is mechanistically linked to asthma pathogenesis (1). In addition to their direct actions, these leukocytes amplify airway inflammation by trafficking into the lung an increased capacity to generate both proinflammatory peptides and lipid mediators, such as Th2 cytokines and cysteinyl leukotrienes (CysLTs) (1). In addition, Th2 cytokines up-regulate the expression of biosynthetic enzymes for eicosanoids, including PGs, LTs, and lipoxins (LXs) with potent immunomodulatory properties (2). Surprising results recently indicated that arachidonic acid (C20:4) is not the only fatty acid precursor converted to potent bioactive mediators during inflammation and resolution. Distinct from their actions on C20:4 and biosynthesis of PGs, LTs, and LXs, new enzymatic pathways, uncovered in inflammatory exudates, convert docosahexaenoic acid (DHA; C22:6, ω-3) in vivo to specific bioactive compounds (3–5). Of interest, DHA levels in the respiratory tract are decreased in asthma and other diseases of excess airway inflammation, such as cystic fibrosis (6). Epidemiological studies suggest that a diet high in marine fatty acids (fish oil) may have beneficial effects on inflammatory conditions including asthma (7, 8), and dietary supplementation with omega-3 fatty acids in children prevents the development of atopic cough, a symptom of allergic airway inflammation (9). The underlying mechanisms for beneficial properties of omega-3 fatty acids in asthma remain to be established.
Natural resolution of acute inflammation (or asthma exacerbation) is driven, in part, by decrements in proinflammatory mediators and removal of inflammatory cells (1, 10). Promotion of resolution is now recognized as an active process with early signaling pathways, for example cyclooxygenase-2-derived PGE2 and PGD2, engaging biosynthetic circuits for the later formation of counterregulatory mediators, such as LXs and the newly identified families of lipid mediators generated from omega-3 fatty acids named resolvins and protectins that can dominate the resolution phase (11). DHA is incorporated into membranes and are rapidly released upon neuronal cell activation (12) for conversion to 17S-hydroxy containing resolvins of the D series (because they are from DHA) and protectin D1 (PD1) (13). The complete stereochemistry for PD1 was recently established as 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid (13). In this study, we report that PD1 is generated from endogenous sources in human asthma and reduces both allergic airway inflammation and hyperresponsiveness.
Materials and Methods
Sensitization and challenge protocols
Five- to 7-wk-old male FvB mice (Charles River Laboratories) were housed in isolation cages under viral Ab-free conditions. Mice were fed a standard diet (Laboratory Rodent Diet 5001; PMI Nutrition International) that contained no less than 4.5% total fat with 0.26% omega-3 fatty acids and <0.01% C20:4. After Harvard Medical Area Institutional Review Board approval (Protocol 02570), mice were sensitized with i.p. injections of OVA (Grade III; Sigma Chemical) (10 μg) plus 1 mg of aluminum hydroxide (J.T. Baker Chemical) as adjuvant in 0.2 ml of PBS on days 0 and 7. On days 14, 15, 16, and 17, the mice received PD1 (2, 20, or 200 ng) (a product of biogenic synthesis; Ref. 3) or PBS with 1.6 mM CaCl2 and 1.6 mM MgCl2 (0.1 ml) by i.v. injection 30 min before an aerosol challenge containing either PBS or 6% OVA for 25 min per day. Matching experiments were performed with 20 ng of PD1 prepared by total organic synthesis (13).
On day 18, 24 h after the last aerosol challenge, either airway responsiveness to aerosolized methacholine (0, 20, 50, and 75 mg, 10 s) was measured, bilateral bronchoalveolar lavage (BAL) (2 aliquots of 1 ml of PBS plus 0.6 mM EDTA) was performed, or tissues were harvested for histological analysis. Lung resistance was measured using a Flexivent ventilator (SCIREQ). Resistance was measured as a function of time for each animal, and peak and average values for each dose of methacholine were recorded. No BAL or histological analysis was performed on those animals undergoing airway hyperresponsiveness or lipid extraction studies.
In select experiments, animals were sensitized and aeroallergen challenged for 4 days before receiving PD1 (20 ng) or PBS by i.v. injection (0.1 ml) on days 18, 19, and 20. On day 21, bilateral BAL was performed.
