

Abstract
Proteins targeted to the mitochondrial matrix are translocated through the outer and the inner mitochondrial membranes by two protein complexes, the translocase of the outer membrane (TOM) and one of the translocases of the inner membrane (TIM23). The protein Tim23, the core component of TIM23, consists of an N-terminal, soluble domain in the intermembrane space (IMS) and a C-terminal domain that forms the import pore across the inner membrane. Before translocation proceeds, precursor proteins are recognized by the N-terminal domain of Tim23, Tim23N (residues 1–96). By using NMR spectroscopy, we show that Tim23N is a monomeric protein belonging to the family of intrinsically disordered proteins. Titrations of Tim23N with two presequences revealed a distinct binding region of Tim23N formed by residues 71–84. In a charge-hydropathy plot containing all soluble domains of TOM and TIM23, Tim23N was found to be the only domain with more than 40 residues in the IMS that is predicted to be intrinsically disordered, suggesting that Tim23N might function as hub in the mitochondrial import machinery protein network.
Keywords: protein import, mitochondria, Tim23, NMR spectroscopy
Introduction
More than 99% of all mitochondrial proteins are encoded by nuclear DNA, and it is only after translation that they traverse the mitochondrial membranes to reach their final destination (for a review, see Ref.1). Protein import is mediated by the translocases of the mitochondrial membranes. The translocases are heterooligomeric protein complexes that act as highly dynamic molecular machines. Their constituent proteins can be divided into receptor proteins, pore proteins, and proteins necessary for complex assembly and complex stability. Mitochondrial protein precursors are either recognized by their internal targeting sequence or their positively charged, N-terminal presequence forming an amphipathic helix.2 After recognition, they are translocated through the outer and the inner mitochondrial membranes by two protein complexes, the translocase of the outer membrane (TOM) and one of the translocases of the inner membrane (TIM23). The TIM23 complex of yeast Saccharomyces cerevisiae contains the proteins Tim50, Tim17, Tim23, and Tim21.3 Tim23, Tim21, and Tim50 have hydrophilic domains exposed in the intermembrane space (IMS) that promote precursor transfer from TOM to TIM23.
Tim23 is the central component of TIM23. It consists of 222 amino acids and can be divided into an N-(1–96) and a C-terminal (97–222) domain.4 The topology of Tim23 is schematically shown in Figure 1(A). The C-terminal part contains four predicted transmembrane (TM) helices spanning the inner membrane. They form a hydrophilic translocation pore with a diameter in the range of 13–14 Å.2,5,6 The channel is just large enough to accommodate a single α-helix. Therefore, proteins need to be unfolded before translocation across the inner membrane can occur. Before translocation through the hydrophilic channel proceeds, the presequences interact with the soluble, N-terminal domain of Tim23. Its first half, residues 1–50, mediates precursor import, whereas its second half (50–97) serves as a presequence receptor domain.3
Figure 1.
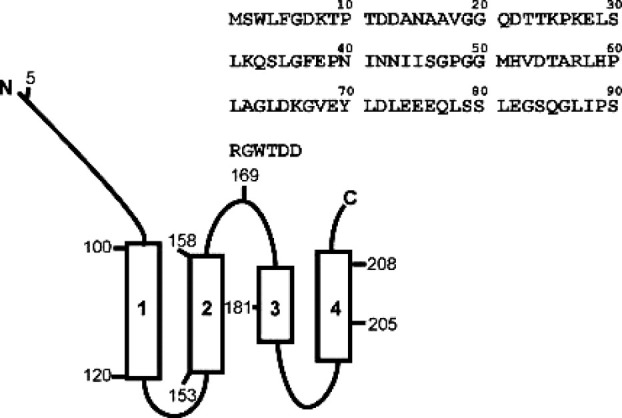
Schematic representation of Tim23. Tim23 consists of a C-terminal domain spanning the inner membrane four times and an N-terminal domain which is exposed to the intermembrane space. The numbers correspond to the residues in the protein sequence. The primary sequence of the N-terminal domain (residues 1–96) of Tim23N is also shown.
Bauer et al.7 reported that the second part of the N-terminal domain (residues 50–100) dimerizes in the absence of a presequence. Upon presequence binding, the dimer is believed to dissociate and trigger channel opening and protein import. As the membrane potential is one of the driving forces for translocation, it is essential that there is no leakage through an open import pore. In the absence of any presequence, the C-terminal IMS domain of Tim50 interacts with Tim23, thus preventing the loss of the proton gradient across the IM. In this way, the channel is only activated by presequence binding.8
Here, we have characterized the structural properties of the N-terminal domain of Tim23 (residues 1–96) and its binding to presequences with single-residue resolution using NMR spectroscopy.
