

Abstract
In most lipases, a mobile lid covers the substrate binding site. In this closed structure, the lipase is assumed to be inactive. Upon activation of the lipase by contact with a hydrophobic solvent or at a hydrophobic interface, the lid opens. In its open structure, the substrate binding site is accessible and the lipase is active. The molecular mechanism of this interfacial activation was studied for three lipases (from Candida rugosa, Rhizomucor miehei, and Thermomyces lanuginosa) by multiple molecular dynamics simulations for 25 ns without applying restraints or external forces. As initial structures of the simulations, the closed and open structures of the lipases were used. Both the closed and the open structure were simulated in water and in an organic solvent, toluene. In simulations of the closed lipases in water, no conformational transition was observed. However, in three independent simulations of the closed lipases in toluene the lid gradually opened. Thus, pathways of the conformational transitions were investigated and possible kinetic bottlenecks were suggested. The open structures in toluene were stable, but in water the lid of all three lipases moved towards the closed structure and partially unfolded. Thus, in all three lipases opening and closing was driven by the solvent and independent of a bound substrate molecule.
Keywords: interfacial activation, lipases, organic solvent, molecular dynamics simulation, activation pathway, lid movement
Introduction
Lipases belong to the family of serine hydrolases and catalyze the hydrolysis of carboxylic esters in water and the reverse reaction, the acylation of alcohols with carboxylic acids, in organic solvents.1 They share a common α/β-hydrolase fold and an active site with the catalytic triad, consisting of Ser, His, and Asp/Glu, and the oxyanion hole.2 Depending on a signature sequence near the oxyanion hole, two classes of lipases were identified; the GX and the GGGX class.3 The characteristic of most lipases is their activation upon binding to a hydrophobic substrate interface, the interfacial activation.4 In water, the active site is covered by a mobile element, the lid, which opens upon binding of the lipase to a hydrophobic interface.5 The inside of the lid is hydrophobic, whereas the outside is hydrophilic.6–8 Upon opening, the inner hydrophobic surface is exposed, and the solvent-accessible surface area increases drastically.9 Therefore, the closed form is supposed to be predominant in water, while the open form is expected to be stabilized in organic solvent.
The transition pathway from the closed to the open structure is mostly unknown. Depending on the structure of the lid, different transition mechanisms were proposed. In lipases with a lid consisting of only one helix, the transition was suggested to be a fast rigid body movement.10 In contrast, in lipases with a more complex lid like the Candida rugosa lipase the secondary structure of the lid changes upon opening, therefore a partial refolding is expected8 which might be a kinetic bottleneck. Understanding these bottlenecks can help in designing more efficient lipases. Computational studies on the lid of lipases revealed aspects of the molecular mechanism. Molecular dynamics with external forces applied on three lipases with a lid consisting of a single short helix revealed varying energy levels upon activation that depend on the distribution of the charged amino acids in the lid.10 Simulations of a lipase of Rhizomucor miehei for 1–2 ns without an external force showed an increased opening of the active site in the presence of a lipid patch when compared to simulations in aqueous environment.11 Studies on Candida rugosa lipase in water and carbon tetrachloride as solvent revealed a reduced flexibility of polar side chains and an increased stability of the lid in the organic solvent which is caused by two salt bridges and a hydrogen bond.12 Further studies on the closed structure of Candida rugosa lipase at three different alkane-water interfaces for 4 ns each showed partial openings of 7–13 Å with the opening time and opening distance depending on the used organic solvent.13 Previously, in two independent simulations a complete pathway of the conformational transition from the closed to the open structure of Burkholderia cepacia lipase in organic solvent was observed, while water as a solvent led to a closing of the open structure.14,15
In this study, we compared three different lipases with two different types of the lid: Candida rugosa lipase (CRL) and the two homologues fungal lipases of Rhizomucor miehei (RML) and Thermomyces lanuginosa (TLL). CRL belongs to the GGGX class and has a large and complex lid (residues 66–92), consisting of a short and a long α-helix.8 A comparison of the closed and open crystal structure revealed that the lid has to refold partially. For this reason, a slower opening and closing when compared to RML and TLL was expected. RML and TLL belong to the GX class and have a short lid helix (RML: residues 82–96, TLL: residues 82–96), connected to the remaining part of the lipase with two hinges.6,16 For these lipases, a fast rigid body movement of the lid was suggested.10 We performed multiple molecular dynamics simulations of the open and closed structure of CRL, RML, and TLL in water and toluene, with a duration of 20–25 ns each. The results were analyzed to identify the proposed lids and to investigate the conformational pathway with kinetic bottlenecks upon opening and closing of the lid.