Allergen-initiated respiratory inflammation
Murine lungs were fixed in 10% buffered formalin and paraffin embedded for H&E and periodic acid-Schiff staining (Sigma-Aldrich). Tissue morphometry was performed by a member (K. Haley) of the Lung Histopathology Core Laboratory at Brigham and Women’s Hospital who was blinded to the experimental conditions before histological analyses. Three fields per slide were examined at ×200 magnification for vessels, and large airways and alveoli with EOS were counted at ×400 magnification in randomly assigned fields. Vessels were identified by perivascular smooth muscle, and large airways were identified by at least one-half their diameter either cuboidal or columnar epithelia. Measurement of inflammatory mediators was determined in cell-free BAL fluid (BALF) (2000 × g, 10 min) by sensitive and specific ELISAs, in tandem, for IL-5, IL-12, IL-13, PGD2 (R&D Systems), CysLTs, (Cayman Chemical), and LXA4 (Neogen). Cells in BALF were resuspended in PBS, enumerated by hemocytometer, and concentrated onto microscope slides by cytocentrifuge (STATspin) (265 × g). Cells were stained with a Wright-Giemsa stain (Sigma-Aldrich) to determine leukocyte differentials (after counting ≥200 cells).
PD1 extraction and identification by liquid chromatography (LC)-tandem mass spectrometry (MS)-MS
Exhaled breath condensates (EBC) were collected by R-tube (Respiratory Research) from volunteer subjects who had given written informed consent to a protocol approved by the Brigham and Women’s Hospital Committee for the Protection of Human Subjects in Research. Samples were collected during 10 min of tidal breathing. Characteristics of the subjects are provided in Table I. For samples of murine lung, blood was flushed from the pulmonary circulation with 2 ml of PBS, and whole murine lungs were removed from OVA-sensitized/OVA-challenged and control mice on day 18. Using a manual dounce, lungs were gently homogenized for direct lipid extraction in MeOH or, in some cases, were warmed (5 min, 37°C) in PBS, and incubated (40 min, 37°C) in the absence or presence of DHA (100 μg). Reactions were stopped with 10 vol of iced MeOH and stored at −20°C overnight.
Table I.
Characteristics of subjectsa
| Healthy | Asthma Exacerbation | |
|---|---|---|
| Sample size (n) | 3 | 4 |
| Age (years) | 28 ± 1 | 41 ± 6 |
| M:F | 1:2 | 2:2 |
| Race | 3 other | 2 Caucasian, 1 African American, 1 other |
| Current cigarette smoker | 0 | 2 |
EBC was collected from individuals in the emergency department with an acute asthma exacerbation and a control group of healthy subjects (see Materials and Methods). Plus-minus values are means ± SD.
Lipids in EBCs or murine lung were extracted using C18 cartridges (Alltech) and deuterium-labeled d4-PGE2 as an internal standard to correct for losses during extraction (3). Materials eluting in the methyl formate fraction were taken to HPLC coupled to a photo-diode-array detector and tandem MS (LC-PDA-MS-MS; ThermoFinnigan) for lipidomic analyses. PD1 was identified using recently published criteria (13) that include retention time, coelution with authentic 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid, UV absorbance in methanol (λmax 270 nm with shoulders at 261 and 281 nm, a triple band of absorption consistent with a conjugated triene structure) and at least 5 diagnostic MS-MS ions (m/z 359 [M-H], 341 ([M-H]-H2O; base peak), 323 ([M-H]-2H2O), 315 ([M-H]-CO2), 297 ([M-H]-H2O,-CO2), and 277 ([M-H]-H2-2H2O-CO2) plus additional ions defining the presence of the C10 and C17 hydroxyl (i.e., m/z 289, 261, 243 (261-H2O), 217 (261-CO2), 205, 181, 163 (181-H2O), and 153) (Fig. 1). The quantitation of PD1 was determined following LC-MS-MS analyses using a calibration curve (r2 = 0.991), and the chromatographic peak areas were obtained via selective ion monitoring.
FIGURE 1.

