

Abstract
Guanosine triphosphate (GTP) binding and hydrolysis events often act as molecular switches in proteins, modulating conformational changes between active and inactive states in many signaling molecules and transport systems. The P element transposase of Drosophila melanogaster requires GTP binding to proceed along its reaction pathway, following initial site-specific DNA binding. GTP binding is unique to P elements and may represent a novel form of transpositional regulation, allowing the bound transposase to find a second site, looping the transposon DNA for strand cleavage and excision. The GTP-binding activity has been previously mapped to the central portion of the transposase protein; however, the P element transposase contains little sequence identity with known GTP-binding folds. To identify soluble, active transposase domains, a GFP solubility screen was used testing the solubility of random P element gene fragments in E. coli. The screen produced a single clone spanning known GTP-binding residues in the central portion of the transposase coding region. This clone, amino acids 275–409 in the P element transposase, was soluble, highly expressed in E.coli and active for GTP-binding activity, therefore is a candidate for future biochemical and structural studies. In addition, the chimeric screen revealed a minimal N-terminal THAP DNA-binding domain attached to an extended leucine zipper coiled-coil dimerization domain in the P element transposase, precisely delineating the DNA-binding and dimerization activities on the primary sequence. This study highlights the use of a GFP-based solubility screen on a large multidomain protein to identify highly expressed, soluble truncated domain subregions.
Keywords: green fluorescent protein, solubility, screen, domain, P element, THAP domain, guanosine triphosphate
Introduction
The P element transposase (TNP) is a multidomain protein consisting of an N-terminal thanatos-associated protein (THAP) DNA binding domain and adjacent leucine-zipper oligomerization domain,1,2 a central GTP-binding domain,3,4 and a C-terminal catalytic domain used for strand cleavage and integration. The N-terminal site-specific DNA-binding domain is a prototypical C2CH zinc-coordinating THAP domain,1 which is adjacent to an oligomerization motif consisting of a leucine-zipper followed by an extended coiled-coil region. Often found in the extensive THAP protein family, two THAP domains can form homodimers via an adjacent leucine zipper.1,2 Previous characterization of the P element THAP domain included the original identification of the domain on the N terminus of the P-element transposase by deletion studies,1,2 the DNA binding sequence by DNAseI footprinting,5 and affinity towards multiple P-element end sequences as both a monomeric and dimeric species.1,2
The C-terminus of the P-element transposase protein contains many acidic residues and is thought to belong to the RNase H-like superfamily of polynucleotidyl transferases, including the bacterial Tn5 transposase,6 the Mos1 transposase,7 the HIV-1 integrase,8 the phage Mu transposase,9 the Holliday junction nuclease RuvC,9 and the RAG1 V(D)J recombinase.10 Although there is generally low sequence identity (<20%) between the members of this protein superfamily, the Structural Classification of Proteins database11 lists nine members whose domain architecture is structurally very similar. These enzymes use a two-metal ion catalysis for phosphodiester bond hydrolysis and transesterification, utilizing a pocket of conserved acidic amino acids to position divalent metal ions similar to RNase H.12,13 Mutagenesis in the P-element transposase revealed four acidic amino acids in the C-terminus that modulate activity in vivo and in vitro and could potentially contain residues equivalent to a DDE or RNase H-like motif (Tang and Rio, unpublished results).
The 87 kD P element transposase of Drosophila melanogaster has a unique requirement for guanosine triphosphate (GTP) binding that distinguishes this family from smaller transposases, such as Tn5 and Mu, although Mu does require an ATP-binding accessory protein MuB.14 The transposase/integrase members of the RNase H-like superfamily can perform excision and integration transesterification reactions using either water or DNA 3′ OH groups as nucleophiles, without exogenous cofactor or nucleotide binding12,13 and are thus isoenergetic, not requiring high energy bond hydrolysis as a source of energy. In vitro studies showed that the P element transposition reaction required GTP and magnesium,3 yet the triphosphate guanosine nucleotides namely GTP, deoxyGTP, dideoxyGTP, or the nonhydrolyzable GTP analogues GTP-γ-S, GMP-PNP, and GMP-PCP fully substitute for GTP in transposase excision and transposition assays. GTP is therefore considered to be an allosteric effector required for proper folding and domain positioning in the P element transposase.
Single molecule atomic force microscopy using recombinant P element transposase and a linear P element DNA substrate15,16 showed pre-formed transposase tetramers in solution without exogenous GTP or DNA. When transposase is bound to P element DNA in the absence of GTP, tetramers bound to only one transposon end. The N-terminal DNA-binding domain of the P element transposase can bind DNA as a monomer containing only the N-terminus without the GTP-binding or dimerization domains.1,2 Also, the full-length protein has been shown to footprint in the absence of GTP.5 These data suggest that GTP is not needed in the early DNA binding events. The addition of GTP promotes the formation of stable synaptic complexes, allowing a single-end tethered transposase tetramer to find the second transposase binding site at the opposite P element end,16 forming a DNA loop that can now proceed to strand excision. The GTP binding domain of the P element transposase has noncanonical versions of motifs from the GTPase superfamily, including DXXG and NKXD motifs,4 yet previous attempts to create soluble fragments of this domain have been unsuccessful (unpublished data).