Results
Association state of Tim23N
Gel filtration and blue native polyacrylamide gel electrophoresis were used to compare the size of Tim23N to other intrinsically disordered proteins (Supporting Information Fig. 1). Monomeric Tim23N (Mw = 10.4 kDa) is expected to have a larger retention time on the gel filtration column than K32, a monomeric 198-residue fragment of the protein Tau with Mw = 21.0 kDa.9 Dimeric Tim23N (Mw = 20.8 kDa) should behave in a similar way to K32 but have a shorter retention time than α-synuclein (Mw = 14.4 kDa), another monomeric intrinsically disordered protein.10,11 The observed retention times are in agreement with a purely monomeric Tim23N: K32 is the biggest protein with the shortest retention time on the gel filtration column, followed by α-synuclein and Tim23N. Also, there is only one peak in the elution profile of Tim23N in disagreement with a monomer–dimer equilibrium. Blue native polyacrylamide gel electrophoresis showed a single band for Tim23N at different concentrations supporting the results from gel filtration (Supporting Information Fig. 1).
As the hydrodynamic radius, Rh, of a protein is directly related to its size, it can be used to characterize its aggregation state. Here, we used dynamic light scattering (DLS) and diffusion-ordered spectroscopy (DOSY) to estimate Rh of Tim23N at different concentrations (Fig. 2). DLS measurements detected only a single species in solution with Rh = 2.6 ± 0.2 nm [Fig. 2(A)]. The peak width remained constant at all concentrations, indicating that no significant concentration-dependent changes in polydispersity occurred. Similarly, in DOSY NMR measurements,12 no concentration dependence was observed with an average Rh value of 2.7 ± 0.1 nm [Fig. 2(B)]. Hence, the hydrodynamic radii obtained from DOSY experiments and DLS measurements are identical within error limits. For comparison, the hydrodynamic radius of the 140-residue monomeric, intrinsically disordered protein α-synuclein is 3.2 nm.13
Figure 2.

Hydrodynamic radius of Tim23N at different concentrations. (A) Scattering intensity versus hydrodynamic radius observed by dynamic light scattering. The concentration of Tim23N was changed from 0.05 to 1.0 mM. (B) Hydrodynamic radii at different Tim23N concentrations as determined by NMR diffusion measurements. Error bars represent the standard deviations obtained from repeat measurements. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
For a quantitative interpretation of the hydrodynamic radius Rh, Flory's equation, Rh = R0Nν, is commonly used, where R0 and ν are constants that are determined empirically (note that for unfolded proteins the radius of gyration is about 1.2–1.5 times larger than the hydrodynamic radius14). Three different parameter sets for intrinsically disordered proteins (Wilkins et al., Bernadó et al., and Marsh et al.) were used to calculate the expected hydrodynamic radius of Tim23N. All three parameter sets predict Rh values for monomeric Tim23N that are in agreement with the experimental data (Table I).
Table I.
Hydrodynamic Radius, Rh, Predicted for Tim23N Using Flory's Equation for Different Parameters R0 and ν
Structural and dynamic properties of Tim23N
To gain insight into the secondary structure content of Tim23N, circular dichroism spectra were recorded at 5, 10, and 25 μM protein concentration. The spectra showed the typical random coil profile [Fig. 3(A)]. In agreement with the circular dichroism spectra, a two-dimensional 1H-15N heteronuclear single-quantum coherence spectrum (HSQC) showed only little chemical shift dispersion in the proton dimension [Fig. 3(B)] indicative of the absence of rigid secondary structure. Moreover, upon addition of 8M urea, the hydrodynamic radius of Tim23N increased only slightly to 2.9 ± 0.1 nm as estimated by DOSY NMR measurements (Supporting Information Fig. 2). Thus, Tim23N is an intrinsically disordered protein that rapidly exchanges between a large number of different conformations.
Figure 3.
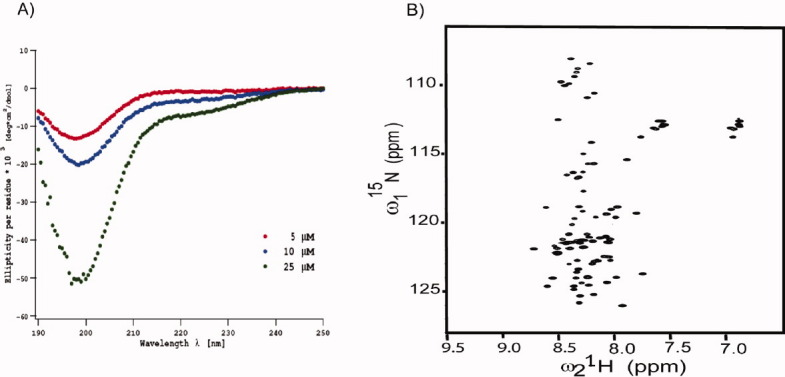
Tim23N is intrinsically disordered. (A) Circular dichroism spectra of Tim23N at 5 μM, 10 μM and 25 μM (from top to bottom) in phosphate buffer. (B) Two-dimensional 1H-15N HSQC spectrum of Tim23N. The spectrum shows only a limited dispersion of chemical shifts typical of intrinsically disordered proteins. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
For the characterization of the structure and dynamics of Tim23N with single-residue resolution, the sequence-specific assignment of the NMR resonances was required. Nearly complete backbone assignment was achieved using three-dimensional (HA)CANNH17 and HNN experiments.18 NMR chemical shifts, in particular of Cα and C′ atoms, are very sensitive probes of secondary structure both in globular and intrinsically disordered proteins.19,20 Cα secondary chemical shifts of about three and minus two are expected for rigid α-helices and β-sheets, respectively.19 In Tim23N, the magnitude of averaged Cα and C′ secondary chemical shifts is mostly below 0.5 ppm [Fig. 4(A)], indicating that no rigid structure is present. In the residue fragment 72–85 secondary chemical shifts are predominantly positive, pointing toward a slight propensity for helical structure.