Results
The three lipases CRL, RML, and TLL were examined in simulations with water or toluene as explicit solvent. Both the open and the closed structure were used for CRL and the homologues RML and TLL. All six structures were solvated with each of the two solvents and three simulations of 20–25 ns with different starting velocities were performed for all systems, resulting in a total number of 36 simulations.
The core of all proteins was stable during all simulations and showed no partial unfolding or changes in secondary structure. The lids in the open structure in water and in the closed structure in toluene were highly flexible, whereas the closed structure in water and the open structure in toluene were generally less flexible. Two simulations of open CRL in water showed a partial unfolding at the lid. In RML a complete conformational transition from the closed to the open structure in toluene could be observed, in CRL a partial opening of the lid in toluene.
CRL—a lipase with a complex lid
All simulated structures of CRL showed a core RMSD (defined as the root mean square deviations of all backbone atoms except for the lid) to the initial crystal structures in the range of 1.5 to 2.0 Å. (Fig. S1) The core structure deviated from the initial crystal structure during the equilibration phase and did not substantially change during the further simulation. All simulations of CRL in water showed a significantly larger solvent accessible surface (SAS) area compared to the simulations in toluene (CRL open: 21500 Å2 in water and 19600 Å2 in toluene, CRL closed: 20500 Å2 in water and 19000 Å2 in toluene).
Closed CRL in water
In all three simulations of the closed structure of CRL in water, small movements of the lid were observed, but they were not sufficiently large to open an access channel from the bulk solvent to the active center. The area of the hydrophobic SAS increased from 6200 Å2 to 6700 Å2 but did not reach the area of the open CRL of 7000–7600 Å2. (Fig. S6). The lid RMSD, defined as the root mean square deviations of all backbone atoms placed in the lid (residues Gly67-Ser91), to the initial crystal structure was less than 3 Å and stable in all three simulations (Fig. S1). The secondary structure of both lid helices remained stable.
Open CRL in toluene
In all three simulations of the open conformation of CRL in toluene, the lid remained close to the crystal structure during the simulations. The access channel from the bulk solvent to the active center remained open in all three simulations, indicated by a nearly constant area of the hydrophobic SAS (7200–7700Å2, Fig. S2). The lid RMSD was stable in all simulations (1–2.5 Å) (Fig. S1). The deviations were mainly caused by movements of the small lid helix (Val86-Pro92), while the long lid helix (Leu73-Ser84) did not change relative to the crystal structure.
Closed CRL in toluene
In all three simulations conformational changes of the lid towards its open crystal structure were observed. These structural changes can be seen in the plot of the hydrophobic SAS, which increased from 6000 Å2 to 7000–7300 Å2 (Fig. S2). However, the conformational changes were not large enough to completely open the access channel to the active center.
The lid RMSD of 4–9 Å indicated a large movement of the lid in all three simulations (Fig. S1). In two simulations, the lid movement started already after 1.5 ns and the lid gradually opened during the simulation as indicated by a movement of Lys75 by 4.5 and 7.5 Å, which is placed between the two helices of the lid. In one simulation the lid movement started only after 14 ns and showed a stepwise opening with a maximum movement at Lys75 by 10.5 Å. In a first step the lid opened by 1.5 Å during 1 ns and stayed during 1.5 ns in this conformation. In a second step, the lid further opened to a RMSD of 3.5 Å during 1 ns and moved towards the open structure. This second step was followed by 5.5 ns without movement. It continued with a third step of 2 ns and a further opening of 4 Å. Until the end of the simulation, no further movement can be seen. Val86, that is found in the lid, was tightly bound into a hydrophobic pocket and appeared to be blocked by the surrounding residues, but has to be released to continue the opening of the lid, which did not happen during the remaining simulation time. In all three simulations, the short helix (Tyr69-Asn72) unfolded, and the long helix (Lys75-Ser84) started to unfold at Lys75 and, in two simulations, up to Leu78. In one simulation, a helical folding of Ser84-Glu88 was observed.
Open CRL in water
In two simulations a conformational change of the lid and an unfolding of the secondary structure were observed. However, the closing movement did lead to a partial closure of the access channel to the active center before an unfolding started. In one simulation the lid RMSD increased to 7.5 Å during 12 ns (Fig. S1), and the lid moved towards the closed crystal structure with a maximum movement of Leu73 by 9 Å. After this closing movement the lid unfolded in the remaining 13 ns. A second simulation showed no movements in the first 5 ns, where the lid was close to the crystal structure with a lid RMSD of 2 Å, followed by an unfolding of the secondary structure. In a third simulation, the lid RMSD increased only slightly (1–3 Å) and the structure and position of the lid remained close to the open crystal structure.