Biochemical and structural analysis of protein domains or truncations is often precluded by the production of soluble and active protein. Approaches to modulating exogenous gene expression in E. coli include optimization of growth conditions,17 such as variation of growth temperature and protein induction levels, empirical screenings of growth media from minimal to rich media, coexpression with chaperones,18 fusion to maltose-binding protein,19 dihydrofolate reductase20 or chloramphenicol acetyl transferase.21 Furthermore, mutagenesis and/or evolution-selection studies can lead to finding protein characteristics that can potentially improve solubility of recombinant proteins.22 Here, we report a chimeric green fluorescent protein (GFP) solubility screen in E. coli, using full-length P element transposase (amino acids 1–751) as a template.
In this study, random N- and C-terminal boundaries of our target protein, the P element transposase, are generated by randomly primed PCR.23 The multidomain transposase was thought to be amenable to a randomized solubility study because of potentially modular domains in the primary sequence. The correct folding of putative domain fragments were screened in E. coli by subsequent fusion and expression as C-terminal chimeras to green fluorescent protein.23–25 GFP fusion screening for solubility has also been used successfully for membrane proteins, from multiple organisms.26 The screen reported here precisely defined both the DNA and dimerization regions in the P element transposase, producing N-terminal structural targets of amino acids 1–77 and 1–170, whose functions had been approximately mapped previously onto the primary sequence as 1–88 and 1–150.1,2 Moreover, a single clone (amino acids 275–409) which contained the GTP binding motifs4 was expressed and found to be soluble. Biochemical validation of the screening results confirms the finding that these soluble protein fragments were also active for either DNA or GTP binding, revealing a more precise definition of the domain organization of the P element transposase, and multiple single domain targets for future structural studies.
Results
To conduct a solubility screen on candidate P element transposase protein fragments, a truncation DNA library was made by random-primed PCR,23 using a full-length P element transposase cDNA as a template. The cDNA had been previously codon-optimized for E. coli. The library of random PCR fragments was cloned into a GFP-containing vector, pPROGFP,23 creating randomized protein chimeras with GFP attached at the N-terminus. The chimeras were transformed into E. coli strain DH10B and allowed to express protein as a bacterial colony on an agar plate under antibiotic drug selection. GFP-positive colonies (usually ˜ 1%) were selected by visualization using UV light at 366 nm24 (Fig. 1), and isolated plasmids sequenced to identify the corresponding transposase fragment boundaries that folded correctly and subsequently allowed for proper GFP folding. The screen yielded ˜100 soluble fragments (Fig. 2, Supporting information, Table 1), that could be grouped into three main categories; (1) N-terminal DNA-binding and dimerization domains (up to amino acid 170), (2) a central GTP-binding domain and (3) a C-terminal catalytic domain. The earliest bright GFP-positive colonies were visible after 14–18 h when grown at 37°C and were marked on the plates for further analysis. After ˜48 h most colonies were GFP-positive and indistinguishable from the original bright GFP clones, thus only early GFP-positive colonies were selected for study. Using a simple screening condition of 37°C, a fixed growth time of 3 h and IPTG induction of 1 mM, >90% of the GFP-positive colonies produced overexpressed protein in the pPROGFP vector (data not shown). Potentially soluble fragments were then subcloned into the expression vector pRSETA lacking GFP, for further expression and solubility testing (Supporting information, Table 2). After sequencing ˜100 clones, only one spanned the potential GTP-binding domain in the central region of the protein; clone 275–409 (Fig. 2). The majority of the clones obtained carried the DNA-binding and adjacent dimerization domains, including fragments 1–74 and 1–80. Cross referencing the sequencing results presented here with structural, bioinformatic, and biochemical data on THAP domains produced two soluble structural candidates from that region; amino acids 1–77 and 1–170 in the P element transposase N-terminus (Fig. 2).
Figure 1.