Figure 4.
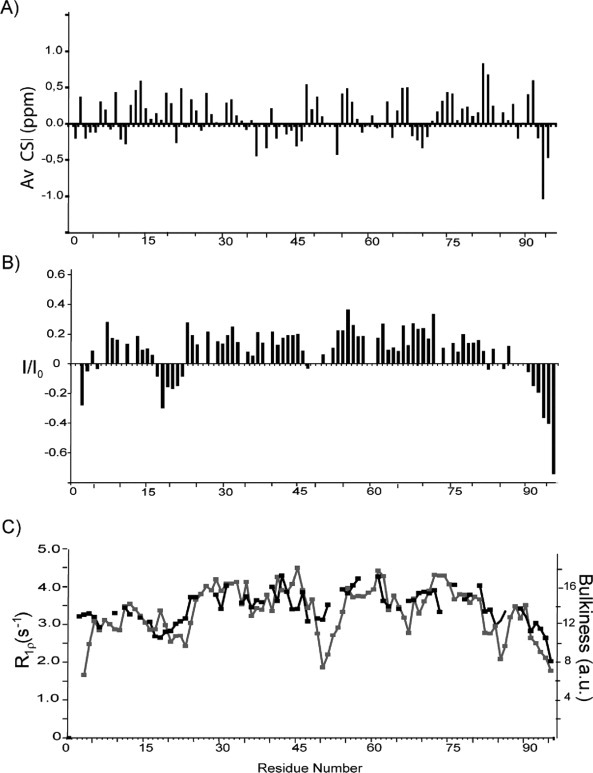
Structural properties of Tim23N. (A) Averaged Cα/CO secondary chemical shifts observed in Tim23N as a function of residue number. Negative (positive) secondary chemical shifts spanning several residues indicate propensity for β-structure (α-helix). (B) Steady-state heteronuclear 15N-[1H]-NOEs in Tim23N at 15°C. (C) 15N R1ρ spin relaxation rates of backbone resonances of Tim23N (black). The experimental relaxation rates follow the bulkiness profile expected from the primary sequence (gray).21
To probe the backbone dynamics of Tim23N, we measured 15N spin relaxation times.22 Folded proteins show steady-state heteronuclear 1H,15N NOEs in the range of 0.8–1.0, whereas disordered proteins have NOEs below 0.5. In Tim23N, heteronuclear 1H,15N NOEs do not exceed 0.4 [Fig. 4(B)]. Besides the two termini, residues 17–22 are characterized by negative steady-state NOE values and comprise the part of the Tim23N backbone with the highest flexibility on the pico- to nanosecond time scale. In addition, 15N R1ρ spin relaxation rates, which are sensitive to motions of the backbone occurring on the micro- to millisecond time scale,22 closely follow the variation in the bulkiness of amino acids along the chain [Fig. 4(C)]. Deviations at the N-terminus might be caused by the two additional residues in Tim23N as a result of cloning that are not shown. The average 15N R1ρ relaxation rate was 3.4 s−1.
Interaction of Tim23N with presequences
The binding of presequences to Tim23N was probed using NMR spectroscopy.23 In NMR titration experiments, residues that are part of the binding interface or experience changes in the chemical environment as a result of ligand binding undergo changes in signal position and potentially signal intensity, whereas unaffected residues experience little or no changes in chemical shifts. The presequences of retinal aldehyde dehydrogenase (rALDH) and cytochrome c oxidase subunit IV (CoxIV)24 were titrated to 15N-labeled Tim23N. During the titrations, neither signal broadening nor additional peaks were observed, indicating that the binding process is fast on the NMR time scale, that is, rapid association and dissociation of the Tim23N-presequence-complex resulting in averaged, gradually shifting peaks of residues at the binding interface. Figure 5(A) shows a superposition of a section of 1H-15N HSQC spectra of Tim23N in the absence and presence of the rALDH presequence. The addition of the rALDH presequence caused a shift of the peak positions for several residues, e.g., L71, L78, or E76, whereas other peaks, e.g., A14 and E38, were not affected.
Figure 5.