RML and TLL—lipases with a simple lid
All simulated structures of RML and TLL showed a slight increase of the core RMSD to 0.9 to 1.2 Å when compared to the initial crystal structures during the equilibration phase (Figs. S3 and S4). An additional increase by up to 1.0 and 0.5 Å during the simulation time of 25 ns were observed in RML and TLL, respectively. The cores of the proteins were stable without unfolding of their secondary structure. The changes in the core RMSD of RML were mainly caused by a flexible loop (residues Ser56-Asn63 in RML and Ser58-Val63 in TLL). The highest flexibility of the loop was found in water with a displacement of up to 7 Å. All simulations of TLL and RML in water showed a significantly larger SAS area when compared to the simulations in toluene (TLL and RML open: 12200 Å2 in water and 11500 Å2 in toluene, and RML and TLL closed: 12000 Å2 in water and 11200 Å2 in toluene).
Closed RML in water
In two simulations the lid remained close to the initial crystal structure with a lid RMSD of less than 1 Å, while one simulation showed movements in the lid (Fig. S3). The channel to the active center remained closed in all three simulations as indicated by a nearly constant area of the hydrophobic SAS of 3800–4000 Å2 (Fig. S2).
The lid behavior differed in all three simulations. In one simulation, the helix was intact, while in the other simulation, the helix unfolded at its N-cap region (Ile81-Trp84). In the third simulation, the lid changed its orientation (lid RMSD 1.75 Å) during the first ns of the simulation and then was stable during the simulation.
Open RML in toluene
In all three simulations of the open conformation of RML in toluene, the lid remained close to the initial crystal structure. The access channel to the active center remained open in all three simulations, shown by a constant area of the hydrophobic SAS of 4300 Å2 (Fig. S2). The lid RMSD was less than 1.2 Å (Fig. S3) and the secondary structure remained the same as in the initial open crystal structure.
Closed RML in toluene
In one simulation a large conformational transition from the closed to the open structure of the lid was observed, and an access channel to the active center was formed. The area of the hydrophobic SAS showed a significant increase from 3700 to 4500 Å2, which is more than the area of the hydrophobic SAS of 4300 Å2 of the open RML structure in toluene (Fig. S2). In a second simulation the lid opened slightly and a narrow access channel to the active center was formed, as indicated by a small increase of the hydrophobic SAS from 3700 to 4100 Å2. In the third simulation, during 30 ns only small movements and a constant hydrophobic SAS were observed, and the lid remained in a closed state.
During the lid opening, the lid RMSD increased up to 5.5 Å (Fig. S3). The movement started after 4 ns and was completed after 8 ns. It consisted in a rigid body movement of the lid helix without changing its secondary structure (residues Ser80-Asp87). Two hinges were identified at position Gly77 and Phe90 by comparing the open and closed structures during the simulations. The open structure of the lipase differed from the open crystal structure by only 1.5 Å.
In the second simulation, the lid RMSD increased to 3.5 Å and a movement of 3.7 Å at one end of the lid helix (Ser80) towards the open crystal structure was observed. In a third simulation, the lid RMSD changed by 2.5 Å, and the lid helix showed a closing movement by 3 Å at Ile85 and Ala86.
Open RML in water
In all three simulations of the open conformation of RML in water, the lid remained close to the initial open crystal structure. The channel to the active center remained open in all three simulations and the area of the hydrophobic SAS showed only small changes from 4200 to 4300 Å2 (Fig. S2). The lid RMSD was less than 1.3 Å (Fig. S3) and the secondary structure did not change except of an unfolding of the first and last amino acid of the lid helix.
Closed TLL in water
In all simulations of the closed conformation of TLL in water, a further closing of the lid was observed. The channel towards the active center remained closed in all simulations, as indicated by a nearly constant area of the hydrophobic SAS of 3100 to 3300 Å2 (Fig. S2).
The lid RMSD increased to 2.0–3.7 Å caused by a movement of the lid towards a more closed structure in all simulations (Fig. S4). During this process, the lid was stable and moved as a rigid body by 1.8 Å at Trp89, 3.1 Å at Glu87, and 4.1 Å at Gly91 in the three simulations. In all three simulations the closing movements started immediately after the equilibration and lasted for 4–18 ns. In the remaining simulation time the lipases were stable and no conformational changes could be observed.
