

Abstract
Tobacco vein mottling virus (TVMV) is a member of the Potyviridae, one of the largest families of plant viruses. The TVMV genome is translated into a single large polyprotein that is subsequently processed by three virally encoded proteases. Seven of the nine cleavage events are carried out by the NIa protease. Its homolog from the tobacco etch virus (TEV) is a widely used reagent for the removal of affinity tags from recombinant proteins. Although TVMV protease is a close relative of TEV protease, they exhibit distinct sequence specificities. We report here the crystal structure of a catalytically inactive mutant TVMV protease (K65A/K67A/C151A) in complex with a canonical peptide substrate (Ac-RETVRFQSD) at 1.7-Å resolution. As observed in several crystal structures of TEV protease, the C-terminus (∼20 residues) of TVMV protease is disordered. Unexpectedly, although deleting the disordered residues from TEV protease reduces its catalytic activity by ∼10-fold, an analogous truncation mutant of TVMV protease is significantly more active. Comparison of the structures of TEV and TVMV protease in complex with their respective canonical substrate peptides reveals that the S3 and S4 pockets are mainly responsible for the differing substrate specificities. The structure of TVMV protease suggests that it is less tolerant of variation at the P1′ position than TEV protease. This conjecture was confirmed experimentally by determining kinetic parameters kcat and Km for a series of oligopeptide substrates. Also, as predicted by the cocrystal structure, we confirm that substitutions in the P6 position are more readily tolerated by TVMV than TEV protease.
Keywords: tobacco vein mottling virus, tobacco etch virus, crystal structure, protease
Introduction
Tobacco vein mottling virus (TVMV) is a member of the Potyviridae family, which composes one large branch of the Picornaviridae superfamily.1 Like other positive-sense, single-stranded RNA viruses, the TVMV genome is initially translated into a single large polyprotein that is subsequently processed into individual proteins by three viral proteases: protein 1 (P1), helper component protease (HC-Pro), and nuclear inclusion-a protease (NIa-pro).2,3 Seven of the nine cleavage events are carried out by the NIa protease.4 NIa proteases adopt a chymotrypsin-like fold but use a cysteine residue instead of a serine as the active-site nucleophile in the catalytic triad.5
The potyviral TEV and rhinoviral 3C proteases are widely used as reagents for endoproteolytic removal of affinity tags from recombinant proteins because of their stringent substrate specificity. TVMV protease, a close relative of TEV protease, has also been used for this purpose.6–10 Although they share a high degree of sequence identity (52%), these two proteases have distinct substrate specificities and do not cleave each other's canonical recognition sites. Consequently, TVMV protease may be a useful alternative to TEV protease when a recombinant protein happens to contain a sequence that is similar to a TEV protease recognition site or for protein expression strategies that involve the use of more than one protease.11 Seeking to understand the structural basis for the differing sequence specificities of TEV and TVMV proteases, we have crystallized the latter enzyme in complex with a peptide substrate and determined its structure at a resolution of 1.7 Å. Several intriguing features of the cocrystal structure were investigated in greater detail by characterizing a mutant form of TVMV protease and variants of a canonical TVMV oligopeptide substrate.
Results and Discussion
Crystallization and structure determination
Wild-type and mutant forms of the TVMV protease catalytic domain were overproduced in Escherichia coli and purified as described.9 To cocrystallize the enzyme with a canonical peptide substrate, a catalytically inactive mutant was constructed by replacing the nucleophilic active-site cysteine with an alanine (C151A). However, no crystals of this mutant protease were ever obtained. Therefore, we used the technique of surface entropy reduction mutagenesis12; two lysine residues were replaced by alanines to create the triple mutant K65A/K67A/C151A. These additional mutations were selected by examining a homology model of the TVMV protease structure that was derived from the structure of TEV protease.10 Additionally, the C-terminus of the TVMV mutant was trimmed by six residues to remove the P6–P1 sites of the natural polyprotein processing site.