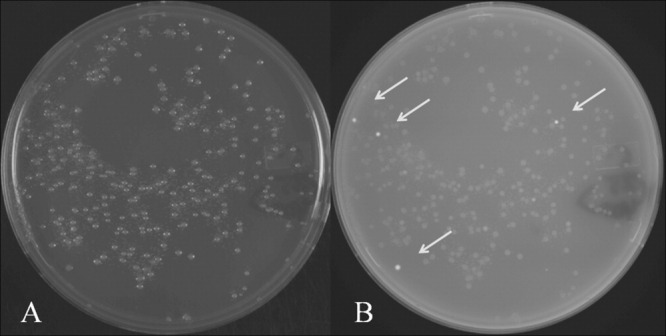
A typical chimeric-GFP solubility screening experiment. (A) white light image, (B) transilluminating UV (365 nm) image of an agar plate used to select GFP-positive colonies in the solubility screen. Electrocompetent DH10B cells were transformed with a transposase fragment library, which was subcloned as a chimera into the GFP-containing vector pPROGFP. The right image contains four “bright” GFP-positive colonies (arrows) which are selected for expression testing, sequencing, and solubility studies. The sensitivity of the screen can be modulated by early or late detection of GFP-positive clones.
Figure 2.
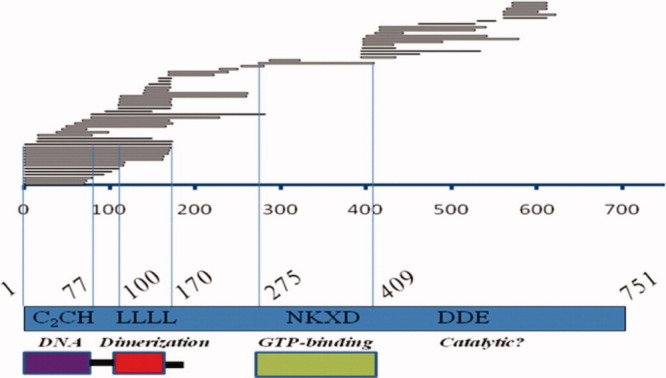
Chimeric-GFP solubility screen results displayed over the P element transposase primary sequence. The P element coding sequence (amino acids 1–751) is depicted in blue, with experimentally verified sequence motifs highlighted left to right; (C2CH) THAP domain, (LLLL) leucine zipper, (NKXD) GTP-binding and (DDE) potential catalytic residues. The DNA sequencing results of individual GFP-positive colonies in the screen are displayed schematically as horizontal bars over the P element sequence. Below are depictions of soluble, active domains found using this screen. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
P-element transposase N-terminal domains: DNA-binding and dimerization
In this study, N-terminal transposase fragments of length 1–74 and 1–80 were found to be potentially soluble, and fragments extending to amino acid 170. On the basis of these sequencing results, and analysis of homologous THAP domain boundaries, it was believed that amino acids 1–77 in the P element transposase would be active for DNA binding (see discussion). Therefore, amino acids 1–77 and 1–170 were subcloned into expression vectors without GFP and tested for solubility as non-GFP-fused proteins in E.coli (Fig. 3). Both constructs produced protein that was readily purified using His6 affinity chromatography, which was tested for DNA binding activity. The nearly identical left and right 10bp high-affinity P element transposon DNA binding sites are internal and adjacent to the 31 base pair terminal P element inverted repeats. In DNase I footprinting protection experiments, we showed that the minimal THAP domain, amino acids 1–77 in the P-element transposase, specifically binds to the 10 base pair specific TNP site, as does the 1–170 fragment [Fig. 4(a,b)], showing that the truncated protein fragments were biochemically active. Indeed, the 1–77 fragment was recently cocrystallized with the high affinity transposase binding site and the structure of the complex solved by X-ray crystallography,27 revealing a novel mode of specific protein-DNA interactions. Moreover, the recombinant protein fragment, amino acids 1–170, was shown to be a dimer by chemical protein crosslinking [Fig. 4(c,d)], and by gel filtration chromatography (data not shown), indicating this soluble fragment retains both DNA binding and dimerization functions of the larger transposase. Besides the original cocrystal structure, the fragment 1–77 is currently being crystallized with additional DNA oligonucleotides corresponding to internal 11 base pair enhancers of transposition, while the fragment 1–170 is currently being investigated structurally by small-angle X-ray scattering to visualize a potential dimeric THAP complex with DNA.
Figure 3.

Solubility testing of recombinant amino acid fragments 1–77 and 1–170 from the P element transposase, expressed in vector pRSETA in BL21 (DE3) cells. Left to right are uninduced (U), induced (I), total cell lysate (Tcl), and soluble fraction (Sol) for both amino acid segments 1–77 and 1–170. Both fragments are highly expressed and soluble (arrows), amenable for structural studies. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 4.