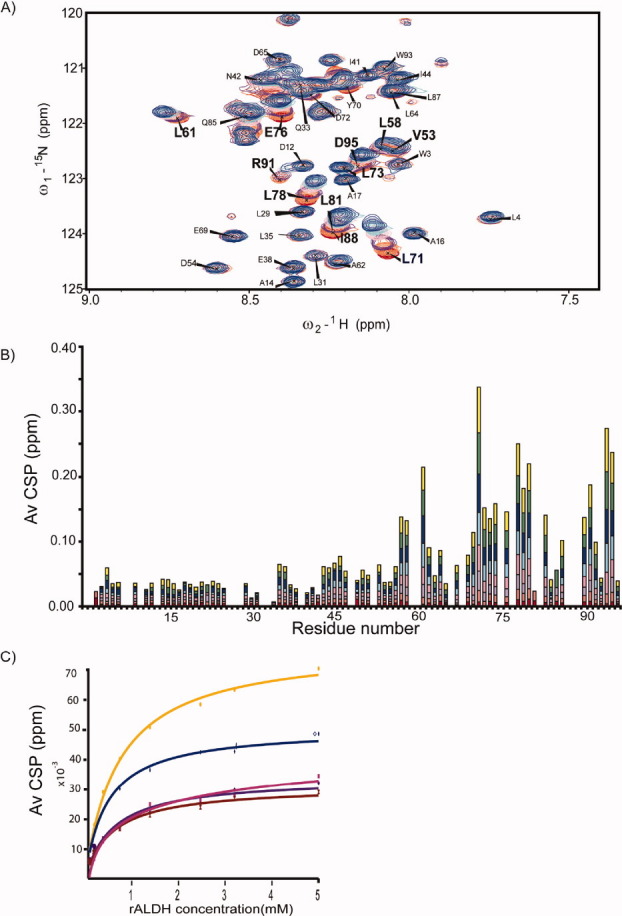
Binding of the presequence of rALDH to Tim23N. (A) Superposition of 2D 1H-15N HSQC spectra of Tim23N without rALDH (red) and at 32-fold excess of rALDH (blue). (B) Averaged 1H,15N chemical shift deviation observed for backbone resonances of Tim23N in 2D 1H-15N HSQC NMR spectra upon addition of rALDH at molar ratios (Tim23N:rALDH) of 1:1 (red), 1:2 (orange), 1:4 (purple), 1:8 (pink), 1:16 (cyan), 1:32 (blue), 1:64 (green), and 1:100 (yellow). (C) Chemical shift perturbation as a function of rALDH concentration for residues 71 (orange), 72 (pink), 74 (purple), 76 (blue), and 78 (brown). Solid lines show the binding curves obtained by fitting the chemical shift changes to a single binding-site model. A weighted average of the dissociation constant of 0.47 ± 0.02 mM was obtained.
Plotting the chemical shift perturbations along the chain shows that the rALDH presequence induced the strongest chemical shift changes in the region L71-S84 of Tim23N [Fig. 5(B)]. Furthermore, the C-terminus, G90-D96, undergoes significant changes in chemical shift values. A third region that experiences weaker chemical shift perturbation includes I41-V68. A similar perturbation profile was found for the titration of CoxIV to Tim23N, with changes within L71-S84 and at the C-terminus (Supporting Information Fig. 3). However, already at a molar ratio of 1:4, precipitation was observed in the NMR tube, pointing to solubility problems of the protein–peptide mixture.
A synthetic peptide (SynB2) with a similar amino acid composition as CoxIV but no function as specific targeting signal24 was titrated as a control to Tim23N. At an eightfold excess only very small chemical shift perturbations were observed, much smaller than those induced by rALDH (Supporting Information Fig. 4). The weak chemical shift changes localized mainly to the region L71-S84 similar to the titrations with the two presequences. Detailed comparison, however, reveals that the patterns within the binding region are quite different. SynB2 interacts most strongly with E76, whereas the presequences undergo the largest chemical shift changes at L71. Moreover, the effects experienced by SynB2 at positions D72 and E74 are distinct from the changes observed in CoxIV and rALDH. SynB2 has a similar amino acid composition to CoxIV and is therefore able to interact very weakly and unspecifically, that is, purely electrostatically with the negative charges of three glutamates (residues E74–E76) in Tim23N.
The rALDH-induced chemical shift perturbations for different residues of Tim23N were plotted as a function of ligand concentration and fitted to a Hill equation [Fig. 5(C)]. The residues experiencing the largest chemical shift changes during the titration could be fitted well to binding curves, thus confirming the postulated binding region. Residues with minor chemical shift perturbations gave only poor fits to the Hill equation. Assuming a 1:1 binding stoichiometry, the dissociation constant KD of the rALDH-Tim23N complex was estimated for different residues (Table II), resulting in a weighted average value of KD = 0.47 ± 0.02 mM. The low affinity of the presequence–Tim23N interaction agrees with the necessary transient nature of the interactions responsible for protein translocation through the inner membrane. Presequences have to interact with Tim23N but subsequently dissociate in order to be transferred to the central pore of TIM23.
