

Abstract
Novel acetone and aldimine covalent adducts were identified on the N-termini and lysine side chains of recombinant monoclonal antibodies. Photochemical degradation of citrate buffers, in the presence of trace levels of iron, is demonstrated as the source of these modifications. The link between degradation of citrate and the observed protein modifications was conclusively established by tracking the citrate decomposition products and protein adducts resulting from photochemical degradation of isotope labeled 13C citrate by mass spectrometry. The structure of the acetone modification was determined by nuclear magnetic resonance (NMR) spectroscopy on modified–free glycine and found to correspond to acetone linked to the N-terminus of the amino acid through a methyl carbon. Results from mass spectrometric fragmentation of glycine modified with an acetone adduct derived from 13C labeled citrate indicated that the three central carbons of citrate are incorporated onto protein amines in the presence of iron and light. While citrate is known to stoichiometrically decompose to acetone and CO2 through various intermediates in photochemical systems, it has never been shown to be a causative agent in protein carbonylation. Our results point to a previously unknown source for the generation of reactive carbonyl species. This work also highlights the potential deleterious impact of trace metals on recombinant protein therapeutics formulated in citrate buffers.
Keywords: acetonation, protein-acetone adducts, protein-aldimine adducts, citrate decarboxylation, photo fenton, transition metal, iron, carbonylation
Introduction
Optimization of recombinant protein manufacturing processes is critical for maintaining product integrity. There are many examples of potency loss in therapeutic proteins resulting from a single chemical modification on a discrete amino acid residue.1–3 While degradation events related to storage conditions are routinely tracked during formulation studies, chemical modifications resulting from manufacturing process impurities are more difficult to define because they typically involve the degradation of standard process reagents to reactive chemical intermediates, which are difficult to track in a systematic fashion. Transition metals, which may be present at trace amounts during recombinant protein manufacturing, may catalyze the formation of reactive chemical intermediates, which ultimately result in protein degradation.
Protein carbonylation is a highly deleterious degradation event that progresses via the formation of reactive oxygen species (ROS) and aldehyde intermediates. The addition of aldehyde and ketone groups to proteins can result from several biological and nonbiological processes and is typically used as a general marker for oxidative stress.4–6 In the case of protein therapeutics, carbonylation may be preceded by a thermal degradation event, which hydrolyzes sucrose, a commonly used excipient, into its constituent monosaccharides glucose and fructose.7–10 Radical attack of free glucose then results in several aldehyde degradation products as well as peroxides, which can subsequently modify nucleophilic centers of proteins via Maillard type reactions.11–14 Carbohydrates can undergo heat induced degradation whereby free glucose modifies lysine side chains at the reducing end of the sugar—commonly referred to as glycation. Subsequent radical attack of the protein bound monosaccharide results in a carbon centered radical at the fourth position of the sugar, which induces cleavage of the sugar backbone and the creation of carboxymethyl-lysine.15,16 Peroxidation of partially unsaturated fatty acids has also been shown to result in the creation of several reactive Type 2 alkenes, which contain a carbon to carbon double bond α-β to the carbonyl.17–20 Type 2 alkenes are highly reactive because of the presence of polarizable outer shell π electrons and the electronegative carbonyl oxygen. The carbonyl oxygen exists in a conjugated system with the alkene carbons and thus draws electron density from the alkene leading to electron deficiency at the β alkene carbon.21 Modification of protein electrophiles then proceeds through Maillard type reactions at the carbonyl or a Michael addition at the β alkene carbon.22–26 Once modified, carbonylated proteins tend to form aggregates due to increased hydrophobicity and through covalent cross-links that result from further modification via the unreacted moiety of the adducted—bifunctional electrophile.27 In addition to being a marker for general oxidative stress, protein carbonylation is also associated with several disease states including Alzheimer's disease, chronic renal failure, diabetes, and other inflammatory conditions.28–31 Potential palliative treatments for diseases associated with protein carbonylation typically involve the in vivo administration of sacrificial nucleophiles, which are modified preferentially over endogenous proteins.32–34
Here we present evidence for a novel modification occurring on proteins; acetonation of protein amines through the methyl carbon of acetone. The modification results from reactive intermediates that are created during the photochemical breakdown of citrate. We have deduced a potential chemical mechanism for the formation of acetone modified products that is consistent with experimental results obtained from the incorporation of carbon from13C labeled citrate buffers. Our findings expand on the known repertoire of protein modifications and elucidate a completely novel pathway for the accumulation of carbonylated proteins.
Results
Covalent modifications of a recombinant monoclonal antibody resulting in 56 and 38 Da adducts were observed during manufacturing process development. These signature masses suggested that the toxic electrophile—acrolein could be present and potentially modifying protein nucleophiles.35,36 We systematically investigated the different steps of the manufacturing process and found that isobaric acrolein-like adducts were generated when the monoclonal antibody was solubilized in citrate buffer and exposed to ambient light. This report describes a unique mechanism of covalent adduct formation resulting from iron catalyzed photodegradation of citrate.
Acetonation of intact antibodies
Antibody samples were exposed to light in the presence of citrate buffer and Fe(II) or acetone, acetate buffer and Fe(II). These experiments addressed the question of whether protein adducts were formed from reactive breakdown products from citrate or as a consequence of free acetone—as formation of acetone from the photochemical degradation of citrate has been previously documented.37 The mass spectrum of the light exposed antibody in the absence of Fe(II) revealed no significant degradation occurring on the heavy chain (HC) and light chain (LC), a result similar to the control sample, which was not exposed to light (Fig. 1, panels C and A, respectively). Addition of Fe(II) in the absence of light exposure resulted in some slight broadening of the rp-HPLC (reversed phase-high performance liquid chromatography) HC peak (Fig. 1, panel B) and the mass spectrum of the HC indicated that a low level of oxidation was occurring, however, there were no significant modifications observed in the mass spectrum of the LC. Results from the analysis of the antibody sample, which was exposed to light in the presence of citrate and Fe(II), indicated that severe degradation was occurring to the molecule (Fig. 1, panel D). Antibody HC was highly degraded as evidenced by the broad rp-HPLC peak, which is an indicator of extreme heterogeneity resulting from covalent chemical modifications. The LC was also highly degraded and the mass spectrum was differentiated from the other samples by the presence of mass adducts of 38 and 56 Da. The absence of these adducts in the LC mass spectrum from the sample incubated in acetone/acetate/Fe(II) (Fig. 1, panel E) suggested that the 38 and 56 Da modifications were resulting from reactive intermediates generated from the photochemical degradation of citrate. The experiments were repeated with the recombinant antibody exposed to light in the presence of 13C isotope labeled citrate and Fe(II) to establish a direct link between the degradation of citrate and the resulting protein modifications. Adducts with a mass of 38 and 56 Da were observed in the mass spectrums of antibody HC and LC, which was exposed to light in unlabeled citrate or 1,5-13C2 labeled citrate and Fe(II) (Fig. 2, panels A and B). However, analysis of the HC and LC mass spectrums of the light exposed recombinant antibody in 2,4-13C2 citrate and Fe(II) revealed the presence of mass adducts which were 2 Da heavier than those that were observed on the light exposed antibody in unlabeled and 1,5-13C2 labeled citrate buffers (Fig. 2, panel C). This result indicated that carbons 2 and 4 of citrate were incorporated into the antibody—potentially on the observed modifications.