Generation of PD1 in asthma. EBC were obtained from volunteer subjects in the emergency department during a clinical exacerbation of asthma. Lipids were extracted and subjected to analysis by LC-PDA-MS-MS. a, LC chromatogram plotted for ms/ms at m/z 343 and corresponding MS profile were diagnostic for 17S-hydroxy-DHA (i.e., 17S-hydroxy-docosa-4Z, 7Z,11E,13E,15Z,19Z-hexaenoic acid) (b). Material was also present in the lipid extracts with LC chromatogram for m/z 217 of MS-MS at m/z 359 (c), UV absorbance spectrum (d, inset, left), and mass spectrum diagnostic for authentic PD1 (i.e., 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z, 19Z-hexaenoic acid). Insets, the fragmentation ions are denoted for 17S-hydroxy-DHA (b) and PD1 (d). Results are representative of n = 3.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical significance of differences was assessed by Student’s t test and Kruskal-Wallis non-parametric one-way ANOVA. A p value <0.05 was set as the level of significance.
Results
PD1 is present in asthma and endogenously generated in allergic lung
To determine whether DHA-derived products are generated in respiratory tissues, we analyzed lipid extracts from EBCs that were collected from healthy volunteer subjects and patients in the emergency department during a clinical exacerbation of asthma (Table I). PD1 and its biosynthetic precursor, 17S-hydroxy-DHA were present in these human respiratory tract secretions (Fig. 1). Levels of PD1 were significantly lower in EBCs from subjects with status asthmaticus (trace amounts) compared with healthy individuals (2.23 ± 1.55 ng PD1/ml EBC; mean ± SEM; p < 0.05). These results indicate that asthma exacerbation is associated with reduced airway levels of the counterregulatory lipid mediator PD1.
To investigate potential roles for PD1 in airway inflammation, we next turned to an experimental animal model of allergic asthma. After sensitization and aerosol challenge with allergen, murine lungs generated PD1 from endogenous sources (73.9 ± 35.6 ng PD1; mean ± SEM for n = 3). Of note, PD1 levels in the inflamed lungs were not significantly different from those in healthy murine lungs (45.8 ng of PD1). Similar to results with human EBCs, 17S-hydroxy-DHA was also present in murine lungs. Addition of exogenous DHA to a homogenate of the inflamed murine lungs ex vivo significantly increased mean PD1 levels by 5.8-fold to 431.6 ± 69.3 ng PD1 (mean ± SEM; n = 3; p < 0.02). These findings indicate that during airway inflammation, respiratory tissues can convert DHA to 17S-hydroxy-DHA and PD1.
Allergic airway inflammation decreases with PD1
To determine the impact of PD1 on airway inflammation, we administered physiologically relevant quantities (2, 20, or 200 ng) by i.v. injection to allergen-sensitized animals just prior (30 min) to each aerosol challenge. For these experiments, PD1 was produced via biogenic synthesis and matching studies were performed with PD1 that was prepared by total organic synthesis (13). Animals receiving PD1 had substantially less EOS and Lymphs in the peribronchial regions and airspaces compared with control mice that received only vehicle (Fig. 2). PD1 also reduced goblet cell hyperplasia and airway mucus as determined by periodic acid Schiff stain (Fig. 3). Morphometric analyses identified significant decreases in EOS tissue infiltration around vessels and in the large and peripheral airways (Fig. 4a). In BALF, PD1 decreased total leukocytes, EOS, and Lymphs in a concentration-dependent manner (Fig. 4b), and levels of peptide and lipid proinflammatory mediators were selectively reduced (Fig. 5). PD1 administration blocked allergen-induced increases in IL-13, CysLTs, and PGD2, all of which have been assigned pivotal roles in asthma pathobiology (14 –16). Of note, PD1 did not significantly impact IL-5 or IL-12 levels in BALF. In conjunction with decreased airway inflammation, levels of the counter-regulatory eicosanoid LXA4 were diminished in the presence of PD1 (Fig. 5b). No behavioral or physical signs of toxicity with PD1 treatment were observed. Together, these results indicate that PD1, in nanogram quantities, significantly reduced allergic pulmonary inflammation, and suggests that its mechanism of action is distinct from LXs.
FIGURE 2.