Table II.
KD Values with Standard Deviations for the Main rALDH Binding Region in Tim23N
| Residue | KD (mM) | sKD (mM) |
|---|---|---|
| L71 | 0.49 | 0.04 |
| L73 | 0.42 | 0.04 |
| E74 | 0.45 | 0.04 |
| E76 | 0.44 | 0.03 |
| Q77 | 0.60 | 0.05 |
| L78 | 0.48 | 0.05 |
| S79 | 0.48 | 0.05 |
| S80 | 0.51 | 0.1 |
Isothermal calorimetry was used as an alternative method to determine the binding constant of presequence binding to Tim23N (data not shown). The reaction showed only small endothermic peaks for the addition of rALDH to Tim23N that could not be fitted. Most likely, the binding is too weak and causes too little changes in heat to be detectable by isothermal calorimetry.
Dynamics in the complex of Tim23N with the rALDH presequence
Upon addition of the rALDH presequence, the signal intensities observed for the residues in the binding region in 2D 15N-1H HSQC spectra decreased (data not shown), indicating that this region of Tim23N becomes either less flexible or is affected by chemical exchange during complex formation. To directly probe the backbone dynamics of Tim23N in the complex, we determined steady-state heteronuclear 1H,15N NOEs in Tim23N in the presence of a 64-fold excess of rALDH (Fig. 6). In the presence of the rALDH presequence, NOE values reached up to 0.6–0.7 for residues L71-S84, indicating that pico-to-nanosecond dynamics are significantly reduced. In addition, residues S90-D96, which were highly flexible in the unbound state, were retained and showed NOE values of 0.3–0.4 in the presence of rALDH. Thus, L71-S84, possibly extended to S96, form the binding site for presequences.
Figure 6.

Transient nature of the Tim23N-rALDH complex. (A) Helical wheel projection of the rALDH presequence. Hydrophilic residues, hydrophobic residues, negatively charged, and positively charged residues are represented by circles, diamonds, triangles, and pentagons, respectively. (B) Helical wheel projection of Tim23N (71–84). (C) Steady-state heteronuclear 15N-[1H]-NOEs observed in Tim23N without (black) and with a 64-fold excess of the rALDH presequence (gray).
Discussion
When previously expressed as glutathione S-transferase (GST) fusion protein, truncation experiments and analysis by gel filtration showed dimerization of Tim23N-GST via the second half of the N-terminal domain (residues 50–100).7 Here, we used a variety of methods to probe the aggregation of Tim23N in aqueous buffer in the absence of a GST-tag. No dimer could be detected by comparison to other unfolded proteins. DLS and DOSY confirmed that free Tim23N is monomeric up to 1 mM concentration. In addition, 15N-HSQC spectra observed for 100 μM Tim23N showed no changes in chemical shift when compared with spectra of 1 mM Tim23N (data not shown). Higher concentrations of Tim23N should lead to an increase in dimer population resulting in chemical shift changes at the dimer interface. GST is known to form dimers in its native state25 and might therefore promote dimerization when part of a fusion protein.
NMR spectroscopy demonstrated that monomeric Tim23N is intrinsically disordered in agreement with previous CD measurements (Gevorkyan-Airapetov et al., JBC 2009). The backbone is highly dynamic on different time scales with a pronounced flexibility at its termini and in the region A17-T23. A very low helical propensity was present throughout the remaining sequence. The close match between the 15N R1ρ relaxation rates and the bulkiness profile [Fig. 4(C)] is in agreement with our previous finding that although various types of intramolecular interactions, such as electrostatic and solvent interactions, play important roles, simple considerations of the side-chain volume of amino acids predict a major component of diverse parameters dependent on the local conformation and dynamics of intrinsically disordered proteins.21
To obtain insight into the importance of protein disorder in the context of the protein import machinery, the mean hydropathy and net charge of all soluble domains in the cytosol, the IMS or the matrix from subunits of TOM and TIM23 were analyzed (Fig. 7). All protein regions larger than 10 residues were chosen that had neither predicted TM helices nor predicted TM β-barrels. TM helices were predicted using HMMTOP,26 whereas β-barrels were predicted by TRAMPLE.27 The mean hydropathies of the domains were calculated from the normalized hydropathies described by Kyte and Doolittle.28 The boundary between intrinsically disordered and native proteins was used as empirically determined by Uversky et al.29 In agreement with our NMR data, the hydropathy plot classifies Tim23N as an intrinsically disordered protein. Noteworthy, Tim23N is the largest disordered domain of TOM and TIM23 found in the IMS. Its intrinsic disorder is expected to be beneficial to act as a hub in the mitochondrial import machinery protein network.
Figure 7.