The purified inactive triple mutant TVMV protease (K65A/K67A/C151A) was mixed with a fivefold molar excess of peptide substrate (Ac-RETVRFQSD) before crystallization trials. The crystal used for data collection grown from a solution consisting of 0.2M potassium formate and 20% PEG 3350, belongs to space group P212121 and contains two monomers per asymmetric unit. The structure was solved by molecular replacement, using the crystal structure of TEV protease (Protein Data Bank (PDB) code: 1Q31) as a search model. The final model was refined to a resolution of 1.7 Å with an Rwork of 17.5% and an Rfree of 21.0%. It is noteworthy that, as is frequently the case when surface entropy reduction mutants are crystallized,12 the K65A and K67A mutations in TVMV protease are located at an interface between two symmetry-related molecules in the crystal lattice.
Overall structure of TVMV protease and comparison with TEV protease
As expected, TVMV protease adopts a typical chymotrypsin-like fold, which consists of two β-barrel domains that pack together to form a shallow peptide-binding cleft with the catalytic triad residues His46, Asp81, and Cys151 located at the interface [Fig. 1(A)]. The two molecules in the asymmetric unit form a dimer that bears a superficial resemblance to the one observed in structure of the S219D mutant of TEV protease14 [Fig. 1(B)]. However, neither TVMV nor TEV protease has been reported to form dimers in solution, suggesting that the intermolecular interactions observed in the crystals are purely the result of crystal packing.
Figure 1.
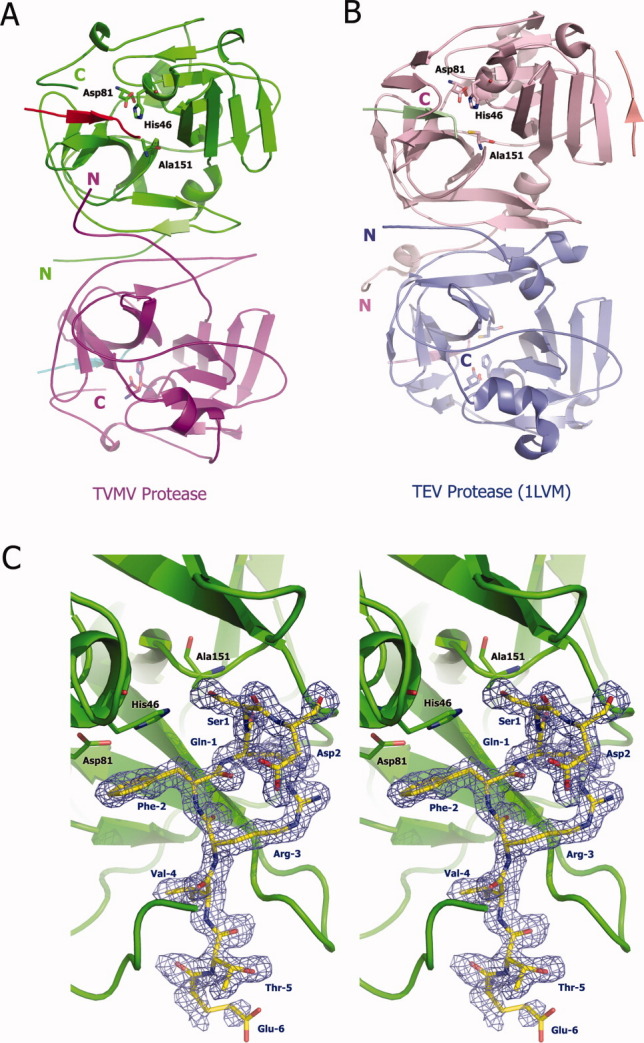
Three-dimensional structure of TVMV protease. (A) Overall structure of TVMV protease in one asymmetric unit. Chain A (1–217, purple) and Chain B (3–216, green) bound to peptide substrates Chain C (residues −6 to 1, cyan) and Chain D (residues −6 to 2, red), respectively. The N- and C-termini are labeled with the letters N and C. The catalytic triad residues are shown in ball-and-stick representation. (B) Crystal structure of TEV protease (PDB ID: 1LVM) viewed from the same perspective. Chains A to E are colored in pink, blue, green, pale pink, and salmon, respectively. (C) Stereoview of the peptide substrate (Chain D, yellow) bound to inactive TVMV protease (Chain B, green). The catalytic triad residues [H46, D81, and A151 (normally C151 in wild-type TVMV protease)] are shown in ball-and-stick representation. The peptide substrate is also displayed in a ball-and-stick format and covered by an omit map contoured at 1.0σ. The composite omit map was calculated at 1.7 Å resolution by CNS,13 with an omit rations of 7.5%.