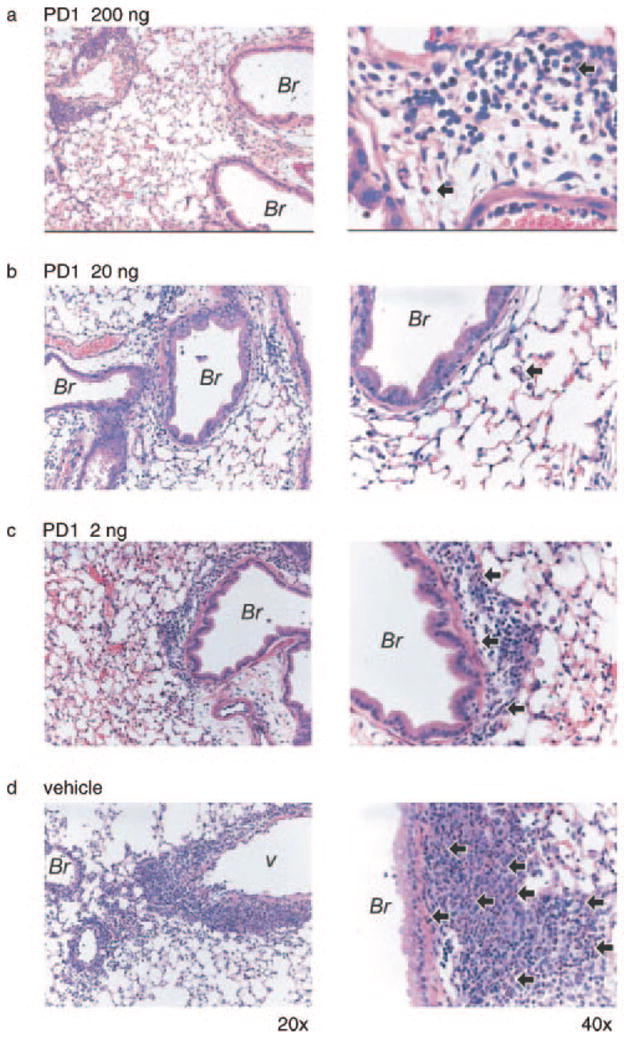
Lung histopathology from mice given PD1. Mice were sensitized and aerosol challenged with OVA in the presence of PD1 (200 ng (a), 20 ng (b), 2 ng (c)) or vehicle (d). Representative (n ≥ 3) lung tissue sections (original magnifications, ×20 (left column) and ×40 (right column)) were obtained from fixed, paraffin-embedded lung tissue, prepared, and stained with H&E. Arrows denote representative EOS; Br, bronchus; v, vessel.
FIGURE 3.
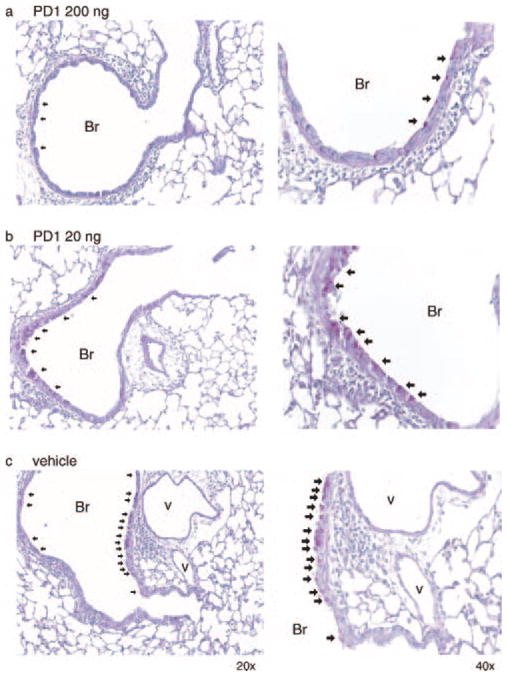
PD1 decreases airway mucus. Representative lung tissue sections from mice given PD1 (200 ng (a), 20 ng (b)) or vehicle (c) were stained with periodic acid Schiff (original magnifications, ×20 (left column) and ×40 (right column). Arrows indicate representative mucus (magenta) containing goblet cells.
FIGURE 4.

PD1 sharply reduces leukocyte infiltration. a, Tissue morphometric analyses were performed to determine the impact of PD1 on EOS accumulation in pulmonary vessels (V-EOS), large airways (Aw-EOS), and alveoli (Alv-EOS). b, BALFs were obtained from OVA-sensitized and challenged mice. Leukocytes in BALF were enumerated and identified after Wright-Giemsa stain. Results are expressed as mean ± SEM (n ≥ 3).*, p < 0.05 by Student’s t test compared with control animals.
