

Abstract
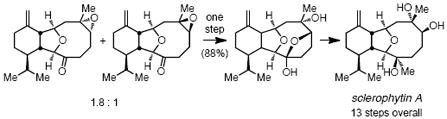
The cytotoxic natural product (-)-sclerophytin A was constructed in 13 steps from geranial. Highlights from the synthesis are a stereoselective Oshima-Utimoto reaction, a Shibata-Baba indium-promoted radical cyclization, and a novel stereoconvergent epoxide hydrolysis.
The oxygenated cembrane diterpenes are a large class of natural products comprising cladiellins, briarellins, asbestinins, and sarcodictyins. Their intricate chemical structures have captivated many in the synthetic chemistry community, with recent successes in the crafting of polyanthellin A1, asbestinins2, briarellins E and F3, vigulariol4, and astrogorgin5 being highly notable recent achievements.6 In addition to intriguing structures, these compounds also tend to possess potent biological activity. Amongst the cladiellins, sclerophytin A (Scheme 1) is a particularly striking compound; it was isolated from the marine soft coral Sclerophytum capitalis and found to exhibit remarkable potency against mouse leukemia cells (cytotoxic at 1 ng/ml versus L1210 cell line), yet its original structural formulation was incorrect.7,8 Total syntheses by the Paquette and Overman groups established the correct formulation of sclerophytin A to be that depicted in Scheme 1.9 As a means to examine the utility of the stereoselective Oshima-Utimoto reaction10 in chemical synthesis, we’ve been attracted to the target sclerophytin A and its structural relatives.11 Herein, we describe a short enantioselective route to ketone 1 (Scheme 1), a key intermediate in a prior synthesis of vigulariol,4b and show that 1 can be readily converted to sclerophytin A by a three step sequence involving a novel stereoconverging epoxide hydrolysis.
Scheme 1.
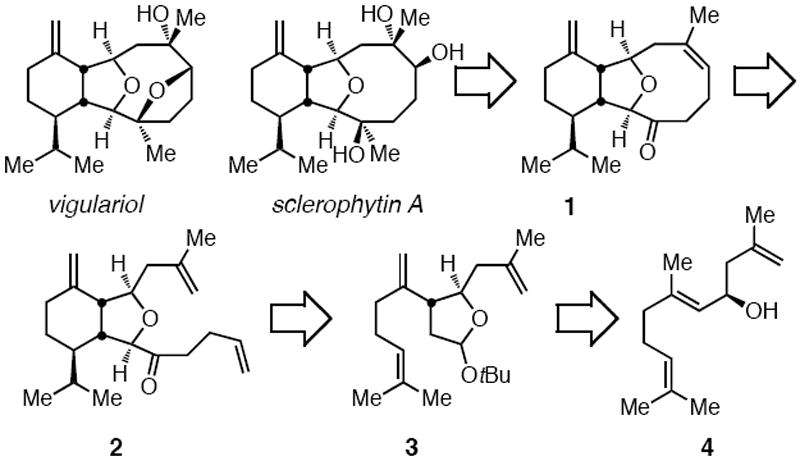
In terms of retrosynthetic analysis (Scheme 1), we considered that ketone 1 might arise by ring-closing olefin metathesis of triene 2 and that 2 might be obtained from intermediate 3 by a 6-exo-trig radical cyclization of an appropriately modified derivative. The Oshima-Utimoto reaction of the allylic alcohol embedded in 4, if it occurs without interference from the adjacent alkenes, was proposed to convert 4 to cyclic acetal 3, stereoselectively. Notably, appropriately functionalized substrate 4 is readily available in a single step from geranial.