Open TLL in toluene
In all three simulations of the open conformation of TLL a slight movement in the lid towards a more closed conformation was observed and the lid RMSD increased to 3 Å (Fig. S4). However, the substrate channel remained open and the area of the hydrophobic SAS was 3800 to 4300 Å2 (Fig. S2). The movement of the lid occurred in the first 4–6 ns with a maximum movement by 2.4 Å at Ile86, 3.9 Å at Ser85, and 4 Å at Leu93 in the three simulations. The lid moved as a rigid body without changes of its secondary structure. After the movement, all simulations were stable during the remaining simulation time.
Closed TLL in toluene
In one simulation the lid moved towards the open structure combined with a partial unfolding of the helix, while an access tunnel towards the catalytic center opened and the area of the hydrophobic SAS increased from 3500 to 4000 Å2 (Fig. S2). In the second simulation, the lid moved two times towards the open crystal structure (lid RMSD up to 3.2 Å). However, this conformation was temporary and the lid returned to the closed structure. In the third simulation the lid helix started to unfold at Asn92 and parts of the lid (residues Ile90-Asp96) moved by 6.2 Å towards the open crystal structure, leading to an increase of the hydrophobic SAS from 3300 to 3700 Å2.
Open TLL in water
In all three simulations the lid moved towards the closed crystal structure, narrowing the access channel to the active site, but with a nearly constant area of the hydrophobic SAS of 3700 Å2 (Fig. S2). The lid RMSD increased to 3 Å during the first 4 ns (Fig. S4). All simulations of the open structure of TLL in water showed an unfolding of the lid helix at Ile86 and simultaneously a movement at Arg84-Ile86. The positions Trp89-Leu97 remained near the initial position, while the region Arg 84-Asn88 moved 4.5–8.6 Å towards the closed crystal structure.
Discussion
The transition between closed and open structures is a rare event and therefore long simulation times and multiple simulations are necessary to improve statistics. Hence, this study compares the movement of three lipases with a complex lid and two simple lids in different conformational states using three independent and unrestrained simulations with duration of 20–25 ns each. However, this number of simulations is not sufficient to evaluate statistical probabilities of the opening and closing event. Therefore, this study focuses on possible transition pathways. In previous studies on these lipases, either external forces on the lid11 or short simulation times13 were used; therefore no complete and unbiased transition pathway has yet been observed. Complete pathways are still not accessible in experiments and were demonstrated by molecular dynamics only for a lipase from Burkholderia cepacia.14,15
In our simulations, a complete lid opening pathway for RML and partial pathways for CRL were observed. Additionally, the lids could easily be identified by their movement, and the hinges could be assigned.
Structure and flexibility in water and organic solvent
The core of the lipases was stable in all 36 simulations, with changes in the RMSD of the backbone atoms of less than 1 Å in water and 0.5 Å in toluene. This difference in the core RMSD indicates that the protein is less flexible in organic solvents, which is in agreement with previous studies17,18 and with our analysis of the SAS. The SAS of all lipases in water was significantly higher than in toluene. In addition to the changes in the core RMSD, side chains on the surface of the lipases were found to be less solvent-exposed in toluene than in water as it was shown before.12
The lids of the closed structures in water and of the open structures in toluene were assumed to remain unchanged. However, the closed structure of CRL in water showed an unexpected slight opening movement and both the open conformation of TLL in toluene and the closed conformation of TLL in water showed a further closing movement by 3–4 Å, but then remained stable (Fig. S5). We assume that the position of the lid in the crystal structure might be mediated by crystal contacts. In contrast, the simulations of the open structure of RML in toluene and the closed structure of RML in water remained close to the initial structures.
Additionally, a loop was identified in RML at Ser56-Asn63 which was highly flexible in water and in toluene (Fig. S6). The respective loop in TLL (Ser58-Val63) was highly flexible in water, but less flexible in toluene (Fig. S6). The movements of this loop caused the observed changes in the core RMSD, but were independent of the lid opening or closing. In both lipases the loop is placed near the lid and points towards a potential interface. Based on the hydrophobic amino acids (RML: leucin, isoleucine, TLL: glycine,valine, glycine) in the loop, we assume an anchoring function at hydrophobic interfaces.
Conformational transitions
In contrast to the low flexibility of the core of all lipases, the lids were found to be more flexible. As suggested previously,6 one of our simulations of the closed structure of RML in toluene showed a rigid body movement of the lid. However, the pathway ended shortly before the open crystal structure was achieved (Fig. 1). We assume that the open crystal structure might not exactly match the open structure of RML in toluene. In water, however, the open structure of RML did not change. Thus, the closing transition either is a very rare event or the transition pathway in water is different from the pathway in organic solvent.