Biochemical analysis of recombinant fragments of amino acids 1–77 and 1–170 of the P element transposase. DNaseI protection footprinting analysis on the high-affinity, specific transposase binding site using increasing amounts of recombinant protein: (A) amino acids 1–77 or (B) amino acids 1–170, identified from the GFP solubility screen. Lanes 1–4 are chemical DNA sequencing markers; Lanes 5 and 10 have no protein; and Lanes 6–9 have increasing amounts of recombinant protein. The recombinant fragment amino acids 1–170 was shown to be a dimer by EGS chemical crosslinking. 375 pmoles of protein are in each lane with 0, 0.18, 0.36, 0.75, 1.5, or 3 mM crosslinking reagent in a 15 μL volume. (C) shows an 18% SDS-P.A.G.E. protein gel stained with coomassie blue. (D) an immunoblot, probed with anti-His6 antibody (Santa Cruz Biotechnology). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Cloning, expression, and activity testing of GTP-binding and catalytic domains
To test several domain boundaries centered around the GTP-binding domain, amino acid positions from the chimeric-GFP solubility screen were used to create protein fragments to test for solubility as nonfused proteins (Supporting information, Table 2). Several potential central transposase domains were highly expressed in BL21 (DE3) cells when induced at 37°C (Supporting information, Table 2); however, the only highly expressed fragment that was soluble was the original clone from the screen, 275–409 (Fig. 5). Screening tens of potential fragments around the GTP-binding residues did not reveal any further constructs that were soluble. However, the 275–409 fragment from this study did express well and purify as a single peak using size-exclusion chromatography (data not shown). When tested biochemically, the recombinant 275–409 protein did bind and retain GTP on a nitrocellulose filter-binding assay [Fig. 6(e,f)], suggesting the GTP-binding activity of the P-element transposase is encoded as a single domain. Mutation of the proposed NKXD motif (D379N) reduced the overall GTP binding [Fig. 6(j,k)], similar to what had been observed for the full-length protein.4 Additionally, combinations of fragment termini from the screen (for example creating a truncation from 1–409) failed to produce soluble protein material in E.coli (Supporting information, Table 2). All fragments tested with C-terminal transposase boundaries were also insoluble (Supporting information, Table 2).
Figure 5.
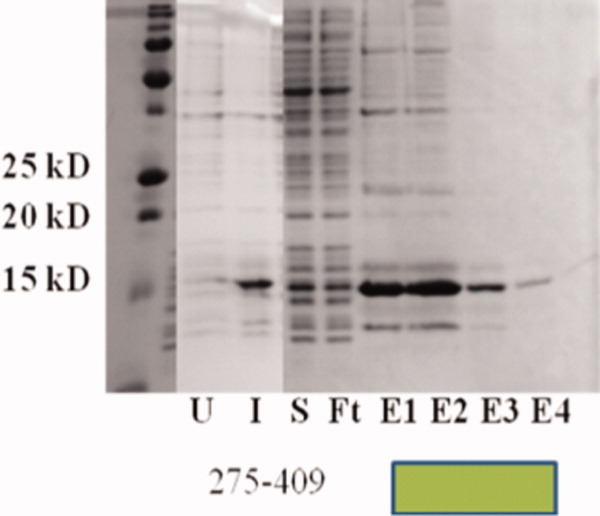
Solubility, expression, and purification on the transposase fragment, amino acids 275–409, centered around the GTP-binding domain. Shown is a coomassie blue stained 18% polyacrylamide gel. Left to right: marker, uninduced cells, induced cells, soluble protein fraction, chelating His6 affinity column flow-through, and elutions. The fragment revealed in the solubility screen (275–409) was amenable to high levels of expression and purification. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 6.
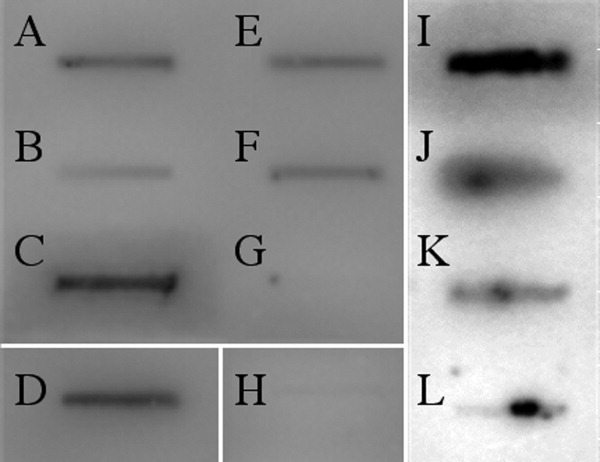
The P element transposase fragment 275–409 binds GTP. Radioactive GTP (2.5 pmoles) was incubated with the purified full-length or truncated P element transposase proteins, prepared in a variety of expression systems; (A) Baculovirus MBP-full length transposase (+ control), (B) Drosophila L2 cells, full-length TNP (+ control), (C) human RAS (+ control), (D) Bacterial MBP-TNP-(88–751) (+ control), (E) and (F) different preparations of fragment 275–409, (G) GTP alone (−control), and (H) THAP domain (−control). All proteins used 35 pmoles per reaction, except for RAS and the THAP domain, which use 350 pmoles. (I) Recombinant human RAS was used as a positive control compared to (L) GTP alone. (K) The NKXD GTP-binding mutant D379N is reduced in GTP binding compared to the 275–409 fragment in (J). The panels (I-L) highlight the difference between the 275–409 fragment in (J) with the mutant in (K). All complexes were spotted on nitrocellulose, filtered, and washed. The nitrocellulose-bound complex was then visualized using a Typhoon Phosphoimager system.