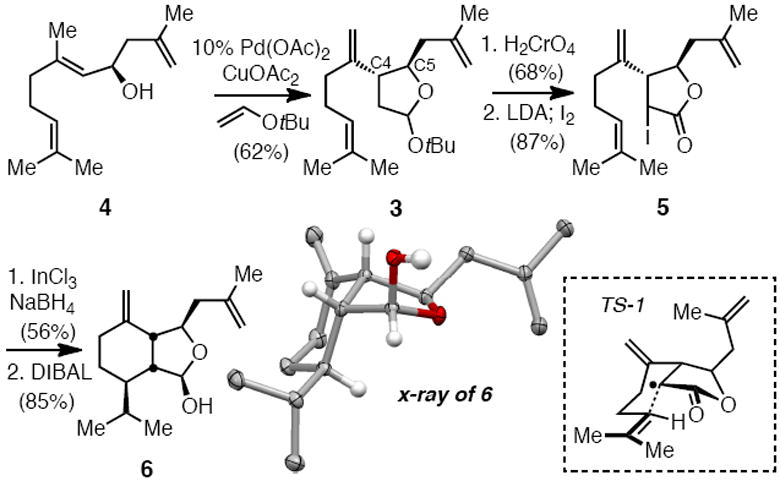
To begin the synthesis, allylic alcohol (E)-4 was prepared in 92% yield and 98% enantiomeric excess by Brown methallylation12 of geranial and was then subjected to the Oshima-Utimoto reaction. This sequence delivered 3 as a mixture of epimers in 62% yield (Scheme 2). Subsequent Jones oxidation of the Oshima-Utimoto product 3 to the derived lactone revealed >20:1 anti:syn (C4:C5) stereoselection from the Oshima-Utimoto reaction.13 After α-iodination to give 5, we examined the tin-free radical reactions pioneered by Shibata and Baba for reductive radical-mediated transformation.14 Thus, treatment of 5 with InCl3 and NaBH4 delivered the reductive radical cyclization product in 56% yield. The intermediate lactone was subsequently reduced with DIBAL-H affording lactol 6 in 85% yield. Of note, the radical ring closure proceeded with excellent stereoselectivity (>10:1; relative configuration established by x-ray analysis, see Scheme 2), presumably a consequence of reaction through transition structure TS-1. Also of note is that the In-promoted radical reaction proceeded in comparable efficiency as optimized tin-based reaction conditions (Ph3SnH/AIBN).
Scheme 2.
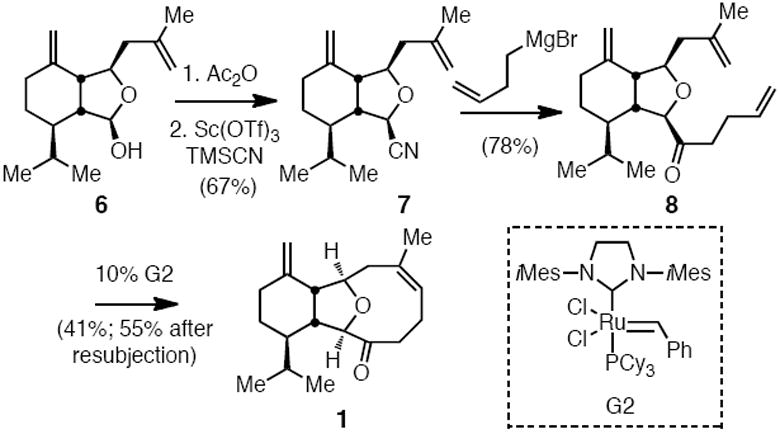
Conversion of 6 to ketone 1 was accomplished in four straightforward synthesis steps (Scheme 3). First the alcohol was acylated and then displaced with cyanide under the aegis of scandium triflate (→ 7). Addition of butenylmagnesium bromide, followed by aqueous work-up, delivered ketone 8 which was converted to ketone 1 by ring-closing metathesis.15
Scheme 3.
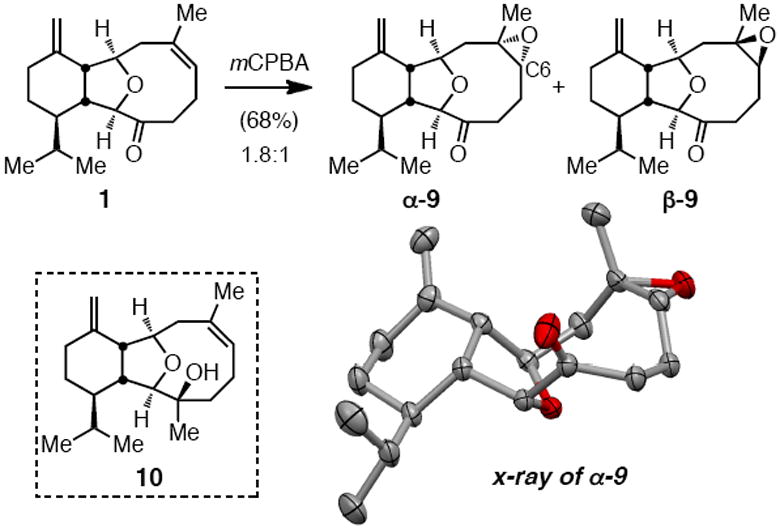
Conversion of ketone 1 to sclerophytin A requires trans dioxygenation of the trisubstituted alkene. It was considered that this might be accomplished by hydrolytic ring opening of an intermediate epoxide. As depicted in Scheme 4, peracid epoxidation occurs regioselectively but without stereocontrol, and therefore provides a mixture of both α-9 and β-9. While epoxidation of the tertiary alcohol 10 (derived from 1 by addition of MeMgBr) could be accomplished stereoselectively as ascertained by Clark,4b spontaneous cyclization to vigulariol could not be avoided (efforts to protect alcohol 10 were futile). Examination of the x-ray structure of epoxy ketone α-9 (see Scheme 4), however, inspired a solution. The structural feature of consequence is a near linear alignment (160°) of the carbonyl oxygen, C6 of the epoxide (eunicellin numbering), and the epoxide oxygen. While the carbonyl oxygen and C6 are separated by 3.68 Å in α-9, it was reasoned that proximity would be increased, perhaps facilitating intramolecular oxirane rupture, upon conversion of the carbonyl to the tetrahedral hydrate. This reactivity would be unique to stereoisomer α-9 and would lead to oxygenation of the alkene in a manner that is stereochemically-appropriate to the target.