Figure 1.

Reversed-phase separation of reduced antibody HC and LC and corresponding mass spectrums of the main LC peak. An IgG2 antibody was incubated in the dark for 48 h in the presence of 100 mM citrate buffer pH 5.0 in the absence and presence of 0.2 mM Fe(II) (A and B, respectively). Analysis of the same samples exposed to light for 48 h is shown in panels C and D, respectively. Antibody exposed to light in 90 mM acetate buffer pH 5.0 and 10% acetone in the presence of Fe(II) is shown in Panel E.
Figure 2.
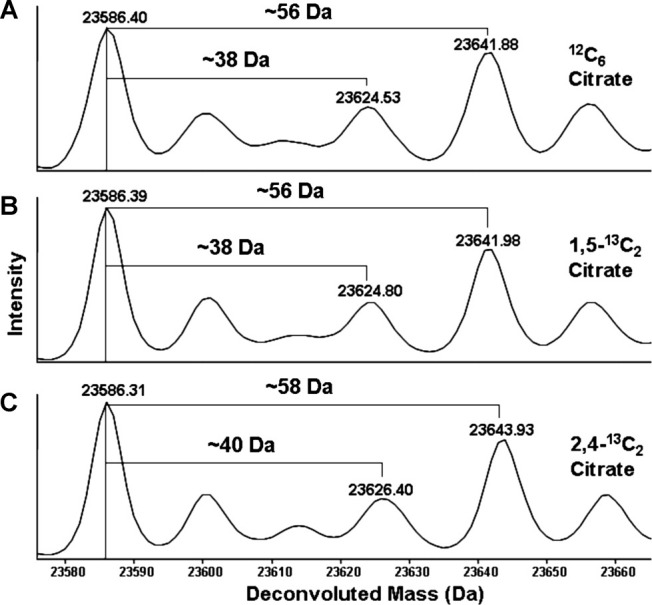
Mass spectrums of reduced antibody LC obtained from samples exposed to light for 48 h in the presence of unlabeled or isotope-labeled (1,5-13C2 and 2,4-13C2) citrate buffer at pH 5.0 with 0.2 mM Fe(II) (panels A−C respectively).
Sites of antibody-covalent acetone adducts
A comparison of the tryptic peptide map profiles obtained from recombinant antibody samples exposed to light for 48 h in the presence of citrate buffer and Fe(II) and nonstressed antibody samples revealed several apparent sites of covalent modification resulting in the incorporation of 56 Da mass adducts. The modification was found predominantly on the N-terminus of HC and LC (Supporting Information Figs. S1 and S2, respectively). Quantification of the 56 Da modification in the light stressed sample using the [M + 2H]2+ and [M + 3H]3+ ions for the modified and unmodified species indicated that the modification was occurring on HC and LC at levels of 24.8 and 8.9%, respectively (data not shown). Lower levels of the 56 Da modifications were also observed on internal lysine residues and resulted in a missed tryptic cleavage at the site of incorporation (data not shown).
Detection and quantitation of free acetone
The observation of 56 and 38 Da mass adducts on the reduced antibody HC and LC and the incorporation of 56 Da on the antibody N-termini and internal lysine residues was consistent with propionaldehyde and aldimine adducts which are commonly associated with acrolein. Thus, a rp-HPLC assay was developed to detect the dinitrophenylhydrazine (DNPH) derivatives of free aldehydes and ketones present in light exposed citrate/Fe(II) solutions. Analysis of a light exposed solution of 100 mM citrate, 50 mM glycine and 0.2 mM Fe(II) indicated that while acetone was formed at a rate of 0.16 mM /h, there was no detectable formation of acrolein (Supporting Information Fig. S3, panel A). Stable isotope labeled citrate was used to determine if the pattern of carbon incorporation into acetone during photochemical degradation of citrate was similar to that which was observed in the mass spectrum of the reduced antibody LC. The [M − H]1− ion for the DNPH-acetone hydrazone generated from the incubation of glycine and Fe(II) in unlabeled citrate and 1,5-13C2 labeled citrate was observed at m/z = 237.17 which was consistent with the expected theoretical [M − H]1− value (Table I). However, use of 2,4-13C2 and 13C6 citrate in the above reaction resulted in the formation of DNPH-acetone hydrazone [M − H]1− ions at m/z of 239.17 and 240.17, respectively (Table I). These results indicated that carbons from positions 2 and 4 of photochemically degraded citrate were incorporated into acetone. This result was in agreement with the carbon incorporation observed in antibody LC as a consequence of photochemical breakdown of citrate.
Table I.
Theoretical and Observed Mass Values for Unabled and 13C Isotope-Labled Citric acid and Glycine Used in Photochemical Degradation Studies and Observed Mass Values for the Reaction Products
| Citric Acid (Isotope) | MW (Theor.) | Glycine (Isotope) | MW (Theor.) | DNPH-AC [M-H]− | Gly-AC [M+H]+ (Obs.) | Gly-AC MS2 [M+H]+ (Obs.) | Gly-AC MS3 [M+H]+ (Obs.) |
|---|---|---|---|---|---|---|---|
| 13C6 | 198.05 | 12C2 | 75.04 | 240.17 | 135.08 | 89.08 | 60.08 |
| 2,4-13C2 | 194.03 | 12C2 | 75.04 | 239.17 | 134.08 | 88.08 | 60.08 |
| 1,5-13C2 | 194.03 | 12C2 | 75.04 | 237.17 | 132.08 | 86.08 | 58.08 |
| 12C6 | 192.03 | 12C2 | 75.04 | 237.17 | 132.08 | 86.08 | 58.08 |
| 12C6 | 192.03 | 13C2 | 77.05 | NA | 134.08 | 87.08 | 59.08 |
| 12C6 | 192.03 | 2-13C | 76.05 | 133.08 | 87.08 | 59.08 | |
| 12C6/13C6 | 192.03, 198.05 | 12C2 | 75.04 | 132.08, 135.08 | 86.08, 89.08 | 58.08, 60.08 | |
| 12C6/2,4-13C2 | 192.03, 194.03 | 12C2 | 75.04 | 132.08, 134.08 | 86.08, 88.08 | 58.08, 60.08 |
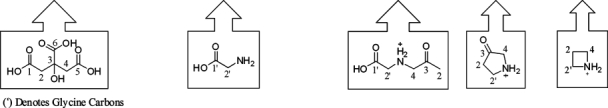
The DNPH-acetone hydrazone (DNPH-AC) observed [M-H]1− m/z are shown as well as the [M+H]1+ m/z for acetone modified glycine (Gly-AC) and the corresponding products ions observed in CID-MS2/MS3 scans. The chemical structures of citrate, glycine, glycine-acetone, the corresponding glycine-acetone product ions and the labeled carbons associated with the structures are shown below the table.