Discussion
Using an E. coli codon-optimized P element transposase cDNA as a template, the solubility screen described here defined ˜100 colonies as candidates for solubility studies. To date, similar GFP-chimeric solubility studies have analyzed fourteen23 and twenty seven24 colonies, respectively, on a roughly equally sized template using similar techniques. The use of an E.coli codon-optimized cDNA template may have increased the success rate of finding soluble protein fragments using E.coli as an expression system. Additionally, our hypothesis was that this solubility screening approach would be viable with the P element transposase because the protein is large (751 amino acids) and has a modular functional organization distributed among several regions in the primary sequence.
In this study, two screening fragments of 1–74 and 1–80 were used as a rough boundary for the DNA-binding activity. Previous biochemical, structural, and bioinformatics studies on homologous THAP domains suggested that amino acids up to ˜77 were important for THAP domain DNA binding function. This region in the N-terminus of the P element THAP domain contains the conserved AVPTIF motif ending at amino acid 77,28 with the proline residue at position 74 being 100% conserved among THAP family members. The conservation after the AVPTIF motif is very poor,28 indicating the functional significance of the conserved amino acids and a potential domain boundary. The boundary at amino acid 77 also correlated well with the NMR structures29,30 and DNA-binding studies29 of C. elegans CTBP, human THAP1 and human THAP2, which have unstructured or “floppy” tails after the amino acid corresponding to residue 77 in the P element sequence. The transposase fragment of 1–77 was therefore expressed, purified, and crystallized with the specific P element transposase binding site and the structure of the complex solved by X-ray diffraction.27
Many potential DNA binding domain boundaries were found in this screen, sometimes redundantly, as either the start or end of a GFP positive clone (Fig. 2). However, the sequencing results did not produce a clone with the exact length of the expressed and crystallized Drosophila melanogaster THAP domain (amino acids 1–77). Instead, the sequencing results were used as a rough (2–3 amino acid) estimate of the end of domains, then cross-referenced against THAP homologue structures, amino acid alignment conservation, and biochemistry to conclude that 1–77 looked structurally stable in other THAPs, and had a strong conservation up to amino acid 77. The number of sequenced colonies (˜100) suggested that our assay could not identify every possible soluble truncation in the P element coding sequence. If sampling numbers of bright GFP-positive plasmids were much higher, a similar screen could cover the an entire coding sequence with redundancy, yet in this case biological data combined with the results from the solubility screen provided a suitable construct for further structural studies.
The full-length P element transposase contains a well-defined leucine-zipper motif from amino acids 100–128, shown to mediate dimerization in a number of proteins,31 yet no structural candidates existed to study this part of protein. There are several transposase repressor proteins that arise from P element internal DNA deletion events that create proteins of ˜200 amino acids in length that retain DNA binding properties of the full-length transposase but cause transpositional repression.1 The naturally occurring construct the KP protein, amino acids 1–207, is not amenable to structural studies due to its insolubility when using E. coli as an expression system.1,2 By contrast the construct found in this study has similar biochemical activities and is a highly expressed soluble structural target.
Although many soluble clones were found in the N terminus of the transposase protein, only one was found in the central portion of the protein, encompassing the NKXD motif that had been shown to be involved in GTP-binding.4 No common GTP-binding motifs are found when using traditional computational search methods (data not shown). However, the GFP-solubility screen returned a promising colony sequence, amino acids 275–409 in the P element transposase, that binds GTP and which may be amenable for further structural studies. Perhaps only finding one soluble clone obtained from the central region of transposase suggests a lack of stability compared to the N-terminal DNA and oligomerization domains. BLAST results with the P element transposase sequence amino acids 275–409 retrieved other P-element sequences, from multiple fly species, and human, zebrafish, and primate THAP9 genes, and a distant homology to the GTP-binding protein Dynamin (data not shown). THAP9 is the closest human/vertebrate homologue to the entire P element coding region, yet its function remains unknown. The distantly related THAP9 proteins may share functions, such as GTP-binding and/or DNA cleavage activity aside from the common DNA binding properties shared between THAP proteins and P element transposase. THAP proteins have been shown to be transcriptional regulators important in human health and development, thus understanding the evolutionary relationships and domain organization between the THAP9 family members and the P element transposase may illuminate the regulation of a novel family of transcription factors. In conclusion, the fragments amino acids 1–77, 1–170, and 275–409 are novel structural targets from the P element transposase resulting from this solubility screen. Although appearing to express well, it is unclear why no C-terminal catalytic fragments were found soluble by this screen (Supporting information, Table 2). Perhaps the catalytic domain can not be made as a single polypeptide, or perhaps the nature of the screen, which adds GFP N-terminally to potential domains, disrupts the proper folding or function of the fused protein fragment. C-terminal fragments from the P element transposase are currently being tested for expression and activity as fusions to maltose-binding protein.