FIGURE 5.

PD1 selectively decreases airway inflammatory mediators. In the presence or absence of PD1, the mediator profile in BALF was determined in materials from OVA-sensitized and challenged mice for specific cytokines (IL-13, IL-5, IL-12) (a ), and lipid mediators (CysLTs, LXA4, and PGD2) (b). Results are expressed as mean ± SEM (n ≥ 5, determinations = 2).*, p < 0.05 by Student’s t test compared with control animals.
PD1 blocks airway hyperresponsiveness
Because increased airway reactivity is a diagnostic hallmark of asthma, we also determined whether PD1 regulated airway hyperresponsiveness to inhaled methacholine. After allergen sensitization and aerosol challenge in the presence of PD1 (0 –200 ng), animals were ventilated and exposed (10 s) to increasing concentrations of inhaled methacholine. Consistent with the regulation of airway inflammation, PD1 also decreased both peak and average lung resistance in response to methacholine (Fig. 6). The log ED200 for the mean airway resistance for all three doses of PD1 (2, 20, and 200 ng) was significantly increased compared with vehicle (Fig. 6b). There was a bell-shaped dose response with maximal protection for PD1 on airway hyperresponsiveness to methacholine apparent with lower amounts (2 and 20 ng). PD1 displayed no significant impact on the airway responses of control animals that received PBS rather than allergen (Fig. 6b). In addition, no significant changes in lung elastance or compliance were observed with PD1 following allergen challenge (data not shown). These results indicate that methacholine-induced bronchoconstriction is significantly reduced by administration of PD1.
FIGURE 6.

PD1 reduces airway hyperresponsiveness. a, OVA-sensitized mice were treated with PD1 20 ng (□), 200 ng (○), or vehicle (▲) before OVA aerosol challenge. Airway reactivity was determined by methacholine-dependent change in peak lung resistance. Results are expressed as mean ± SEM (n ≥ 5).*, p < 0.05 by one-way ANOVA compared with control animals. b, ED200 was determined for methacholine-dependent changes in mean lung resistance for OVA-sensitized animals receiving PD1 (0, 2, 20, or 200 ng) before OVA aerosol challenge and for control animals receiving buffer (PBS) instead of OVA during sensitization and challenge phases of the model.*, p < 0.05 by Student’s t test compared with control animals.
Impact of PD1 treatment on airway inflammation
To more closely mimic the clinical scenario of asthma exacerbation, we next determined whether PD1 could dampen established airway inflammation by administration after aeroallergen challenge. Mice were sensitized and allergen challenged on four consecutive days. PD1 (20 ng, i.v.) or vehicle (0.9% saline) was then given once a day for three additional days, and BAL was performed to enumerate cellular infiltration into the lung. Despite no further aeroallergen challenges, animals receiving vehicle still carry a substantial number of EOS and Lymphs in BALF at day 21 in our protocol (Fig. 7). In sharp contrast, PD1 administration led to significant decrements in the numbers of total leukocytes, EOS, and Lymphs in BALFs (Fig. 7). These findings indicated that PD1 has the capacity to accelerate resolution of allergic airway inflammation.
FIGURE 7.

PD1 treatment promotes resolution of allergen-driven leukocytes in mouse lung. BALFs were obtained from OVA-sensitized and challenged mice that received either PD1 (20 ng, ▨) or vehicle (0.9% saline, ■) for three consecutive days before study. Leukocytes in BALF were enumerated and identified after Wright-Giemsa stain. Results are expressed as mean ± SEM (n ≥ 3).*, p < 0.05 by Student’s t test compared with control animals.