Acetone adducts result from incorporation of the axial citrate carbons 2–4
Photochemical degradation of citrate was carried out in the presence of free glycine to provide a model system for the structural elucidation of the adduct and to investigate the mechanism of citrate degradation which was producing a 56 Da modification on protein amines. Unlabeled glycine or glycine with a 13C incorporated in the α carbon (2-13C) or both carbons (13C2) was incubated with unlabeled citrate or 13C labeled citrate/Fe(II) buffer as described above and exposed to light for 48 h. The degradation of glycine was determined at the beginning and end of the light exposure study and found to have decreased from an initial concentration of 50 to 42 mM (data not shown). The formation of the 56 Da adduct on glycine was monitored by extracted ion current integration of the normal phase-high performance liquid chromatography (np-HPLC) separated [M + H]1+ ion at m/z = 132.08. As was the case for photochemical induced formation of acetone from citrate, the adduction of glycine was found to occur linearly over time at a rate of 0.17 mM/h; assuming that the decrease in glycine was due solely to incorporation of the 56 Da modification (Supporting Information Fig. S3, panel B).
The dependence of the 56 Da modification on hydrogen peroxide (H2O2) and hydroxyl radicals (.OH) observed in photo Fenton reactions was probed by adding radical scavengers to the citrate/Gly/Fe(II) system during light exposure and assessing the level of modified glycine by np-HPLC-MS. Addition of the .OH scavenger isopropanol (IPA) decreased the observed level of glycine + 56 Da by 94%, indicating that .OH was necessary for citrate mediated modification of glycine (Fig. 3). There was no appreciable accumulation of modified glycine when Fe(II) or Fe(III) was replaced with Cu(II), Co(II) or H2O2.
Figure 3.
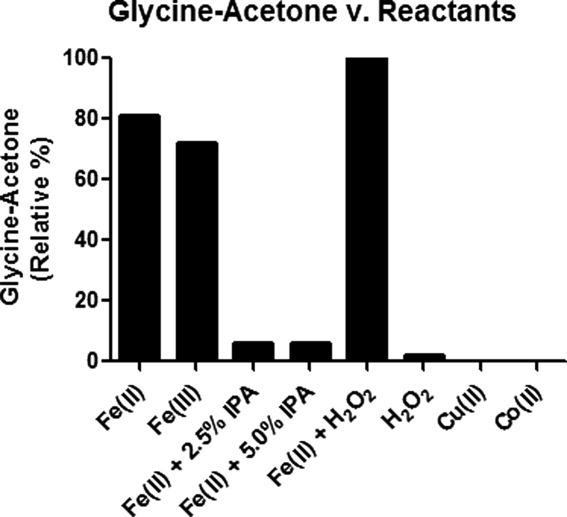
Relative quantitation of the formation of acetone adducted glycine in the presence of citrate and various transition metals and radical scavengers.
Results from the mass spectrum of modified glycine resulting from incubation with 13C labeled citrate and summarized in Table I indicated that carbons 2 and 4 of citrate were incorporated onto glycine as evidenced by a 2 Da mass increase in the modified product resulting from incubation with 2,4-13C2 citrate. The reaction product from incubation of glycine with 13C6 labeled citrate was increased 3 Da over the reaction product from unlabeled citrate clearly indicating that the modification involved the net addition of 3 carbons onto free glycine. The elemental composition of modified glycine was confirmed from the measurement of the [M + H]1+ ion at m/z = 132.06526, which was within 2.0 ppm of the theoretical value for the molecular formula C5H9O3N (data not shown). Modified glycine was also analyzed by collision induced dissociation-mass spectrometry (CID-MS)2 to provide structural insights into the mechanism of citrate degradation and subsequent amino acid modification. The m/z of the product ion that resulted from fragmentation of unlabeled glycine modified with the 56 Da adduct was consistent with the neutral loss of the carboxylate group (46 Da) on glycine. This assignment was confirmed by mass measurement of the product ion from modified-13C2 labeled glycine, which showed a loss of 47 Da, clearly indicating that the carbon loss was occurring from the glycine C-terminal carboxylate and not from the 56 Da modification (Table I). CID-MS3 analysis of the major ion resulting from fragmentation of modified glycine resulted in a dominant product ion, which was 28 Da lower in mass than the precursor selected from the MS2 scan; a mass decrease consistent with the loss of a carbon and an oxygen atom. Results from the structural characterization of the dominant CID-MS3 product ion that resulted from fragmentation of modified 2-13C and 13C2 glycine indicated that the 13C labeled α-carbon was maintained on the dominant MS3 product ion. This was clear evidence that the neutral loss of 28 Da was occurring on the N-terminal portion of modified glycine, from fragmentation of the 56 Da modification, not the glycine α-carbon (Table I). The CID-MS3 product ions resulting from fragmentation of modified glycine generated from photochemical degradation in 2,4-13C2 and 13C6 citrate had the same m/z and were shifted 2 Da higher relative to the MS3 product resulting from unlabeled citrate. The neutral loss of 29 Da observed during CID-MS3 of modified glycine generated from 13C6 citrate clearly indicated that a 13C carbon derived from citrate was lost from the modification. However, if citrate carbons maintained their relative order along the backbone of the molecule during photochemical degradation, then the m/z of the modified glycine MS3 product from 2,4-13C2 citrate should be intermediate to that obtained for 13C6 citrate and unlabeled citrate. The results obtained from MS3 fragmentation of modified glycine generated from 2,4-13C2 and 13C6 citrate are elucidated by assuming that the product of MS2 fragmentation was the five membered ring, 3-oxopyrrolidinium. The product ion spectrum resulting from CID-MS2 analysis of 3-oxopyrrolidinium was identical to the spectrum obtained from MS3 analysis of the major ion resulting from MS2 fragmentation of modified glycine (Supporting Information Fig. S4). The subsequent loss of CO from 3-oxopyrollidinium is consistent with typical ketone fragmentation pathways involving α-cleavage, that is, cleavage of the covalent bond between the keto carbonyl and the alpha (with respect to the ketone) carbon. Fragmentation of one α-keto carbon bond results in the creation of an acylium ion and precipitates the second α-cleavage and concomitant release of CO. In light of the cyclic structure of the MS2 intermediate, the loss of the keto carbon from modified glycine during MS3 fragmentation is to be expected. The results from fragmentation of modified glycine were consistent with an incorporation of the axial citrate carbons 2, 3, and 4 onto glycine with no apparent rearrangement. Incubation of glycine in equimolar concentrations of unlabeled and 13C labeled citrate resulted in glycine modified with 56 and 58 Da adducts in the case of unlabeled and 2,4-13C2 labeled citrate and 56 and 59 Da adducts when the reaction mixture was composed with unlabeled and 13C6 labeled citrate.