Scheme 4.
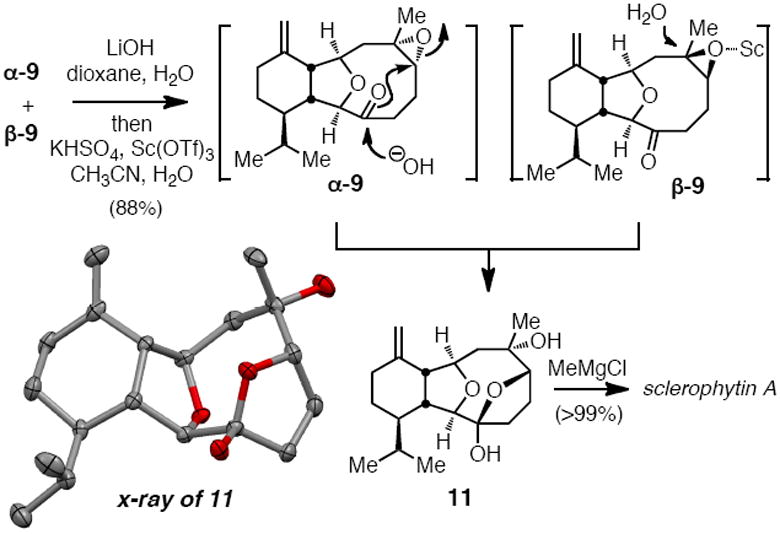
In the event, treatment of a 1.8:1 mixture of α-9 and β-9 with aqueous LiOH at room temperature resulted in rapid hydrolysis of α-9 to give 11 (Scheme 5). That an intramolecular advantage may operate in this hydrolytic epoxide opening is supported by three lines of evidence: first, NMR and x-ray analysis (Scheme 5) of the product shows that it exists as the stable hemiketal 11; second, analogous ring-opening of 1-methylcyclohexene oxide under the same reaction conditions occurs in <5% conversion; third, β-9 is unreactive under these conditions and could be recovered from the reaction mixture. While resolution of α-9 and β-9 is an attractive feature of the basic reaction conditions, it was considered that β-9 might also be converted to the target under acidic conditions where the regiochemical preference for epoxide opening would be opposite to that observed with LiOH. Ultimately, it was found that this stereoconvergent epoxide hydrolysis could be accomplished in a single-pot process. First, the mixture of α-9 and β-9 was subjected to LiOH until TLC analysis indicated complete consumption of α-9, then the mixture was neutralized with KHSO4, diluted with CH3CN, and treated with Sc(OTf)3. Under these conditions, the product hemiketal 11 was isolated in 88% yield as a single product stereoisomer. Finally, subjection of 11 to MeMgCl delivered sclerophytin A in excellent yield and in agreement with characterization data.7,9f
Scheme 5.
In conclusion, we have described an enantioselective synthesis of (-)-sclerophytin A that occurs in 13 steps from geranial. The versatility of radical reactions and the Oshima-Utimoto reaction suggests that the synthesis strategy described herein may be transferable to a number of other eunicellin-derived natural products and these will be reported in due course.
Supplementary Material
Acknowledgments
We thank Dr. Bo Li (Boston College) for X-ray structures. We thank the NIGMS (GM-59417) and the NSF (DBI-0619576; BC Mass Spectrometry Center) for support.
Footnotes
Supporting Information Available: Characterization and procedures. This material is available free of charge through the internet at http://pubs.acs.org.
References
- 1.(a) Campbell MJ, Johnson JS. J Am Chem Soc. 2009;131:10370. doi: 10.1021/ja904136q. [DOI] [PubMed] [Google Scholar]; (b) Kim H, Lee H, Kim J, Kim S, Kim D. J Am Chem Soc. 2006;128:15851. doi: 10.1021/ja065782w. [DOI] [PubMed] [Google Scholar]
- 2.Crimmins MT, Ellis JM. J Org Chem. 2008;73:1649. doi: 10.1021/jo0712695. [DOI] [PubMed] [Google Scholar]
- 3.Corminboeuf O, Overman LE, Pennington LD. J Org Chem. 2009;74:5458. doi: 10.1021/jo9010156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Becker J, Bergander K, Frohlich R, Hoppe D. Angew Chem, Int Ed. 2008;47:1654. doi: 10.1002/anie.200704678. [DOI] [PubMed] [Google Scholar]; (b) Clark JS, Hayes ST, Wilson C, Gobbi L. Angew Chem Int Ed. 2007;46:437. doi: 10.1002/anie.200603880. [DOI] [PubMed] [Google Scholar]
- 5.Crimmins MT, Brown BH, Plake HR. J Am Chem Soc. 2006;128:1371. doi: 10.1021/ja056334b. [DOI] [PubMed] [Google Scholar]
- 6.For an excellent review of completed and partial syntheses of this target class, see: Ellis JM, Crimmins MT. Chem Rev. 2008;108:5278. doi: 10.1021/cr078117u.