The two TVMV protease molecules in the asymmetric unit are quite similar, with an overall RMSD of 1.36 Å. The principal differences are located in four loops, which occur between β1/β2, 310-helix-A/β4, β5/310-helix-B, and β8/β9. None of these loops are close to the active site of the enzyme. In molecule A, the electron density for the bound substrate (Chain C, Ac-RETVRFQSD) is well defined except for the N-terminal Arg and C-terminal Asp residues. In molecule B, on the other hand, the C-terminal Asp residue of the substrate is clearly visible in the electron density map [Fig. 1(C)]. In both protease molecules, the peptide substrates are bound in an extended conformation in the active site.
TVMV protease shares 52% amino acid sequence identity with TEV protease. Consequently, as one might expect, the two proteases exhibit a great deal of structural similarity with an overall RMSD of 2.96 Å for 209 Cα atoms (Fig. 2). The catalytic triad residues align very well, but as a result of substantive structural differences in the substrate-binding sites of the two enzymes, the backbone of the bound peptide in TVMV protease is shifted about 1 Å from the position that the corresponding peptide occupies in TEV protease [Fig. 2(A)]. The major differences between the two proteases are located in some of the loops, such as the loop between strands β8 and β9 and especially the loop between β12 and β13, which is involved in the formation of the substrate-binding pocket. In particular, the conformations of the C-termini of the two proteases are extremely different. In TEV protease, the two C-terminal β-strands (β15 and β16) form a small antiparallel β-sheet. By contrast, the corresponding region of TVMV protease forms a 310-helix (helix C) and a short loop.
Figure 2.
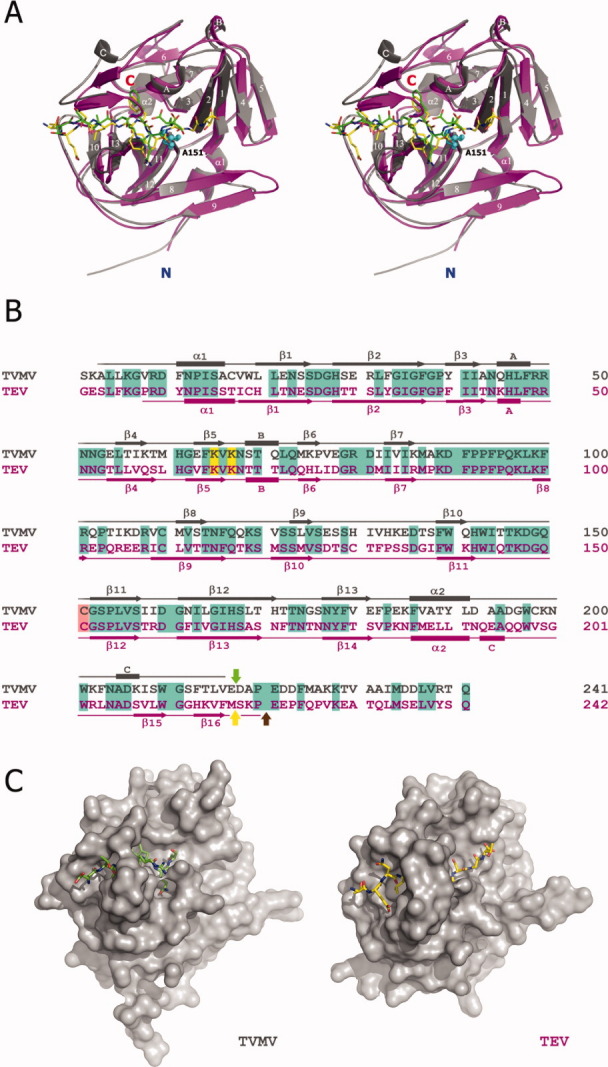
Comparison between TVMV and TEV proteases. (A) Stereoview of superposition of the crystal structures of TVMV (Chain B, gray) and TEV (PDB ID: 1LVB, Chain A, magenta) proteases with their peptide substrates. Secondary structure elements are labeled according to the TVMV structure. Canonical peptide substrates bound to TVMV and TEV proteases are shown in green and yellow, respectively. The N- and C-termini of the proteases are labeled in blue and red, respectively. The side chains of the mutated catalytic residue (C151A in TVMV protease) from both structures are shown as cyan spheres. (B) Sequence alignment of TVMV (gray) and TEV (magenta) proteases. The secondary structures were determined by the results from iMolTalk server (http://i.moltalk.org/). β-strands and α-helices are numbered and 310-helices are labeled A–C. Identical residues are shaded in cyan. Two surface entropy reduction mutation sites (K65&K67) are shaded in yellow. The active-site cysteine residues are shaded in salmon. The self-cleavage site within TEV protease is indicated by the yellow arrow. The C-termini of available crystal structure models of TEV protease (PDB ID: 1LVB and 1LVM) are denoted by the brown arrow. The C-terminus of the truncated TVMV protease (TVMV1–217 protease) investigated in this study is indicated by the green arrow. (C) Surface representation of TVMV (left) and TEV (right) proteases viewed at the same angle.