Detection of citrate degradation intermediates
The photochemically degraded citrate/glycine/Fe(II) samples described above were analyzed by rp-HPLC and the resulting UV chromatograms were compared with UV chromatograms obtained from the separation of several commercially available, standard organic acids. The retention times obtained for 3-oxoglutarate and acetoacetate standards, which are known citrate degradation products,37 were consistent with unknown peaks observed in the photochemically degraded citrate samples (Fig. 4, panel A). It was determined from the UV spectrum associated with the standards that 3-oxoglutarate and acetoacetate had absorbance maxima at 243 and 247 nm, respectively (Fig. 4, panels B and C, respectively). The UV spectrums obtained for photochemically degraded citrate unknowns were found to be consistent with the standard spectrums for 3-oxoglutarate and acetoacetate thus confirming the presence of these chemicals in the citrate/glycine/Fe(II) reactions. These chemicals were not observed at light exposure time 0 but became evident in the 48 and 96 h samples indicating that they were formed as a consequence of photochemical processes.
Figure 4.
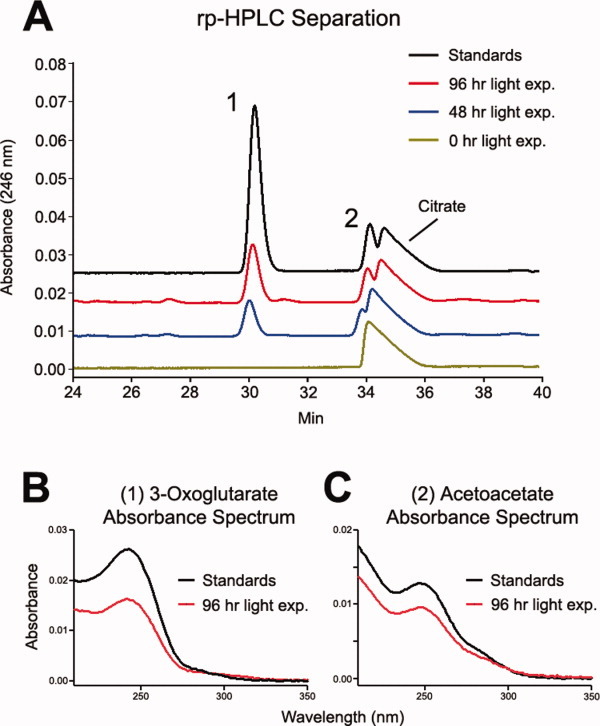
The rp-HPLC separation of the reaction intermediates from the photochemical degradation of citrate in the presence of glycine and Fe(II) is shown in panel A. The reaction intermediates 3-oxoglutarate (1) and acetoacetate (2) are detected by monitoring absorbance at 246 nm. The UV spectrums for 3-oxoglutarate and acetoacetate are shown in panels B and C, respectively.
Structural elucidation of glycine-acetone by NMR spectroscopy
The 1H NMR spectrum of modified glycine isolated by np-HPLC revealed the presence of three singlet aliphatic signals. Integration ratios and one-bond correlation data from a multiplicity edited heteronuclear single quantum coherence (HSQC) spectrum indicated these signals corresponded to two CH2 groups (δ 4.09 and δ 3.51) and one CH3 group (δ 2.15). Also present in the aliphatic region of the 1H spectrum were signals from citric acid (δ 2.55, δ 2.45), acetonitrile (δ 1.94) and ammonium acetate (δ 1.79).
The 1H—13C heteronuclear multiple bond correlation (HMBC) spectrum provided sufficient information to assign the structure of the modified glycine residue as 2-(2-oxopropylamino)acetic acid; acetone covalently linked to the N-terminus of glycine through a methyl carbon. The HMBC spectrum indicated the presence of the expected five carbon chemical shifts. In addition to three aliphatic 13C shifts at δ 54.7 (CH2), δ 48.8 (CH2), and δ 26.4 (CH3), two carbonyl chemical shifts were observed at δ 170.9 and δ 203.2. Analysis of the 1H—13C HMBC spectrum indicated that the C=O signal at δ 170.9 has a single correlation to the upfield CH2 protons at δ 3.51. Chemical shifts for both the C=O and CH2 of this linked carbon pair were consistent with a glycine acid moiety and assigned as C-5 and C-4, respectively (Supporting Information Table SI). The second C=O signal at δ 203.2 correlated to both the singlet CH3 proton signal at δ 2.15 and the downfield singlet CH2 signal at δ 3.57. These two-bond correlations from the aliphatic protons into a keto carbonyl suggest a ketone fragment and were assigned as C-1, C-2, and C-3. This ketone fragment, in turn, correlates to the glycine moiety via a three-bond correlation from the methylene protons of C-3 into carbon C-4. 1H and 13C NMR chemical shift data and structural assignments are given in Supporting Information Table SI.
Discussion
In this study, we have described the covalent addition of acetone to the amino termini and lysine side chains of a recombinant monoclonal antibody as a consequence of the photochemical degradation of citrate buffer. Acetone modification was initially observed during forced degradation studies in the presence of citrate buffer, which was used during manufacturing process development. The modification masses of 56 and 38 Da were consistent with propionaldehyde and aldimine adducts respectively which are typically observed on nucleophiles in the presence of acrolein.38 However, results from MS and NMR for the 56 Da modification on free glycine indicated that the molecular structure of this modification was in fact acetone, linked to the amino terminus through the methyl carbon. Distinguishing between protein modifications caused by citrate degradation from those caused by acrolein is further obfuscated by the virtual coelution of acetone and acrolein hydrazones when analyzed with rp-HPLC, the standard methodology used for detection of the free molecule. While modification of proteins during acetone precipitation has recently been reported,39 the modification we have observed has a different mass and is mechanistically and structurally distinct from that which is described by Simpson et al.