In conclusion, this study characterizes the domain boundaries for DNA binding, oligomerization, and GTP binding activities in the P element transposase primary sequence, which will allow future structural characterization of these domains. The simplicity of the assay and analysis suggests that given a suitable template, one can define multiple domains in a multidomain protein to within a few amino acids to pursue towards structural studies.
Methods
Chimeric-GFP solubility screen
The GFP solubility screen PCR conditions, vector pPROGFP, and primer design were performed as described.23,24 Briefly, an E. coli codon-optimized full-length P-element transposase cDNA was used as a randomly primed PCR template, adding HindIII restriction enzyme sites to each end of the library products. An internal HindIII site in the transposase was therefore previously removed by mutagenesis in the PCR template. Random-length PCR fragments were then cloned adjacent to the GFP coding sequence in the vector pPROGFP, and allowed to express protein. The brightest GFP-positive colonies are believed to contain well-folded chimeric constructs, thus reporting on the solubility of the library. Three rounds of screening were performed, the first using a PCR-amplified coding section for amino acids 1–751, the second with amino acids 150–751, and the third with the coding region for amino acids 370–751. The final template was further modified by removing a second HindIII site in the P-element coding region (a poorly cutting AAGCT at nucleotide 1740) and removing the stop codon at the end of the protein-coding segment. PCR products were purified on 1% agarose gels using a gel purification kit (Qiagen).
Random PCR was performed as described,23,24 using the primer A; gaccatggattacgccaagcttNNNNN NNNNNNNNNN, and primer B; gaccatggattacgccaagctt. Briefly, 100 ng of template is incubated with 100 pmol primer A, 0.2 mM of each dNTP, and 0.2 U TaqHiFi (Invitrogen) with cycling conditions (94°C for 10 min, 10 cycles of 94°C 1 min, 40°C 3 min, and 68°C for 3 min, followed by 68°C for 10 min) for the first PCR, in a final volume of 150 μL total divided into three PCR tubes. PCR kit purification (Qiagen) was done according to the manufacturer's instructions on the pooled material. The second PCR was done in a 50 μL total volume, spiking in 500 pmol primer B, 0.4 U TaqHiFi, and 0.2 mM dNTPs with cycling conditions (94°C for 10 min, 30 cycles of 94°C 1min, 55°C 3 min, and 68°C for 3 min, followed by 68°C for 10 min.) for the second PCR. PCR products were purified using a PCR purification kit (Qiagen). The mixture of products was cleaved with 30 U HindIII (New England Biolabs) overnight at 37°C, and the PCR “smear” purified by agarose gel purification using a gel-purification kit (Qiagen). Empirical amounts (1, 5, 10 μL) of the random PCR fragments were ligated into 100 ng HindIII-digested pPROGFP23 in a 20 μL reaction volume. 2–5 μL of each ligation was transformed into 40 μL electrocompetent DH10B cells (Invitrogen), diluted into 1 mL SOC medium (Invitrogen), then plated onto 5 selective agar plates.
The earliest signs of GFP production were visible after ˜14 h with either a handheld UV light, or an imaging station at 365 nm (Alphaimager, Alpha Innotech). All plates were made without the additive glucose, which is inhibitory to protein expression in the pPROGFP vector. Only the early time point GFP-positive colonies were selected for expression testing and sequencing (ELIM BIO). The GFP-positive colonies were clonally selected and grown in liquid media for DNA extraction (Qiagen).
Small-scale expression testing
The clonal vectors were individually transformed into BL21 (DE3) cells, grown in liquid culture (5 mL LB, ampicillin) overnight, and 200 μL of this overnight suspension used to seed 5 mL of LB the following morning. This growth suspension was grown at 37°C until an OD600 of ˜0.5, and induced with 1 mM IPTG. After 3 h of growth, 0.5 OD600 units of bacteria were pelleted and the media removed. This cell pellet was resuspended in 200 μL 1X SDS-PAGE loading dye. Samples were run on 18% denaturing SDS-PAGE gels for 1 h at 23 amps for expression testing.