Discussion
In this study, we identify PD1 as a natural product of a new C22:6 signaling pathway (3, 5) during respiratory tract inflammation that displays potent counterregulatory actions on key asthma phenotypes, namely airway levels of proinflammatory peptide and lipid mediators, airway mucus, leukocyte accumulation, and hyperresponsiveness. PD1 was first identified in murine exudates and human brain, blood, and glial cells (3). In this study, we present evidence for the first identification of 17S-hydroxy-DHA and PD1 in human asthma. In addition, airway inflammation triggered PD1 formation in vivo and conversion of C22:6 to PD1 in lung tissues. Similar to the inflamed airway, generation of PD1 occurs elsewhere during multicellular host inflammatory responses, including Alzheimer disease, brain ischemia-reperfusion injury, and activated human whole blood (3, 4, 17). The biosynthesis of PD1 proceeds via 15-lipoxygenase-catalyzed conversion of DHA to 17S-hydroperoxy and 16(17)-epoxide intermediates (3, 13) in activated human leukocytes and in Alzheimer’s brain and murine cornea (4, 13, 18, 19). Lipoxygenases and epoxide hydrolases are both prominent classes of enzymes in asthmatic lung that are induced by pivotal regulators of allergy, including specific Th2 cytokines (20 –23). Potential source(s) for PD1 generation include airway epithelial cells, EOS, and other leukocytes, but the definitive cellular and enzymatic source of PD1 in the lung remains to be established in future studies. Our findings indicate the presence of specialized enzyme systems in the lung for this new DHA pathway that convert the omega-3 fatty acid to biologically active chemical mediators during airway inflammation.
Eosinophilic airway inflammation and airway hyperresponsiveness are characteristic features of asthma. EOS recruitment to the lung in asthma is primarily a consequence of Th2 Lymph activation (1). Because PD1 was identified in EBCs in the nanogram range, and this sampling technique likely reflects only a small fraction of total PD1 generated in the lung, we chose to examine the impact of PD1 in physiologically relevant nanogram quantities. After allergen sensitization and aerosol challenge, EOS trafficking was reduced by as little as 2 ng of PD1. Levels of Th2 cytokines in BALF and the number of Lymphs in both BALF and lung tissue were decreased. These findings provide evidence for potent, concentration-dependent reduction of both Th2 Lymphs and EOS responses in vivo.
Lymph and EOS activation in the lung are held to contribute to asthma pathobiology. In addition, neutrophil (PMN) activation contributes to the pathogenesis of asthma exacerbation (24) and severity (25). PD1 promotes T Lymph apoptosis in vitro (18), and PD1 also carries systemic and topical anti-inflammatory actions for PMNs in vivo (3, 17). In the nervous system, PD1 decreases brain leukocyte infiltration, IL-1β-induced NF-κB activation, and cyclooxygenase-2 expression to elicit neuroprotection (12, 17). In this study, PD1 also dampened hyperresponsiveness to methacholine and mucus production in the inflamed airway. The local generation of PD1 in allergic inflammation together with counterregulatory properties in the airway broadens its potential cellular sources and actions in vivo to new leukocyte classes and tissue resident cells and points to a more generalized counterregulatory function as an autacoid in inflammation.
LXs are also generated in asthma and serve as potent inhibitors of both airway inflammation and hyperresponsiveness (26). Although there is some overlap in the pattern of cytokine regulation for PD1 and a LX stable analog in this murine model of asthma, some key differences were observed. First, whereas both mediators blocked IL-13 and CysLT generation and had no significant effect on BAL IL-12 levels (26), the inhibitory concentrations of PD1 were 1 to 2 log orders more potent than the LX analog. Secondly, IL-5 production was reduced by the LX stable analog, but not PD1, suggesting a direct effect of PD1 on EOS, T Lymphs, and other effector cells. Third, administration of PD1 led to decreased airway levels of LXA4, suggesting that the circuit for PD1 formation and actions is distinct from LX signaling in the murine lung. In aggregate, these findings indicate the presence of unique homeostatic pathways for DHA-derived bioactive mediators in the lung.
Formation of counterregulatory LXs is defective in severe inflammatory diseases of the airways, including asthma and cystic fibrosis (27–29). DHA levels in the respiratory tract are decreased in both of these illnesses (6), and in this study, in comparison to healthy controls, we uncovered lower levels of PD1 during human asthma exacerbation. Given its counterregulatory properties, decreased formation of PD1 from low levels of DHA would adversely impact control of airway inflammation and hyperresponsiveness. Of note, PD1 levels in the inflamed murine lungs were not significantly different from those in healthy murine lungs, suggesting that temporal and spatial factors for PD1 generation as well as the lung compartment sampled are important considerations.