Figure 1.

(A) Opening of RML from the closed crystal structure (orange) to the open crystal structure (cyan) in toluene. Yellow: lid position in the most open structure during the simulation. (B) Core RMSD (red), lid RMSD to the closed crystal structure (initial lid RMSD, green), lid RMSD to the open crystal structure (final lid RMSD, blue).
The homologue of RML, TLL, was expected to show a similar movement of the lid as in RML. Instead, a different movement and a partial unfolding of the lid helix were found. Thus, homologues proteins do not necessarily share similar transition pathways. Interestingly, the movement of the open structure of TLL in water started at the N-cap region of the lid (Arg84-Ile86), while it started at the C-cap region (Ile90-Asp96) in the closed structure in toluene. After 25 ns one simulation of each the open and closed structure resulted in almost the same intermediate structure (Fig. 2). Thus, the opening and closing pathways seem to be similar in TLL.
Figure 2.
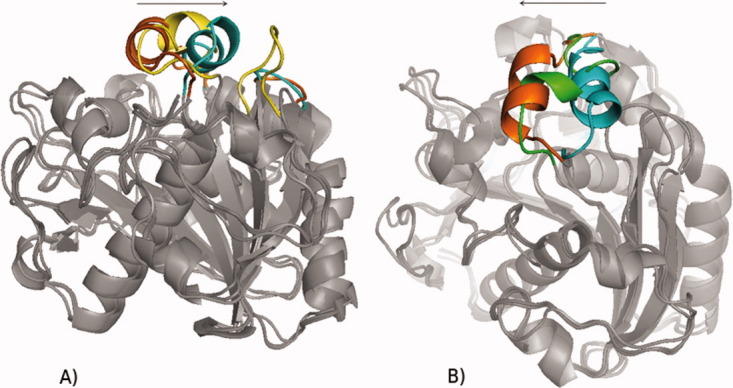
(A) Opening of TLL from the closed crystal structure (orange) to the open crystal structure (blue) in toluene. Yellow: lid position at 25 ns of simulation. Only one end of the lid helix reached the open crystal structure. (B) Closing of TLL from the open crystal structure (blue) to the closed crystal structure (orange). Green: lid position at 25 ns of simulation. Only one end of the lid helix reached the closed crystal structure.
In two simulations of the open structure of CRL in water, the lid helices started to unfold after an initial closing movement, according to the necessary refolding of the two helices in the lid (Fig. S7). In the closed structure of CRL in toluene, the partial refolding was in accordance with the open structure: the helix at position 69–71 unfolded, while a helix started to form at 84–88 (Fig. 3). Because formation of secondary structures is a slow process, we assume that it was the kinetic bottleneck in our simulations, and the movement ended halfway between the closed and the open structure (Fig. 4). In contrast to our simulations of CRL, previous studies of the homologues lipase from Burkholderia cepacia14,15 observed a rigid body movement of both helices of the lid without a refolding of the secondary structure. Again this indicates that homologues structure can show different transition pathways. As second possible bottleneck in the pathway of CRL we assumed that Val86, which was tightly bound into a hydrophobic pocket, has to be released to continue the opening movement. To prove this hypothesis, we mutated Val86 into a smaller amino acid, Gly86. However, in three simulations of the mutant Val86Gly, no change in the opening movement was found.
Figure 3.
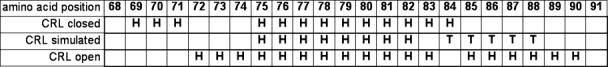
Comparison of the secondary structure of the simulated closed structure in toluene after 12 ns (“CRL simulated”) and the closed and open crystal structures (“CRL closed” and “CRL open”, respectively). Helical amino acids are indicated by “H”, turns by “T” (classification by STRIDE).
Figure 4.

(A) Opening of CRL from the closed crystal structure (orange) towards the open crystal structure (blue) in toluene after 25 ns. Green: lid position in the most open structure during the simulation. (B) Core RMSD (red), lid RMSD to the closed crystal structure (initial lid RMSD, green), lid RMSD to the open crystal structure (final lid RMSD, blue).