Solubility screening
Solubility screening was done by growing ˜50 mL of a given GFP-positive construct and inducing as described above. The bacterial pellet was centrifuged at 3000 rpm for 30 min and resuspended in 10 mL per gram of lysis buffer (25 mM Hepes-NaOH, pH 8.0, 1M NaCl, 10% glycerol, 0.5 mM TCEP, 0.5 μg mL-1 each of leupeptin, pepstatin, aprotinin, antipain, and chymostatin, 1 mM PMSF). This material was spun at 45,000 rpm in a Beckman Ti45 rotor to pellet insoluble material and the soluble protein fraction was retained for further analysis.
Expression and purification of recombinant proteins
Primers for all subcloned protein constructs were designed to introduce a C-terminal hexhistidine tag, a 5′ NDE1 and 3′ NCO1 restriction enzyme sites for cloning into pRSETA (Invitrogen). Primers were synthesized on an ABI model 392. The plasmids were transformed into BL21 (DE3) cells, a single colony was selected for incubating 5 mL overnight growth for expression tests as described above. One liter cultures were grown for each of the well-expressing constructs. Recombinant proteins were purified by 0.5 mL HiTrap fast-flow Ni2+-charged resin using gravity, then buffer exchanged using a 5 mL HiTrap desalting column or a Superdex75, 24 mL column into gel-filtration buffer (10 mM Hepes-KOH, pH 8.0, 50 mM NaCl, and 0.5 mM TCEP). Recombinant proteins were then flash-frozen in liquid nitrogen and stored at −80°C.
In vitro protein–protein crosslinking and DNaseI protection footprinting
Oligomerization was detected using ethylene glycol bis(succinimidyl succinate) (EGS; Pierce) as a crosslinking agent, as described previously.2 A fixed amount of protein, 15 μL of 25 pmol/μL, was mixed with increasing crosslinking reagent, at 0.18–3.0 mM, incubated for 30 minutes at 22°C, stopped using 2 μL of 1 M Tris pH 7.5, and analyzed using SDS-PAGE gel electrophoresis, then stained with Coomassie blue and by immunoblotting. DNaseI protection footprinting was performed as described.1,2,5
Radioactive GTP-binding experiments
Purified proteins were tested for the ability to bind GTP by retention of radiolabelled [alpha -32P] GTP on nitrocellulose filters, as described previously.4 Briefly, 0.2 μL of radiolabelled GTP (800 μci/nmol, 12.5 μM) was mixed in 650 μL of buffer (10 mM Hepes-KOH, pH 8.0, 50 mM NaCl, and 0.5 mM TCEP), with purified protein (0, 35, or 350 pmoles) and applied to a nitrocellulose membrane using a dot-blot chamber under vacuum (Bio-Rad), washed with excess buffer (10 mM Hepes-KOH, pH 8.0, 50 mM NaCl, and 0.5 mM TCEP), dried, and visualized using a Storm phosphoimager system.
Acknowledgments
A.S. and D.C.R. would like to thank Tom Cech (H.H.M.I. and University of Colorado, Boulder) for advice in performing the GFP screen, Jennifer Doudna (H.H.M.I. and University of California, Berkeley) for materials and advice in performing the GFP screen, Jodi Gureasko, and John Kuriyan (H.H.M.I. and University of California, Berkeley) for the gift of human RAS, Sheena Degnan, and Tom Alber (University of California, Berkeley) for use of equipment and materials, and James M. Berger and DavidWemmer for critical reading of the manuscript. Work in the Rio lab was supported by NIH R01 GM61987. A.S. and D.C.R. would also like to thank Rio lab members; Sharmistha Majumdar for assistance with footprinting, Malik Francis for chemical sequencing markers and assistance with footprinting, Rishi Rawat for assistance in cloning and protein purification of the GTP-binding mutants, and Anil George with assistance in DNA sequence processing.