Mass spectrometric analysis of the modification products from light exposure studies using free glycine in isotope labeled citrate/Fe(II) buffer systems point to a mechanism for citrate decomposition, which is consistent with Hofer-Moest type reactions.40,41 While our results were obtained by coincubation of citrate with Fe(II), conversion of Fe(II) to Fe(III) through reaction with .OH or H2O2
| (1) |
| (2) |
(Eqs. (1) and (2), respectively) is expected in a photochemical Fenton system.42 Formation of the acetone modification on glycine was evaluated in the presence of IPA, which is a .OH scavenger. The relative level of glycine-acetone was reduced by 94% with the addition of 2 different concentrations of IPA (Fig. 3), indicating that .OH is mechanistically important for the modifications that we have observed on amino acids and proteins. The inhibitory effect of IPA on the formation of glycine-acetone is consistent with a mechanism of Fe(II) to Fe(III) conversion in which .OH is present on the reactants side of the equation. This result indicates that Eq. (1) is more relevant than Eq. (2) for describing Fe(II) to Fe(III) conversion in the context of acetone modifications that we are reporting. Further elucidation of the role of various radical species to iron cycling using enzymatic scavengers such as catalase and superoxide dismutase was not possible because of rapid photodegradation and catalytic inactivation during light incubation.43
Prior work by Abrahamson et al. indicates that the initial degradant formed by photochemical decomposition of citrate is 3-oxoglutarate.37 Analysis of the chemical intermediates formed during photochemical degradation of citrate confirmed the presence of 3-oxoglutarate and supports this step in the mechanism (Fig. 4). Thus, we propose that the initial step in the degradation of citrate and subsequent adduction of amines is the decarboxylation of citrate carbon C6 that results from 2 electron abstractions by 2Fe(III) (Fig. 5). The loss of CO2 results in the formation of a ketone at C3 yielding 3-oxoglutarate. The formation of free acetone from 3-oxoglutarate likely occurs due to nonoxidative decarboxylation at C1 and C5 proceeding through an iron or hydrogen ion mediated pericyclic transition state, which is a common mechanism for β-ketoacid decarboxylation and summarized in Eq. (3).
Figure 5.

Proposed mechanism for the photochemical degradation of citrate in the presence of Fe(II)/Fe(III) and subsequent modification of the amino terminus of free glycine.
| (3) |
A potential mechanism for the formation of acetone modifications on amino acids and proteins diverges from the mechanism for the creation of free acetone after the formation of 3-oxoglutarate. Modification of protein amines is likely driven by a subsequent abstraction of an electron by Fe(III) resulting in a carbon centered radical on C2 or C4 (Fig. 5). Abstraction of the radical by a second Fe(III) precedes the nucleophilic attack of citrate carbons C2 or C4 by a protein amine resulting in the incorporation of acetoacetate onto proteins. Decarboxylation of the acetoacetate β-ketoacid produces an acetone adducts linked to a protein amine through the methyl carbon of acetone. The mechanism we are proposing is consistent with experimental evidence derived from the CID-MS2/MS3 fragmentation products of glycine that is modified with acetone, which is created during degradation of isotopic 13C citrate. The chemical reaction implied by the mechanism of protein modification due to light exposure in the presence of citrate and Fe(II)/Fe(III) is summarized in Eq. (4).
| (4) |
The +38 Da aldimine adduct was evident on the antibody when exposed to light in the presence of citrate and Fe(II) but was not observed on free glycine in the same system. It is likely that the reversibility of the aldimine—Schiff's base linkage results in this modification being more labile versus the amine linkage found on the acetone modified amino acids. On proteins, the local environment surrounding the Schiff's base may stabilize the modification sufficiently so that it can be observed in the mass spectrums of antibody HC and LC. We have found that the +38 Da aldimine adduct is observable on LC N-termini when off-line chromatographic techniques are used to enrich these populations (data not shown).
In the context of the proposed mechanism (Fig. 5), the 38 Da aldimine adduct may be formed by addition of a hydroxyl to acetoacetate radical forming 2-hydroxy-3-oxobutanoic acid. Decarboxylation of this species results in hydroxyacetone and Yaylayan et al. have demonstrated that hydroxyacetone can undergo enolization to hydroxypropanal, and ultimately acrolein,44,45 which is a known source of aldimine adducts. We have observed low levels of hydroxyacetone resulting from photochemical degradation of citrate, which supports the proposed mechanistic branch point (data not shown) but have not detected free acrolein. Whether this is due to the extreme reactivity of acrolein and the resultant short half-life in solution or, some other as yet unknown mechanism, is under investigation. Regardless of the origination, photochemical degradation of citrate results in aldimine adducts on intact proteins. The presence of C=C bonds on the adduct relegate it to a class of highly reactive electrophiles that are capable of covalent cross-linking.
Citric acid is a weak organic acid consisting of three ionizable carboxylates and one hydroxyl group. At a pH of 6.0, citrate has been shown to chelate divalent metals through coordinate bonds with acidic oxygens on carboxylate carbons 1 and 6 and the hydroxyl group of carbon 3.46–48 Binuclear coordination complexes with citrate and divalent metals have also been reported.49,50 In the course of investigating the mechanism of photochemical citrate degradation, we considered the possibility that carbon swapping might occur between 2 citrate molecules bound to transition metals in a binuclear complex. We addressed this hypothesis by incubating glycine in equimolar concentrations of unlabeled citrate and 2,4-13C2 or 13C6 labeled citrate and determining the m/z of the resulting glycine adducts (Table I). We found that the unlabeled and 2,4-13C2 labeled citrate – glycine mixture yielded [M + H]1+ modification products at m/z = 132.08 and 134.08 and the unlabeled and 13C6 labeled citrate—glycine mixture yielded [M + H]1+ modification products at m/z = 132.08 and 135.08. We observed no modification products of intermediate m/z suggesting that carbons are not swapping between unlabeled and isotope-labeled citrate molecules. Note that these results do not indicate whether citrate as examined in the current system exists as a mononuclear or binuclear chelator of divalent metal, only that the modification product resulting from the photochemical degradation of citrate is unimolecular with respect to citrate. In other words, the data suggests that one citrate molecule results in one acetone modification on an amine.
We have discovered a novel protein modification resulting from the photochemical degradation of citrate. The acetonation reaction as we have described occurs readily under mildly acidic conditions and thus has broad relevance to any chemical process whereby citrate is combined with transition metal and exposed to light in the presence of a nucleophile. While the occurrence of this process in in vivo systems at physiological pH is under evaluation, it is not unreasonable to imagine that acetonation of protein amines might occur at low levels in biological systems undergoing oxidative stress. While the chemistry and structure underlying acetone adducts resulting from citrate is clear, further work is needed to elucidate the effect of these modifications on both protein structure and function.
Materials and Methods
Recombinant antibodies
The IgG2 antibody used in this study was a human recombinant monoclonal antibody stably expressed in Chinese hamster ovary (CHO) cells and purified using conventional techniques.51 Purified antibodies were formulated in sodium acetate buffer at a pH of 5.0.