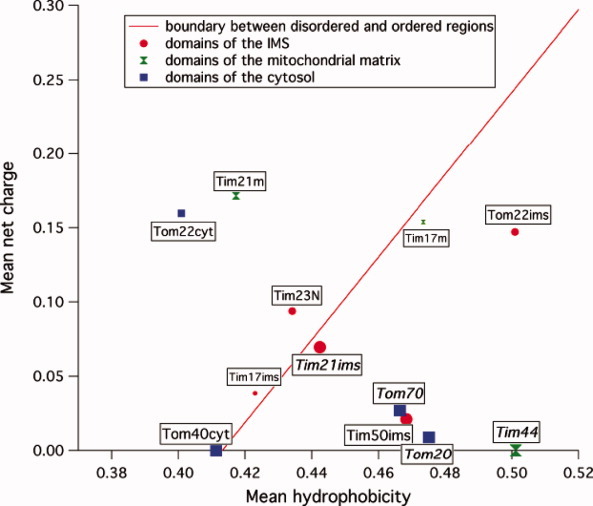
Charge-hydropathy plot of soluble domains of TOM and TIM23. Soluble domains are represented according to their location in the cytosol (blue squares), the intermembrane space (red circles), or the matrix (green diamonds). The order–disorder boundary is shown as red line. The size of the domains correlates with the size of the symbol: <40 residues (small), 40–100 residues (medium), and >100 residues (large). Domains of known structure are labeled italics.
To gain insight into the interactions between the presequences and Tim23N, helical wheel projections of the presequences, the synthetic peptide and the binding region of Tim23N were determined (Fig. 6 and Supporting Information Figs. 3 and 4). Presequences are believed to form amphipathic helices with an accumulation of positively charged residues on one side and hydrophobic residues on the opposite site of the helix, favoring helix–helix-type interactions. Within the binding region of Tim23N, a high density of negatively charged residues and leucines is seen on one side of the helical projection. L71, E74, E75, L78, and L81 are residues located next to each other in the helical projection. They also show the largest chemical shift changes in the presequence titrations. Although presequences and Tim23N are believed to interact electrostatically, Figures 5 and 6 suggest that also hydrophobic interaction in the binding region of Tim23N could be important for presequence binding. Abe et al.30 showed binding of rALDH to a hydrophobic pocket of Tom20. They suggested that the combination of positive charges and hydrophobicity in an amphipathic helix provides different, independent recognition elements. In case of SynB2, the helical wheel projection does not show a hydrophobic face and no clear amphipathic character. It does, however, show two arginines (R9 and R12) and two leucines (L2 and L23) in close proximity. This region might account for the observed weak unspecific interactions of SynB2 with Tim23N.
In conclusion, our study demonstrates that Tim23N is a monomeric protein belonging to the family of intrinsically disordered proteins. Titrations of Tim23N with two presequences revealed a distinct binding region of Tim23N formed by residues 71–84. In a charge-hydropathy plot containing all soluble domains of TOM and TIM23, Tim23N was found to be the only domain with more than 40 residues in the IMS that is predicted to be intrinsically disordered, suggesting that Tim23N might function as hub in the mitochondrial import machinery protein network
Materials and Methods
Primers used for cloning were obtained from Invitrogen (Karlsruhe, Germany). Sequences of newly cloned, purified plasmid DNA were determined by SeqLab (Göttingen, Germany). The cDNA-encoding Tim23N was cloned into a modified pGEX expression vector (GE Healthcare, Uppsala, Sweden) with ampicillin resistance for the production of N-terminal GST-tagged fusion protein with a TEV cleavage site. The expression plasmid was transformed into E. coli BL21 (DE3)RIL cells. Protein expression was carried out at 25°C. TEV-cleavage was performed overnight at room temperature. Cleaved Tim23N protein was dialyzed, concentrated, and purified by gel filtration chromatography. Gel filtration was carried out on an Äkta Purifier system using a Superdex 75 HiLoad column. Tim23N was further purified by RP-HPLC to remove residual GST. Blue native polyacrylamide gel electrophoresis was performed with BioRad Precast Gels (4–15% resolving gel). Protein concentrations for spectroscopic measurements were between 0.05 and 1.0 mM of Tim23N in 20 mM Hepes buffer, 50 mM NaCl, pH 7.2 (NMR buffer).
The rALDH presequence (MLRAALSTARRGP RLSRLLSAA) was synthesized by G. Wolf. CoxIV (MLSLRQSIRFFKPATRTLSSSRY) and SynB2 (MLS RQQSQRQSRQQSQRQSQYLL) were obtained from EMC Microcollections, Tübingen, Germany.
CD measurements were performed using a Jasco J-720 instrument at 15°C. Spectra were measured in Hellma quartz cuvettes with a path length of 0.1 cm. Spectra were obtained by averaging over 5–50 scans using a scan rate of 50 nm min−1. DLS measurements were performed with a Wyatt DynaPro Titan (Wyatt Technology, CA). Samples were filtered with a 0.2-μm syringe filter before the measurement.