- 7.(a) Sharma P, Alam M. J Chem Soc Perkin Trans. 1988;1:2537. [Google Scholar]; (b) Alam M, Sharma P, Zektzer AS, Martin GE, Ji X, van der Helm D. J Org Chem. 1989;54:1896. [Google Scholar]
- 8.For the study of sclerophytin analogs, see: Bateman TD, Joshi AL, Moon K, Galitovskaya EN, Upreti M, Chambers TC, McIntosh MC. Biorg Med Chem Lett. 2009;19:6898. doi: 10.1016/j.bmcl.2009.10.079.Davidson JEP, Gilmour R, Ducki S, Davies JE, Green R, Burton JW, Holmes AB. Synlett. 2004:1434.Jung ME, Pontillo J. J Org Chem. 2002;67:6848. doi: 10.1021/jo016246j.
- 9.(a) Paquette LA, Moradei OM, Bernardelli P, Lange T. Org Lett. 2000;2:1875. doi: 10.1021/ol000083o. [DOI] [PubMed] [Google Scholar]; (b) Friedrich D, Doskotch RW, Paquette LA. Org Lett. 2000;2:1879. doi: 10.1021/ol000084g. [DOI] [PubMed] [Google Scholar]; (c) Bernardelli P, Moradei OM, Friedrich D, Yang J, Gallou F, Dyck BP, Doskotch RW, Lange T, Paquette LA. J Am Chem Soc. 2001;123:9021. doi: 10.1021/ja011285y. [DOI] [PubMed] [Google Scholar]; (d) Overman LE, Pennington LD. Org Lett. 2000;2:2683. doi: 10.1021/ol006236p. [DOI] [PubMed] [Google Scholar]; (e) MacMillan DWC, Overman LE, Pennington LD. J Am Chem Soc. 2001;123:9033. doi: 10.1021/ja016351a. [DOI] [PubMed] [Google Scholar]; (f) Gallou F, MacMillan DWC, Overman LE, Paquette LA, Pennington LD, Yang J. Org Lett. 2001;3:135. doi: 10.1021/ol000345m. [DOI] [PubMed] [Google Scholar]
- 10.Evans MA, Morken JP. Org Lett. 2005;7:3367. doi: 10.1021/ol051275s.Fugami K, Oshima K, Utimoto K. Bull Chem Soc Jpn. 1989;62:2050.Fugami K, Oshima K, Utimoto K. Tetrahedron Lett. 1987;28:809.. For related processes, see: Hafner W, Prigge H, Smidt J. Liebigs Ann Chem. 1966:109.Larock RC, Lee NH. J Am Chem Soc. 1991;113:7815.Larock RC, Stinn DE. Tetrahedron Lett. 1989;30:2767.Kraus GA, Thurston J. J Am Chem Soc. 1989;111:9203.. For prior use in synthesis: Evans MA, Morken JP. Org Lett. 2005;7:3371. doi: 10.1021/ol051276k.Trudeau S, Morken JP. Org Lett. 2005;7:5465. doi: 10.1021/ol0522687.
- 11.For compilations of related natural products, see: Sung P-J, Chen M-C. Heterocycles. 2002;57:1705.Bernardelli P, Paquette LA. Heterocycles. 1998;49:531.
- 12.(a) Racherla US, Brown HC. J Org Chem. 1991;56:401. [Google Scholar]; (b) Brown HC, Jadhav PK, Perumal PT. Tetrahedron Lett. 1984;25:5111. [Google Scholar]; (c) Brown HC, Jadhav PK. J Am Chem Soc. 1983;105:2092. [Google Scholar]
- 13.While the pendant alkenes in 4 appeared inconsequential, a break from prior studies should be noted: the isomeric starting material (Z)-4, derived from neral, delivered mixtures of olefin-isomeric products.
- 14.(a) Inoue K, Sawada A, Shibata I, Baba A. J Am Chem Soc. 2002;124:906. doi: 10.1021/ja017537c. [DOI] [PubMed] [Google Scholar]; (b) Baba A, Shibata I. Chem Rec. 2005;5:323. doi: 10.1002/tcr.20055. [DOI] [PubMed] [Google Scholar]
- 15.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.