Although observational studies have identified an increased risk of asthma in populations with diets low in DHA, interventional trials with DHA supplementation have not consistently improved clinical outcomes (Cochrane Database of Systematic Reviews, www.cochrane.org), despite altering the responses of isolated leukocytes to inflammatory stimuli (30). In contrast to asthma, nutritional supplementation with omega-3 essential fatty acids has proven beneficial in cystic fibrosis and the acute respiratory distress syndrome, clinical disorders of excess PMN-mediated inflammation (Cochrane Database of Systematic Reviews, www.cochrane.org) (31). Because the molecular rationale for these beneficial effects is uncertain, there remain many potential reasons for the lack of clinical success with DHA feeding in asthma, including purity, dose, time course, and difficulties tolerating the ingestion of large amounts of fish oils for extended periods of time (32). After the induction of experimental asthma by aeroallergen challenge, we determined that administration of PD1 promoted the resolution of airway inflammation. Thus, identification of PD1 as a DHA-derived counterregulatory autacoid in the lung opens the door to new mechanism-based therapeutic strategies in airway inflammation.
Our results are the first demonstration of PD1 formation in human asthma in vivo from DHA and identify direct protective and regulatory roles for this novel mediator in allergic inflammation and airway hyperresponsiveness. In light of its ability to strongly reduce both of these key asthma phenotypes, the PD1 pathway may offer new therapeutic approaches for asthma. Moreover, our results indicate that endogenous conversion of DHA to PD1 represents a potential mechanism for the therapeutic benefits derived from diets rich in this omega-3 essential fatty acid in maintaining respiratory homeostasis.
Acknowledgments
We thank GuangLi Zhu and Nazim Khan for expert technical assistance and Christy Schneider for assistance in manuscript preparation.
Footnotes
This work was supported in part by National Institutes of Health Grants HL068669, AI068084, and P50-DE016191.
Abbreviations used in this paper: EOS, eosinophil; Lymph, lymphocyte; CysLT, cysteinyl leukotriene; LX, lipoxin; DHA, docosahexaenoic acid; PD1, protectin D1; BAL, bronchoalveolar lavage; BALF, BAL fluid; LC, liquid chromatography; MS, mass spectrometry; EBC, exhaled breath condensate; PMN, neutrophil.
Disclosures
B. D. Levy and C. N. Serhan, together with Brigham and Women’s Hospital, have a pending patent on the use of docosatrienes in the treatment of airway diseases and asthma.
References
- 1.Busse WW, Lemanske RF., Jr Asthma. N Engl J Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 2.Levy BD. Lipoxins and lipoxin analogs in asthma. Prostaglandins Leukotrienes Essent Fatty Acids. 2005;73:231–237. doi: 10.1016/j.plefa.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells: autacoids in anti-inflammation. J Biol Chem. 2003;278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 4.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freedman SD, Blanco PG, Zaman MM, Shea JC, Ollero M, Hopper IK, Weed DA, Gelrud A, Regan MM, Laposata M, et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med. 2004;350:560–569. doi: 10.1056/NEJMoa021218. [DOI] [PubMed] [Google Scholar]
- 7.Peat JK, Salome CM, Woolcock AJ. Factors associated with bronchial hyperresponsiveness in Australian adults and children. Eur Respir J. 1992;5:921–929. [PubMed] [Google Scholar]
- 8.Schwartz J, Weiss ST. The relationship of dietary fish intake to level of pulmonary function in the first National Health and Nutrition Survey (NHANES I) Eur Respir J. 1994;7:1821–1824. doi: 10.1183/09031936.94.07101821. [DOI] [PubMed] [Google Scholar]
- 9.Peat JK, Mihrshahi S, Kemp AS, Marks GB, Tovey ER, Webb K, Mellis CM, Leeder SR. Three-year outcomes of dietary fatty acid modification and house dust mite reduction in the Childhood Asthma Prevention Study. J Allergy Clin Immunol. 2004;114:807–813. doi: 10.1016/j.jaci.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 10.Henson PM. Dampening inflammation. Nat Immunol. 2005;6:1179–1181. doi: 10.1038/ni1205-1179. [DOI] [PubMed] [Google Scholar]