NMR spectra were recorded at 15°C on Bruker Avance 600 and 700 MHz spectrometers equipped with shielded gradient triple resonance probes. For determining the hydrodynamic radius, NMR samples contained dioxane as an internal radius standard and viscosity probe.12 In DOSY experiments, the gradient strength was linearly increased from 2 to 95% of the maximum gradient strength. For each 1H spectrum, 128 scans and 16 K complex were acquired. The diffusion data (signal intensity vs. gradient strength) were fitted to exponential functions using Igor Pro 5.01 (WaveMetrics). From the apparent diffusion coefficients of Tim23N and dioxane and the known Stokes radius of dioxane (2.12 Å), Stokes radii of Tim23N were calculated.31
For backbone resonance assignment, 3D HNCO, 3D (HA)CANNH,17 and HNN18 experiments were collected. NMR data were processed and analyzed using NMRPipe32 and Sparky 3 (T.D. Goddard and D.G. Kneller, http://www.cgl.ucsf.edu/home/sparky). Secondary shift values were calculated as the differences between measured Cα chemical shifts and the empirical random coil value for the appropriate amino acid type.33 Random coil values for charged residues were taken from Wishart and Sykes,34 as the chemical shifts of these residues are particularly sensitive to pH. Steady-state heteronuclear 1H,15N NOEs and 15N R1ρ relaxation rates (for a review, see Ref.22) were measured at a 1H frequency of 700 MHz. For R1ρ measurements, a spinlock frequency of 2 kHz and relaxation periods of 20, 60, 120, 180, 240, and 350 ms were used. Relaxation times were calculated by fitting an exponential function to the decaying signal integrals.
Samples used for NMR titrations typically contained 0.1 mM 15N-labeled Tim23N in NMR buffer. Chemical shift perturbations were measured using 2D 1H-15N-HSQC experiments. To prevent artifacts caused by changes in buffer upon addition of the ligand, protein and ligand were dialyzed for at least 12 h against the same buffer before titratioñ. Normalized weighted average chemical shift differences for amide 1H and 15N chemical shifts upon presequence binding were calculated using Δav(NH) = [(ΔH2 + (ΔN/5)2)/2]1/2, where ΔH and ΔN are the differences between the free and bound chemical shifts. Titration experiments were evaluated using a single binding-site model35:
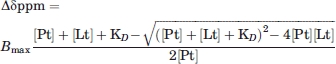 |
where [Pt], [Lt], KD, and Bmax are the total protein concentration, ligand concentration, dissociation constant, and chemical shift deviation at saturation, respectively. [Pt] was treated as a constant, KD and Bmax were the parameters to be fitted. Fits were performed using Igor Pro 5.01 (WaveMetrics).
Acknowledgments
The authors thank Reinhard Albrecht and Kornelius Zeth for help in the initial stages of the project, Christian Griesinger and Peter Rehling for useful discussions, and Ulrich Dürr and Lukasz Skora for help with NMR measurements.
References
- 1.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Cell Dev Biol. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 2.Pfanner N, Chacinska A. The mitochondrial import machinery: preprotein-conducting channels with binding sites for presequences. Biochim Biophys Acta. 2002;1592:15–24. doi: 10.1016/s0167-4889(02)00260-4. [DOI] [PubMed] [Google Scholar]
- 3.van der Laan M, Hutu DP, Rehling P. On the mechanism of preprotein import by the mitochondrial presequence translocase. Biochim Biophys Acta. 2010;1803:732–739. doi: 10.1016/j.bbamcr.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Donzeau M, Kaldi K, Adam A, Paschen S, Wanner G, Guiard B, Bauer MF, Neupert W, Brunner M. Tim23 links the inner and outer mitochondrial membranes. Cell. 2000;101:401–412. doi: 10.1016/s0092-8674(00)80850-8. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz MP, Matouschek A. The dimensions of the protein import channels in the outer and inner mitochondrial membranes. Proc Natl Acad Sci USA. 1999;96:13086–13090. doi: 10.1073/pnas.96.23.13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alder NN, Jensen RE, Johnson AE. Fluorescence mapping of mitochondrial TIM23 complex reveals a water-facing, substrate-interacting helix surface. Cell. 2008;134:439–450. doi: 10.1016/j.cell.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Bauer MF, Sirrenberg C, Neupert W, Brunner M. Role of Tim23 as voltage sensor and presequence receptor in protein import into mitochondria. Cell. 1996;87:33–41. doi: 10.1016/s0092-8674(00)81320-3. [DOI] [PubMed] [Google Scholar]
- 8.Meinecke M, Wagner R, Kovermann P, Guiard B, Mick DU, Hutu DP, Voos W, Truscott KN, Chacinska A, Pfanner N, et al. Tim50 maintains the permeability barrier of the mitochondrial inner membrane. Science. 2006;312:1523–1526. doi: 10.1126/science.1127628. [DOI] [PubMed] [Google Scholar]