References
- 1.Lee CC, Beall EL, Rio DC. DNA binding by the KP repressor protein inhibits P-element transposase activity in vitro. EMBO J. 1998;17:4166–4174. doi: 10.1093/emboj/17.14.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee CC, Mul YM, Rio DC. The Drosophila P-element KP repressor protein dimerizes and interacts with multiple sites on P-element DNA. Mol Cell Biol. 1996;16:5616–5622. doi: 10.1128/mcb.16.10.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman PD, Rio DC. P element transposition in vitro proceeds by a cut-and-paste mechanism and uses GTP as a cofactor. Cell. 1992;69:27–39. doi: 10.1016/0092-8674(92)90116-t. [DOI] [PubMed] [Google Scholar]
- 4.Mul YM, Rio DC. Reprogramming the purine nucleotide cofactor requirement of Drosophila P element transposase in vivo. EMBO J. 1997;16:4441–4447. doi: 10.1093/emboj/16.14.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman PD, Doll RF, Rio DC. Drosophila P element transposase recognizes internal P element DNA sequences. Cell. 1989;59:359–371. doi: 10.1016/0092-8674(89)90297-3. [DOI] [PubMed] [Google Scholar]
- 6.Davies DR, Goryshin IY, Reznikoff WS, Rayment I. Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science. 2000;289:77–85. doi: 10.1126/science.289.5476.77. [DOI] [PubMed] [Google Scholar]
- 7.Richardson JM, Zhang L, Marcos S, Finnegan DJ, Harding MM, Taylor P, Walkinshaw MD. Expression, purification and preliminary crystallographic studies of a single-point mutant of Mos1 mariner transposase. Acta Crystallogr D Biol Crystallogr. 2004;60:962–964. doi: 10.1107/S0907444904003798. [DOI] [PubMed] [Google Scholar]
- 8.Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice P, Mizuuchi K. Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- 10.Kim DR, Dai Y, Mundy CL, Yang W, Oettinger MA. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 1999;13:3070–3080. doi: 10.1101/gad.13.23.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barton GJ. Scop: structural classification of proteins. Trends Biochem Sci. 1994;19:554–555. doi: 10.1016/0968-0004(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 12.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 13.Nowotny M. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 2009;10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schweidenback CT, Baker TA. Dissecting the roles of MuB in Mu transposition: ATP regulation of DNA binding is not essential for target delivery. Proc Natl Acad Sci USA. 2008;105:12101–12107. doi: 10.1073/pnas.0805868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang M, Cecconi C, Bustamante C, Rio DC. Analysis of P element transposase protein-DNA interactions during the early stages of transposition. J Biol Chem. 2007;282:29002–29012. doi: 10.1074/jbc.M704106200. [DOI] [PubMed] [Google Scholar]
- 16.Tang M, Cecconi C, Kim H, Bustamante C, Rio DC. Guanosine triphosphate acts as a cofactor to promote assembly of initial P-element transposase-DNA synaptic complexes. Genes Dev. 2005;19:1422–1425. doi: 10.1101/gad.1317605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgiou G, Valax P. Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotechnol. 1996;7:190–197. doi: 10.1016/s0958-1669(96)80012-7. [DOI] [PubMed] [Google Scholar]
- 18.Baneyx F, Palumbo JL. Improving heterologous protein folding via molecular chaperone and foldase co-expression. Methods Mol Biol. 2003;205:171–197. doi: 10.1385/1-59259-301-1:171. [DOI] [PubMed] [Google Scholar]
- 19.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu JW, Boucher Y, Stokes HW, Ollis DL. Improving protein solubility: the use of the Escherichia coli dihydrofolate reductase gene as a fusion reporter. Protein Expr Purif. 2006;47:258–263. doi: 10.1016/j.pep.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 21.Maxwell KL, Mittermaier AK, Forman-Kay JD, Davidson AR. A simple in vivo assay for increased protein solubility. Protein Sci. 1999;8:1908–1911. doi: 10.1110/ps.8.9.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabantous S, Rogers Y, Terwilliger TC, Waldo GS. New molecular reporters for rapid protein folding assays. PLoS ONE. 2008;3:e2387. doi: 10.1371/journal.pone.0002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawasaki M, Inagaki F. Random PCR-based screening for soluble domains using green fluorescent protein. Biochem Biophys Res Commun. 2001;280:842–844. doi: 10.1006/bbrc.2000.4229. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs SA, Podell ER, Wuttke DS, Cech TR. Soluble domains of telomerase reverse transcriptase identified by high-throughput screening. Protein Sci. 2005;14:2051–2058. doi: 10.1110/ps.051532105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 26.Hammon J, Palanivelu DV, Chen J, Patel C, Minor DL., Jr A green fluorescent protein screen for identification of well-expressed membrane proteins from a cohort of extremophilic organisms. Protein Sci. 2009;18:121–133. doi: 10.1002/pro.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat Struct Mol Biol. 2010;17:117–123. doi: 10.1038/nsmb.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roussigne M, Kossida S, Lavigne AC, Clouaire T, Ecochard V, Glories A, Amalric F, Girard JP. The THAP domaa novel protein motif with similarity to the DNA-binding domain of P element transposase. Trends Biochem Sci. 2003;28:66–69. doi: 10.1016/S0968-0004(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 29.Bessiere D, Lacroix C, Campagne S, Ecochard V, Guillet V, Mourey L, Lopez F, Czaplicki J, Demange P, Milon A, Girard JP, Gervais V. Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J Biol Chem. 2008;283:4352–4363. doi: 10.1074/jbc.M707537200. [DOI] [PubMed] [Google Scholar]
- 30.Liew CK, Crossley M, Mackay JP, Nicholas HR. Solution structure of the THAP domain from Caenorhabditis elegans C-terminal binding protein (CtBP) J Mol Biol. 2007;366:382–390. doi: 10.1016/j.jmb.2006.11.058. [DOI] [PubMed] [Google Scholar]
- 31.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]