Sodium citrate-amino acid solutions
Stable isotope citric acid with 13C incorporated on carbons 2 and 4, 1 and 5 or all citrate carbons (2,4-13C2, 1,5-13C2, or 13C6, respectively) was obtained from Sigma Aldrich (Sigma, St Louis, MO). A citrate structure with annotated carbons is shown in Table I. Stable isotope glycine 2-13C (13C α carbon) and 13C2 (13C α and α carbon) were also obtained from Sigma Aldrich. Citric acid stock solutions were prepared at a concentration of 200 mM and the pH was adjusted to 5.0 with 50% sodium hydroxide. Glycine and Fe(III) chloride salt, Fe(II), Cu(II), or Co(II) sulfate salt were added to the sodium citrate solution and the final concentration of sodium citrate, glycine, and transition metal were 100, 50, and 0.2 mM respectively. The preceding reactions were also carried out in the presence of 2.5 or 5% v:v IPA. Hydrogen peroxide was also added to the citrate-glycine buffer at a concentration of 2 mM in the presence or absence of transition metal. Degradation of sodium citrate was induced by exposing samples in polypropylene tubes 4–6 in. from a fluorescent light source. Light exposure occurred at a constant intensity of ∼10,000 Lux/h, which is 10−15 fold higher than typical ambient laboratory fluorescent light intensities measured at bench level. The free glycine was quantitated before and after light exposure using the Waters AccQ-Tag amino acid analysis kit (Waters, Milford, MA) following the manufacturer's instructions.
Sodium citrate and acetone-acetate antibody solutions
Citrate and stable isotope citrate buffer solutions were prepared as described above. The recombinant human IgG2 antibody was buffer exchanged into water using PD-10 desalting columns (GE Healthcare, Piscataway, NJ) and following the manufacturer's instructions. Desalted antibody was then added to 100 mM stock sodium citrate solutions to a final concentration of 5 mg/mL. Acetone-acetate buffer solutions were prepared by adding concentrated sodium acetate at pH 5.0 to the desalted antibody to a final acetate concentration of 100 mM. Acetone was then added to a final concentration of 10 mM. Fe(II) was added to solutions of antibody in citrate or acetone-acetate to a final concentration of 0.2 mM and light exposure was carried out as described above.
Reversed-phase HPLC-MS of antibody HC and LC
Separation of antibody HC and LC was carried out according to a method based on Ren et al.52 The recombinant antibody was reduced by heating samples to 55°C for 30 min in the presence of 10 mM Dithiothreitol, 4 M guanidine HCl, 57 mM Tris-HCl pH 8.3. 50 μg of reduced antibody was injected onto a (2.0 × 250 mm) Varian diphenyl rp-HPLC column (Varian, Palo Alto, CA) with 3 μm particles with 200 Å pores. The column temperature was maintained at 80°C during the separation and the reduced antibody HC and LC were eluted with an acetonitrile gradient formed by the mixing of A and B solvents (0.1% trifluoroacetic acid and 0.1% trifluoroacetic acid, 80% acetonitrile, respectively). The reduced antibody HC and LC were bound to the column in the presence of 20% solvent B for 5 min at which time the solvent composition was brought to 43% B over 5 min and subsequently from 43 to 55% B over 50 min. The column was then washed in 100% solvent B for 3 min and returned to 20% B and the column was equilibrated for 13 min prior to the next injection. The flow rate was maintained at 0.25 mL/min during the separation. The mass of reduced HC and LC fragments was determined on an Agilent LC-MSD-TOF mass spectrometer. Optimal desolvation of antibody fragments was obtained by adjusting the electrospray ionization source drying gas and nebulizer gas flows to 12 and 35 L/h, respectively. The capillary voltage was set to 4500 volts and the fragmentor and octapole rf voltages were maintained at 350 and 300 volts, respectively. Mass spectral data was deconvoluted using Agilent MassHunter Qualitative Analysis software.
Normal-phase HPLC-MS and CID-MS2/MS3 of glycine+56 Da
A 2.1 × 150 mm 1.7 μm particle Waters BEH amide column attached to a Waters Aquity UPLC system was used for quantitation of modified glycine. Samples were prepared for injection as described above and 20 μL was injected onto the column heated at 40°C and pre-equilibrated in solvent A (described above) at a flow rate of 0.2 mL/min. Following a 5 min hold at the initial conditions, modified and unmodified glycine were eluted with a linear gradient to 21.5% solvent B (described above) over 30 min at which time the column was washed with 100% solvent B for 3 min and subsequently equilibrated in solvent A for 15 min prior to the next injection. Modified and unmodified glycine was detected using a Thermo Finnigan LTQ XL mass spectrometer which was set to monitor the mass range from 50 to 500 Da and perform CID-MS2 fragmentation in a data dependant manner. Detailed CID-MS2/MS3 structural analysis of modified glycine was obtained by collecting 40 sec fractions from the analytical normal phase separation using a Triversa Nanomate as described above. The fraction collection was monitored on-line using a Thermo Finnigan LTQ XL mass spectrometer to aid in identification of fractions of interest. Structural characterization of modified glycine was carried out using the Triversa nanomate in direct infusion mode. The [M + H]1+ ion for modified glycine was isolated using a 2 Da mass window and analyzed by CID-MS2/MS3 using an LTQ XL mass spectrometer with a collision energy of 30 V and mass spectral data was collected in profile mode. Exact mass measurement of modified glycine was performed using electrospray ionization (positive mode) on a hybrid linear ion trap-orbitrap mass spectrometer (Thermo Scientific). The sample was directly infused (∼2 μL/min) using a syringe pump. The instrument was externally calibrated using Na-TFA cluster ions; and the resulting mass accuracy for all calibrant ions was better than 1.5 ppm. Spectra were acquired with a resolution setting of 60,000 at m/z 400 (full width at half-maximum).
Analytical reversed-phase HPLC
The chemical intermediates resulting from photochemical degradation of citrate were separated by rp-HPLC using two 3.0 × 250 mm 3 μm particle Phenomenex Gemini C18 columns (Phenomenex, Torrance, CA) connected in series and attached to Waters Alliance 2695 HPLC. Samples were prepared for injection by diluting 1:10 in 40 mM H2SO4 and 40 μL was injected onto the column heated at 40°C and pre-equilibrated in solvent A (40 mM H2SO4) at a flow rate of 0.15 mL/min. Following a 5 min hold at the initial conditions, the citrate degradation products were eluted with a linear gradient to 50% solvent B (50% acetonitrile) over 90 min at which time the column was washed with 100% solvent B for 40 min at a flow rate of 0.2 mL/min and subsequently equilibrated in solvent A for 63 min prior to the next injection. The reaction products were detected with a Waters Alliance W2996 photodiode array detector. The photochemical degradation products from citrate implicated in protein modification were specifically detected by monitoring UV absorbance at 246 nm. The UV spectrums at the 246 nm peak apex were plotted from the photodiode array data acquired during the rp-HPLC separation. Injections of a standard solution consisting of 100 mM citrate, 4.6 mM 3-oxoglutarate, and 4.6 mM acetoacetate were used to positively identify the above chemicals in the citrate-glycine-Fe(II) samples that were exposed to light.