Since no external force was applied on the lipases, conformational transitions were driven by the surrounding solvent, but no directed driving force by the solvent could be identified, and therefore no detailed mechanistic explanation of the influence of the solvent can be given. However, previous studies showed the influence of hydrogen bonds and salt bridges in the lid regions of Burkholderia cepacia lipase and Candida rugosa lipase.12,14
Material and Methods
Lipases
Crystal structures were taken from the Protein Data Bank (PDB)19: entries 1CRL (resolution 2.06 Å)20 and 1TRH (resolution 2.10 Å)8 for the open and closed structure of Candida rugosa lipase (lid: residues 66–92), 4TGL (resolution 2.60 Å)21 and 3TGL (resolution 1.90 Å)16 for the open and closed structure of Rhizomucor miehei lipase (lid: residues 82–96), and 1DTE (resolution 2.35 Å)6 and 1DT5 (resolution 2.40 Å)6 for the open and closed structure of Thermomyces lanuginosa lipase (lid: residues 82–96) (Fig. 5).
Figure 5.
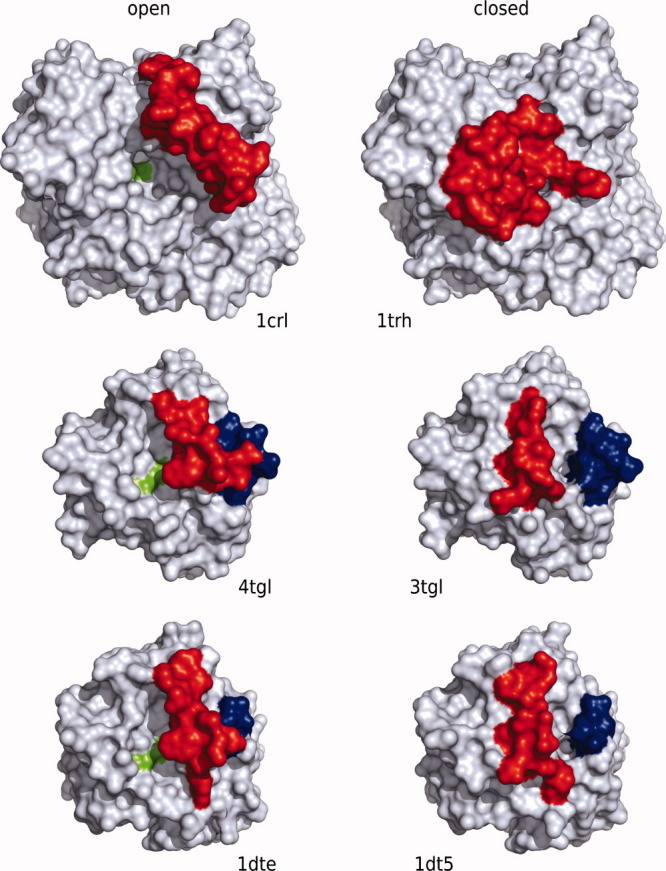
Overview of the open (left) and closed (right) structures of the lipases CRL (top), RML (middle), and TLL (bottom). The lid is marked in red, the catalytic triad in green, and the flexible loop in TLL and RML in blue.
A comparison of the sequences of 3TGL and 4TGL revealed three different positions with two possible amino acids each. 3TGL was mutated according the sequence of 4TGL using SwissPDBViewer.22
Molecular dynamics simulations
pKa values and protonation states of titratable sites Arg, Lys, Asp, Glu, and His at pH 7 were calculated using TITRA.23 In organic solvents, the sites were protonated assuming a pH memory from the protonation states in water,24,25 TLEAP was used to set up the systems. Hydrogens were added and disulfide bridges were built from information in the crystal structure. 8–16 Na+ ions were added as counter ions to neutralize the systems.
The six crystal structures including the crystal water were solvated in a truncated octahedron solvent box with a minimal distance between protein surface and box of 12 and 14 Å for water and toluene, respectively. For water, the TIP3P model26 was used and 6400–12,300 molecules were added to the systems, while toluene was parameterized as described previously27 and 600–1300 molecules were added.
The AMBER program suite and the all-atom force field ff99 were used for all simulations.28 The systems were minimized with the SANDER program using 50 cycles of the steepest descent algorithm followed by 450 cycles of the conjugated gradient algorithm. The PMEMD program was used for molecular dynamics simulations. For each system, three simulations were performed with different initial velocities. The equilibration of the systems was performed at 300 K and 1 bar with restraints. First, the restraining forces were applied to all atoms with 10 kcal/(mol Å2) for 100 ps. In four following steps, the forces were applied to the backbone atoms with 10, 5, 1, and 0.1 kcal/(mol Å2) for 50 ps each. Then, 25 ns of simulation were performed under periodic boundary conditions and without restraints (time step 1 fs). Temperature (300 K) and pressure (1 bar) were controlled by using a weak heat bath coupling29 with a temperature coupling time constant of 1.0 ps and a pressure coupling time constant of 1.2 ps. Nonbonded interactions were calculated using the Particle-Mesh Ewald method with a cut-off of 8 Å.30,31 The SHAKE algorithm32 was used to constrain all bonds involving hydrogen atoms with a tolerance of 0.000001 Å.