- 9.Mukrasch MD, von Bergen M, Biernat J, Fischer D, Griesinger C, Mandelkow E, Zweckstetter M. The “jaws” of the tau-microtubule interaction. J Biol Chem. 2007;282:12230–12239. doi: 10.1074/jbc.M607159200. [DOI] [PubMed] [Google Scholar]
- 10.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 11.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci USA. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 13.Cho MK, Nodet G, Kim HY, Jensen MR, Bernado P, Fernandez CO, Becker S, Blackledge M, Zweckstetter M. Structural characterization of alpha-synuclein in an aggregation prone state. Protein Sci. 2009;18:1840–1846. doi: 10.1002/pro.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherman E, Haran G. Coil-globule transition in the denatured state of a small protein. Proc Natl Acad Sci USA. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernado P, Blackledge M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys J. 2009;97:2839–2845. doi: 10.1016/j.bpj.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsh JA, Forman-Kay JD. Sequence determinants of compaction in intrinsically disordered proteins. Biophys J. 2010;98:2383–2390. doi: 10.1016/j.bpj.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zweckstetter M, Bax A. Single-step determination of protein substructures using dipolar couplings: aid to structural genomics. J Am Chem Soc. 2001;123:9490–9491. doi: 10.1021/ja016496h. [DOI] [PubMed] [Google Scholar]
- 18.Panchal SC, Bhavesh NS, Hosur RV. Improved 3D triple resonance experiments, HNN and HN(C)N, for HN and 15N sequential correlations in (13C, 15N) labeled proteins: application to unfolded proteins. J Biomol NMR. 2001;20:135–147. doi: 10.1023/a:1011239023422. [DOI] [PubMed] [Google Scholar]
- 19.Wishart DS, Sykes BD. Chemical shifts as a tool for structure determination. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 20.Dyson HJ, Wright PE. Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol. 2001;339:258–270. doi: 10.1016/s0076-6879(01)39317-5. [DOI] [PubMed] [Google Scholar]
- 21.Cho MK, Kim HY, Bernado P, Fernandez CO, Blackledge M, Zweckstetter M. Amino acid bulkiness defines the local conformations and dynamics of natively unfolded alpha-synuclein and tau. J Am Chem Soc. 2007;129:3032–3033. doi: 10.1021/ja067482k. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Palmer AG., III Solution NMR methods for quantitative identification of chemical exchange in N-15-labeled proteins. Magn Reson Chem. 2003;41:866–876. [Google Scholar]
- 23.Craik DJ, Wilce JA. Studies of protein-ligand interactions by NMR. Methods Mol Biol. 1997;60:195–232. doi: 10.1385/0-89603-309-0:195. [DOI] [PubMed] [Google Scholar]
- 24.Allison DS, Schatz G. Artificial mitochondrial presequences. Proc Natl Acad Sci USA. 1986;83:9011–9015. doi: 10.1073/pnas.83.23.9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker MW, Lo Bello M, Federici G. Crystallization of glutathione S-transferase from human placenta. J Mol Biol. 1990;213:221–222. doi: 10.1016/s0022-2836(05)80183-4. [DOI] [PubMed] [Google Scholar]
- 26.Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 27.Martelli PL, Fariselli P, Krogh A, Casadio R. A sequence-profile-based HMM for predicting and discriminating beta barrel membrane proteins. Bioinformatics. 2002;18(Suppl 1):S46–S53. doi: 10.1093/bioinformatics/18.suppl_1.s46. [DOI] [PubMed] [Google Scholar]
- 28.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 29.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 30.Abe Y, Shodai T, Muto T, Mihara K, Torii H, Nishikawa S, Endo T, Kohda D. Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell. 2000;100:551–560. doi: 10.1016/s0092-8674(00)80691-1. [DOI] [PubMed] [Google Scholar]
- 31.Jones JA, Wilkins DK, Smith LJ, Dobson CM. Characterisation of protein unfolding by NMR diffusion measurements. J Biomol NMR. 1997;10:199–203. [Google Scholar]
- 32.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 33.Schwarzinger S, Kroon GJ, Foss TR, Chung J, Wright PE, Dyson HJ. Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 34.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 35.Auguin D, Barthe P, Royer C, Stern MH, Noguchi M, Arold ST, Roumestand C. Structural basis for the co-activation of protein kinase B by T-cell leukemia-1 (TCL1) family proto-oncoproteins. J Biol Chem. 2004;279:35890–35902. doi: 10.1074/jbc.M400364200. [DOI] [PubMed] [Google Scholar]
- 36.Gevorkyan-Airapetov L, Zohary K, Popov-Celeketic D, Mapa K, Hell K, Neupert W, Azem A, Mokranjac D. Interaction of Tim23 with Tim50 Is essential for protein translocation by the mitochondrial TIM23 complex. J Biol Chem. 2009;284:4865–48. doi: 10.1074/jbc.M807041200. [DOI] [PubMed] [Google Scholar]