Acknowledgments
We acknowledge Darren Reid, Bruce Kerwin, Christian Schoneich, David Hambly, Catherine Eakin, and Wenge Zhong for insightful discussions regarding potential chemical mechanisms of citrate decomposition and Capucine Thwin, Julia Bach and Sheila Kingrey-Gebe for experimental contributions to the process development aspects of this work. We thank Arvia Morris, Karen Anderson, and Nicole Costa for discussions regarding the chemical components of cell growth media and are grateful to Robert Bailey and Duke Phan for providing the IgG2 antibodies used in these studies. We are grateful to our colleagues at Amgen who evaluated this manuscript; they deserve special recognition for their insightful comments regarding the proposed mechanism of citrate degradation. We also thank Dean Pettit for critical review of the manuscript and continuing support of this work.
Glossary
Abbreviations:
- CID
collision induced dissociation
- DNPH
dinitrophenylhydrazine
- HC
heavy chain
- HMBC
heteronuclear multiple bond correlation
- HSQC
heteronuclear single quantum coherence
- IPA
isopropanol
- LC
light chain
- NMR
nuclear magnetic resonance
- np-HPLC
normal phase-high performance liquid chromatography
- Q-TOF
quadrupole-time of flight
- ROS
reactive oxygen species
- rp-HPLC
reversed phase-high performance liquid chromatography
- UV
ultraviolet.
References
- 1.Cacia J, Keck R, Presta LG, Frenz J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: identification and effect on binding affinity. Biochemistry. 1996;35:1897–1903. doi: 10.1021/bi951526c. [DOI] [PubMed] [Google Scholar]
- 2.Rehder DS, Chelius D, McAuley A, Dillon TM, Xiao G, Crouse-Zeineddini J, Vardanyan L, Perico N, Mukku V, Brems DN, Matsumura M, Bondarenko PV. Isomerization of a single aspartyl residue of anti-epidermal growth factor receptor immunoglobulin gamma2 antibody highlights the role avidity plays in antibody activity. Biochemistry. 2008;47:2518–2530. doi: 10.1021/bi7018223. [DOI] [PubMed] [Google Scholar]
- 3.Yan B, Steen S, Hambly D, Valliere-Douglass J, Vanden Bos T, Smallwood S, Yates Z, Arroll T, Han Y, Gadgil H, Latypov RF, Wallace A, Lim A, Kleemann GR, Wang W, Balland A. Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J Pharm Sci. 2009;98:3509–3521. doi: 10.1002/jps.21655. [DOI] [PubMed] [Google Scholar]
- 4.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28:1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 5.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 6.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metab Rev. 1998;30:225–243. doi: 10.3109/03602539808996310. [DOI] [PubMed] [Google Scholar]
- 7.Banks DD, Hambly DM, Scavezze JL, Siska CC, Stackhouse NL, Gadgil HS. The effect of sucrose hydrolysis on the stability of protein therapeutics during accelerated formulation studies. J Pharm Sci. 2009;98:4501–4510. doi: 10.1002/jps.21749. [DOI] [PubMed] [Google Scholar]
- 8.Brady LJ, Martinez T, Balland A. Characterization of nonenzymatic glycation on a monoclonal antibody. Anal Chem. 2007;79:9403–9413. doi: 10.1021/ac7017469. [DOI] [PubMed] [Google Scholar]
- 9.Smales CM, Pepper DS, James DC. Protein modification during antiviral heat bioprocessing. Biotechnol Bioeng. 2000;67:177–188. doi: 10.1002/(sici)1097-0290(20000120)67:2<177::aid-bit7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Smales CM, Pepper DS, James DC. Protein modifications during antiviral heat bioprocessing and subsequent storage. Biotechnol Prog. 2001;17:974–978. doi: 10.1021/bp010086f. [DOI] [PubMed] [Google Scholar]
- 11.Glomb MA, Monnier VM. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem. 1995;270:10017–10026. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 12.Manini P, La PP, Panzella L, Napolitano A, d'Ischia M. Glyoxal formation by Fenton-induced degradation of carbohydrates and related compounds. Carbohydr Res. 2006;341:1828–1833. doi: 10.1016/j.carres.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 13.Thornalley P, Wolff S, Crabbe J, Stern A. The autoxidation of glyceraldehyde and other simple monosaccharides under physiological conditions catalysed by buffer ions. Biochim Biophys Acta. 1984;797:276–287. doi: 10.1016/0304-4165(84)90131-4. [DOI] [PubMed] [Google Scholar]
- 14.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344(Pt 1):109–116. [PMC free article] [PubMed] [Google Scholar]
- 15.Ledl F, Schleicher E. New aspects of the Maillard reaction in foods and in the human body. Angew Chem Int Ed. 1990;29:565–594. [Google Scholar]
- 16.Spiteller G. Peroxyl radicals are essential reagents in the oxidation steps of the Maillard reaction leading to generation of advanced glycation end products. Ann NY Acad Sci. 2008;1126:128–133. doi: 10.1196/annals.1433.031. [DOI] [PubMed] [Google Scholar]
- 17.Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem. 2008;283:21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider C, Tallman KA, Porter NA, Brash AR. Two distinct pathways of formation of 4-hydroxynonenal. Mechanisms of nonenzymatic transformation of the 9- and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. J Biol Chem. 2001;276:20831–20838. doi: 10.1074/jbc.M101821200. [DOI] [PubMed] [Google Scholar]
- 19.Schneider C, Porter NA, Brash AR. Autoxidative transformation of chiral omega6 hydroxy linoleic and arachidonic acids to chiral 4-hydroxy-2E-nonenal. Chem Res Toxicol. 2004;17:937–941. doi: 10.1021/tx049913n. [DOI] [PubMed] [Google Scholar]
- 20.Spiteller P, Kern W, Reiner J, Spiteller G. Aldehydic lipid peroxidation products derived from linoleic acid. Biochim Biophys Acta. 2001;1531:188–208. doi: 10.1016/s1388-1981(01)00100-7. [DOI] [PubMed] [Google Scholar]