Analysis
The resulting trajectories were analyzed using PTRAJ after fitting the backbone atoms of the core (all amino acids except the lid) of each protein to the initial minimized crystal structure. Three different kinds of root-mean squared deviations (RMSD) were calculated:
The RMSD between all backbone atoms of the simulated protein without the lid and the used crystal structure without the lid (core RMSD).
The RMSD between all backbone atoms of the lid of the simulated protein and the lid of the initial closed or open crystal structure (initial lid RMSD).
The RMSD between all backbone atoms of the lid of the simulated protein and the lid of the final open or closed crystal structure (final lid RMSD).
A comparison of the initial and final lid RMSD indicates the type of movement. If the initial lid RMSD and the final lid RMSD lid are strongly anticorrelated, the lid performs a movement between the open and closed conformation. If both RMSDs increase simultaneously, the lid unfolds.
The solvent accessible surface area was calculated for both the hydrophobic amino acids (Fig. S2) and the complete protein (data not shown) using VMD and a solvent probe radius of 1.4 Å. To identify flexible regions, the simulated B-factors were calculated with PTRAJ over 10–20 ns of the trajectories (Fig. S6).
Distances between amino acids of the crystal structures and simulated structures were measured by PyMOL33 and the SwissPDBViewer.22 The visualization of the trajectories was done by VMD.34 The secondary structure was calculated using PyMOL and the STRIDE method.35
Acknowledgments
We thank the German Science Foundation DFG (Sonderforschungsbereich 716) for financial support and the High Performance Computing Center Stuttgart (HLRS) for kindly providing computational resources.
Glossary
Abbreviations:
- MD
molecular dynamics
- RMSD
root mean square deviation
- SAS
solvent accessible surface.
References
- 1.Schmid RD, Verger R. Lipases: interfacial enzymes with attractive applications. Angew Chem Int Ed. 1998;37:1609–1633. doi: 10.1002/(SICI)1521-3773(19980703)37:12<1608::AID-ANIE1608>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 2.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 3.Fischer M, Pleiss J. The lipase engineering database: a navigation and analysis tool for protein families. Nucleic Acids Res. 2003;31:319–321. doi: 10.1093/nar/gkg015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen M, Jensen TR, Kjaer K, Björnholm T, Mouritsen OG, Peters GH. Orientation and conformation of a lipase at an interface studied by molecular dynamics simulations. Biophys J. 2002;83:98–111. doi: 10.1016/S0006-3495(02)75152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brady L, Brzozowski AM, Derewenda ZS, Dodson E, Dodson G, Tolley S, Turkenburg JP, Christiansen L, Huge-Jensen B, Norskov L, et al. A serine protease triad forms the catalytic center of a triacylglycerol lipase. Nature. 1990;343:767–770. doi: 10.1038/343767a0. [DOI] [PubMed] [Google Scholar]
- 6.Brzozowski AM, Savage H, Verma CS, Turkenburg JP, Lawson DM, Svendsen A, Patkar S. Structural origins of the interfacial activation in Thermomyces (Humicola) lanuginosa lipase. Biochemistry. 2000;39:15071–15082. doi: 10.1021/bi0013905. [DOI] [PubMed] [Google Scholar]
- 7.Brzozowski AM, Derewenda U, Derewenda ZS, Dodson GG, Lawson DM, Turkenburg JP, Bjorkling F, Huge-Jensen B, Patkar SA, Thim L, et al. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature. 1991;351:491–494. doi: 10.1038/351491a0. [DOI] [PubMed] [Google Scholar]
- 8.Grochulskis P, Li Y, Schrags JD, Cygler M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994;3:82–91. doi: 10.1002/pro.5560030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Tilbeurgh H, Egloff M, Martinez C, Rugani N, Verger R, Cambillau C. Interfacial activation of the lipase procolipase complex by mixed micelles revealed by x-ray crystallography. Nature. 1993;362:814–820. doi: 10.1038/362814a0. [DOI] [PubMed] [Google Scholar]
- 10.Peters GH, Toxvaerd S, Olsen OH, Svendsen A. Computational studies of the activation of lipases and the effect of a hydrophobic environment. Protein Eng. 1997;10:137–147. doi: 10.1093/protein/10.2.137. [DOI] [PubMed] [Google Scholar]