- 11.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1199–1205. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee PK, V, Marcheselli L, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, Yang R, Colgan SP, Petasis NA. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. 2006;176:1848–1859. doi: 10.4049/jimmunol.176.3.1848. [DOI] [PubMed] [Google Scholar]
- 14.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 15.Vachier I, Kumlin M, Dahlen SE, Bousquet J, Godard P, Chanez P. High levels of urinary leukotriene E4 excretion in steroid treated patients with severe asthma. Respir Med. 2003;97:1225–1229. doi: 10.1016/s0954-6111(03)00253-1. [DOI] [PubMed] [Google Scholar]
- 16.Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, et al. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- 17.Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–43817. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- 18.Ariel A, Li PL, Wang W, Tang WX, Fredman G, Hong S, Gotlinger KH, Serhan CN. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J Biol Chem. 2005;280:43079–43086. doi: 10.1074/jbc.M509796200. [DOI] [PubMed] [Google Scholar]
- 19.Gronert K, Maheshwari N, Khan N, Hassan IR, Dunn M, Laniado Schwartzman M. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J Biol Chem. 2005;280:15267–15278. doi: 10.1074/jbc.M410638200. [DOI] [PubMed] [Google Scholar]
- 20.Nassar GM, Morrow JD, Roberts LJ, 2nd, Lakkis FG, Badr KF. Induction of 15-lipoxygenase by interleukin-13 in human blood monocytes. J Biol Chem. 1994;269:27631–27634. [PubMed] [Google Scholar]
- 21.Pouliot M, McDonald PP, Khamzina L, Borgeat P, McColl SR. Granulocyte-macrophage colony-stimulating factor enhances 5-lipoxygenase levels in human polymorphonuclear leukocytes. J Immunol. 1994;152:851–858. [PubMed] [Google Scholar]
- 22.Munafo DA, Shindo K, Baker JR, Bigby TD. Leukotriene A4 hydrolase in human bronchoalveolar lavage fluid. J Clin Invest. 1994;93:1042–1050. doi: 10.1172/JCI117053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaitsu M, Hamasaki Y, Matsuo M, Kukita A, Tsuji K, Miyazaki M, Hayasaki R, Muro E, Yamamoto S, Kobayashi I, et al. New induction of leukotriene A4 hydrolase by interleukin-4 and interleukin-13 in human polymorphonuclear leukocytes. Blood. 2000;96:601–609. [PubMed] [Google Scholar]
- 24.Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. 1995;95:843–852. doi: 10.1016/s0091-6749(95)70128-1. [DOI] [PubMed] [Google Scholar]
- 25.Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma: persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. 1997;156:737–743. doi: 10.1164/ajrccm.156.3.9610046. [DOI] [PubMed] [Google Scholar]
- 26.Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, Szczeklik W, Drazen JM, Serhan CN. Multi-pronged inhibition of airway hyperresponsiveness and inflammation by lipoxin A4. Nat Med. 2002;8:1018–1023. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 27.Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, Yang R, Uddin J, Giggino WB, Atabani SF, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol. 2004;5:388–392. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- 28.Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. 2005;172:824–830. doi: 10.1164/rccm.200410-1413OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanak M, Levy BD, Clish CB, Chiang N, Gronert K, Mastalerz L, Serhan CN, Szczeklik A. Aspirin-tolerant asthmatics generate more lipoxins than aspirin-intolerant asthmatics. Eur Respir J. 2000;16:44–49. doi: 10.1034/j.1399-3003.2000.16a08.x. [DOI] [PubMed] [Google Scholar]
- 30.Lee TH, Mencia-Huerta JM, Shih C, Corey EJ, Lewis RA, Austen KF. Effects of exogenous arachidonic, eicosapentaenoic, and docosahexaenoic acids on the generation of 5-lipoxygenase pathway products by ionophore-activated human neutrophils. J Clin Invest. 1984;74:1922–1933. doi: 10.1172/JCI111612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadek JE, DeMichele SJ, Karlstad MD, Pacht ER, Donahoe M, Albertson TE, Van Hoozen C, Wennberg AK, Nelson JL, Noursalehi M. Effect of enteral feeding with eicosapentaenoic acid, γ-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome: Enteral Nutrition in ARDS Study Group. Crit Care Med. 1999;27:1409–1420. doi: 10.1097/00003246-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Spector SL, Surette ME. Diet and asthma: has the role of dietary lipids been overlooked in the management of asthma? Ann Allergy Asthma Immunol. 2003;90:371–377. doi: 10.1016/S1081-1206(10)61817-0. [DOI] [PubMed] [Google Scholar]