- 21.LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carbone DL, Doorn JA, Kiebler Z, Petersen DR. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 23.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 24.Yamada S, Funada T, Shibata N, Kobayashi M, Kawai Y, Tatsuda E, Furuhata A, Uchida K. Protein-bound 4-hydroxy-2-hexenal as a marker of oxidized n-3 polyunsaturated fatty acids. J Lipid Res. 2004;45:626–634. doi: 10.1194/jlr.M300376-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maisonneuve E, Fraysse L, Lignon S, Capron L, Dukan S. Carbonylated proteins are detectable only in a degradation-resistant aggregate state in Escherichia coli. J Bacteriol. 2008;190:6609–6614. doi: 10.1128/JB.00588-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishii T, Yamada T, Mori T, Kumazawa S, Uchida K, Nakayama T. Characterization of acrolein-induced protein cross-links. Free Radic Res. 2007;41:1253–1260. doi: 10.1080/10715760701678652. [DOI] [PubMed] [Google Scholar]
- 28.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 29.Maisonneuve E, Ezraty B, Dukan S. Protein aggregates: an aging factor involved in cell death. J Bacteriol. 2008;190:6070–6075. doi: 10.1128/JB.00736-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Margetis P, Antonelou M, Karababa F, Loutradi A, Margaritis L, Papassideri I. Physiologically important secondary modifications of red cell membrane in hereditary spherocytosis-evidence for in vivo oxidation and lipid rafts protein variations. Blood Cells Mol Dis. 2007;38:210–220. doi: 10.1016/j.bcmd.2006.10.163. [DOI] [PubMed] [Google Scholar]
- 31.Margetis PI, Antonelou MH, Petropoulos IK, Margaritis LH, Papassideri IS. Increased protein carbonylation of red blood cell membrane in diabetic retinopathy. Exp Mol Pathol. 2009;87:76–82. doi: 10.1016/j.yexmp.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Aldini G, Dalle-Donne I, Facino RM, Milzani A, Carini M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med Res Rev. 2007;27:817–868. doi: 10.1002/med.20073. [DOI] [PubMed] [Google Scholar]
- 33.Burcham PC, Raso A, Thompson C, Tan D. Intermolecular protein cross-linking during acrolein toxicity: efficacy of carbonyl scavengers as inhibitors of heat shock protein-90 cross-linking in A549 cells. Chem Res Toxicol. 2007;20:1629–1637. doi: 10.1021/tx700192e. [DOI] [PubMed] [Google Scholar]
- 34.Burcham PC, Raso A, Thompson CA. Toxicity of smoke extracts towards A549 lung cells: role of acrolein and suppression by carbonyl scavengers. Chem-Biol Interact. 2010;183:416–424. doi: 10.1016/j.cbi.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 35.Monteil C, Le PE, Buisson S, Morin JP, Guerbet M, Jouany JM. Acrolein toxicity: comparative in vitro study with lung slices and pneumocytes type II cell line from rats. Toxicology. 1999;133:129–138. doi: 10.1016/s0300-483x(99)00015-3. [DOI] [PubMed] [Google Scholar]
- 36.Thompson CA, Burcham PC. Genome-wide transcriptional responses to acrolein. Chem Res Toxicol. 2008;21:2245–2256. doi: 10.1021/tx8001934. [DOI] [PubMed] [Google Scholar]
- 37.Abrahamson HB, Rezvani AB, Brushmiller JG. Photochemical and spectroscopic studies of complexes of iron(III) with citric acid and other carboxylic acids. Inorg Chim Acta. 1994;226:117–127. [Google Scholar]
- 38.Furuhata A, Ishii T, Kumazawa S, Yamada T, Nakayama T, Uchida K. N(epsilon)-(3-methylpyridinium)lysine, a major antigenic adduct generated in acrolein-modified protein. J Biol Chem. 2003;278:48658–48665. doi: 10.1074/jbc.M309401200. [DOI] [PubMed] [Google Scholar]
- 39.Simpson DM, Beynon RJ. Acetone precipitation of proteins and the modification of peptides. J Proteome Res. 2010;9:444–450. doi: 10.1021/pr900806x. [DOI] [PubMed] [Google Scholar]
- 40.Hofer H, Moest M. Justus Liebigs Ann Chim. 1902;323:284–323. [Google Scholar]
- 41.Stapley JA, Bemiller JN. The Ruff degradation: a review of previously proposed mechanisms with evidence that the reaction proceeds by a Hofer-Moest-type reaction. Carbohydr Res. 2007;342:407–418. doi: 10.1016/j.carres.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 42.Haber F, Willstätter R. Unpaarigheit und radikalketten im reaktion-mechanismus organischer und enzymatischer vorgänge. Chem Ber. 1931;64:2844–2856. [Google Scholar]
- 43.Hakala M, Rantamaki S, Puputti EM, Tyystjarvi T, Tyystjarvi E. Photoinhibition of manganese enzymes: insights into the mechanism of photosystem II photoinhibition. J Exp Bot. 2006;57:1809–1816. doi: 10.1093/jxb/erj189. [DOI] [PubMed] [Google Scholar]
- 44.Yaylayan VA, Harty-Majors S, Ismail AA. Monitoring carbonyl-amine reaction and enolization of 1-hydroxy-2-propanone (Acetol) by FTIR spectroscopy. J Agric Food Chem. 1999;47:2335–2340. doi: 10.1021/jf9812836. [DOI] [PubMed] [Google Scholar]
- 45.Yaylayan VA, Keyhani A. Origin of carbohydrate degradation products in L-Alanine/D-[(13)C]glucose model systems. J Agric Food Chem. 2000;48:2415–2419. doi: 10.1021/jf000004n. [DOI] [PubMed] [Google Scholar]
- 46.Deng YF, Zhou ZH, Wan HL, Ng SW. D-Aqua-S-citrato(2-) manganese(II) Acta Crystallogr. 2003:310–312. [Google Scholar]
- 47.Glusker JP. Citrate conformation and chelation: enzymatic implications. Acc Chem Res. 1980;13:345–352. [Google Scholar]
- 48.Zhang G, Yang G, Ma JS. Versatile framework solids constructed from divalent transition metals and citric acid: syntheses, crystal structures, and thermal behaviors. Cryst Growth Des. 2006:375–381. [Google Scholar]
- 49.Baker EN, Baker HM, Anderson BF, Reeves RD. Chelation of nickel(II) by citrate. The crystal structure of a nickel–citrate complex, K2[Ni(C6H5O7)(H2O)2]24H2O. Inorg Chim Acta. 1983:281–285. [Google Scholar]
- 50.Kotsakis N, Raptopoulou CP, Tangoulis V, Terzis A, Giapintzakis J, Jakusch T, Kiss T, Salifoglou A. Correlations of synthetic, spectroscopic, structural, and speciation studies in the biologically relevant cobalt(II)-citrate system: the tale of the first aqueous dinuclear cobalt(II)-citrate complex. Inorg Chem. 2003;42:22–31. doi: 10.1021/ic011272l. [DOI] [PubMed] [Google Scholar]
- 51.Shukla AA, Hubbard B, Tressel T, Guhan S, Low D. Downstream processing of monoclonal antibodies--application of platform approaches. J Chromatogr B. 2007;848:28–39. doi: 10.1016/j.jchromb.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 52.Ren D, Pipes GD, Hambly DM, Bondarenko PV, Treuheit MJ, Brems DN, Gadgil HS. Reversed-phase liquid chromatography of immunoglobulin G molecules and their fragments with the diphenyl column. J Chromatogr A. 2007;1175:63–68. doi: 10.1016/j.chroma.2007.10.005. [DOI] [PubMed] [Google Scholar]