- 11.Peters GH, Bywater RP. Influence of a lipid interface on protein dynamics in a fungal lipase. Biophys J. 2001;81:3052–3065. doi: 10.1016/S0006-3495(01)75944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tejo BA, Salleh AB, Pleiss J. Structure and dynamics of Candida rugosa lipase: the role of organic solvent. J Mol Model. 2004;10:358–366. doi: 10.1007/s00894-004-0203-z. [DOI] [PubMed] [Google Scholar]
- 13.James JJ, Lakshmi BS, Gautam P. Activation of Candida rugosa lipase at alkane-aqueous interfaces: a molecular dynamics study. FEBS Lett. 2007;581:4377–4383. doi: 10.1016/j.febslet.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Trodler P, Schmid RD, Pleiss J. Modeling of solvent-dependent conformational transitions in Burkholderia cepacia lipase. BMC Struct Biol. 2009;9:38. doi: 10.1186/1472-6807-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbe S, Lafaquière V, Guieysse D, Monsan P, Remaud-Siméon M, André I. Insights into lid movements of Burkholderia cepacia lipase inferred from molecular dynamics simulations. Proteins. 2009;77:509–523. doi: 10.1002/prot.22462. [DOI] [PubMed] [Google Scholar]
- 16.Brzozowski AM, Derewenda ZS, Dodson EJ, Dodson GG, Turkenburg JP. Structure and molecular model refinement of Rhizomucor miehei triacyglyceride lipase: a case study of the use of simulated annealing in partial model refinement. Acta Crystallogr, Sect B: Struct Sci. 1992;48:307–319. [Google Scholar]
- 17.Norin M, Haeffner F, Hult K. Molecular dynamics simulations of an enzyme sorrounded by vacuum, water, or a hydrophobic solvent. Biophys J. 1994;67:548–559. doi: 10.1016/S0006-3495(94)80515-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broos J, Visser A, Engbersen JF, Verboom W, Hoek AV, Reinhoudt DN. Flexibility of enzymes suspended in organic solvents probed by time-resolved fluorescence anisotropy. Evidence that enzyme activity and enantioselectivity are directly related to enzyme flexibility. J Am Chem Soc. 1995;117:12657–12663. [Google Scholar]
- 19.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grochulski P, Li Y, Schrag JD, Bouthillier F, Smith P, Harrison D, Rubin B, Cygler M. Insights into interfacial activation from an open structure of Candida rugosa lipase. J Biol Chem. 1993;268:12843–12847. [PubMed] [Google Scholar]
- 21.Derewenda U, Brzozowski AM, Lawson DM, Derewenda ZS. Catalysis at the interface: the anatomy of a conformational change in a triglyceride lipase. Biochemistry. 1992;31:1532–1541. doi: 10.1021/bi00120a034. [DOI] [PubMed] [Google Scholar]
- 22.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 23.Martel PJ, Baptista A, Petersen S. Protein electrostatics. Biotechnol Ann Rev. 1996;2:315. [Google Scholar]
- 24.Soares CM, Teixeira VH, Baptista AM. Protein structure and dynamics in nonaqueous solvents: insights from molecular dynamics simulation studies. Biophys J. 2003;84:1628–1641. doi: 10.1016/S0006-3495(03)74972-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costantino HR, Griebenow K, Langer R, Klibanov AM. On the pH memory of lyophilized compounds containing protein functional groups. Biotechnol Bioeng. 1997;53:345–348. doi: 10.1002/(SICI)1097-0290(19970205)53:3<345::AID-BIT14>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 27.Trodler P, Pleiss J. Modeling structure and flexibility of Candida antarctica lipase B in organic solvents. BMC Struct Biol. 2008;8:9. doi: 10.1186/1472-6807-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berendsen HJ, Postma JP, van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 30.Walker RC, Crowley MF, Case DA. The implementation of a fast and accurate QM/MM potential method in Amber. J Comput Chem. 2008;29:1019–1031. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 31.Nam K, Gao J, York DM. An efficient linear-scaling Ewald method for long-range electrostatic interactions in combined QM/MM calculations. J Chem Theory Comput. 2005;1:2–13. doi: 10.1021/ct049941i. [DOI] [PubMed] [Google Scholar]
- 32.Ryckaert J, Ciccotti G, Berendsen H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-Alkanes. J Comput Phys. 1977;23:327. [Google Scholar]
- 33.DeLano WL. The PyMOL user's manual. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. VMD—Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]