Enzymatic activity of TVMVWT and TVMV1-217
TEV protease readily undergoes autolysis at a specific site near its C-terminus, giving rise to a truncated enzyme with greatly reduced activity.15,16 Curiously, although they clearly influence the enzymatic activity of the protease, the C-terminal residues that are removed by this truncation are not visible in electron density maps of the full-length enzyme in complex with a peptide substrate or product.14 The closely related TVMV protease does not undergo autolysis.7,9 Even so, those TVMV residues corresponding to the disordered C-terminal residues of TEV protease are also disordered in the cocrystal structure of the TVMV/substrate complex.
To ascertain whether there is any difference between the enzymatic activity of wild-type (TVMVWT) and truncated TVMV proteases, the latter protein (TVMV1–217) was overproduced and purified in the same manner as the other forms of TVMV protease. Kinetic parameters Km and kcat were determined for the full-length and truncated TVMV enzymes using an oligopeptide substrate (Table I). Surprisingly, the truncated TVMV enzyme was substantially more active than the analogous TEV protease construct, exhibiting nearly as much activity as the full-length enzyme. These results were confirmed by conducting assays with an MBP-NusG fusion protein substrate9 in vitro (data not shown).
Table I.
Kinetic Parameters for the Wild-Type (TVMVWT) and Truncated (TVMV1–217) Proteases with the Peptide Substrate TETVRFQSGTRR-NH2
| Enzyme | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | rel. kcat/Km |
|---|---|---|---|---|
| TVMVWT | 0.082 ± 0.020 | 0.092 ± 0.010 | 1.12 ± 0.30 | 1.00 |
| TVMV1–217 | 0.108 ± 0.016 | 0.094 ± 0.006 | 0.87 ± 0.14 | 0.78 |
Structural determinants of substrate specificity
Previous studies have shown that seven residues surrounding their cleavage sites (positions P6–P1′) comprise the specificity determinants for TEV and TVMV proteases.8,17 The most efficient substrates for TEV and TVMV proteases are ENLYFQS and ETVRFQS, respectively.8,17 The availability of crystal structures of both proteases in complex with oligopeptide substrates containing their optimal (canonical) recognition sequences provides substantial insight into the structural determinants of substrate specificity for both enzymes.
The P6 glutamic acid is an important specificity determinant for TEV protease.10,18 As shown in Figure 3(A,B), P6 Glu interacts with the loop between β12 and β13A in TEV protease, whereas the side chain of P6 Glu rotates 90° and points into the solvent in the TVMV protease/peptide cocrystal structure. As shown in Figure 3(C), the only interaction made by the side chain of P6 Glu in the TVMV/peptide structure is a hydrogen bond between its O-ɛ1 atom and the O-γ atom of P5 Thr. Protease residues make only main-chain contacts with P6 Glu, which should have little or no impact on sequence specificity. In TEV protease, the interactions with P6 Glu are far more extensive. As shown in Figure 3(D), atom O-ɛ1 and atom O-ɛ2 of P6 Glu are within hydrogen-bonding distance of N-δ2 of Asn171, N-δ2 of Asn176, and O-η of Tyr178. In addition, its main-chain O atom makes a hydrogen bond with N-δ1 of His214. These observations suggest that the P6 position is a more important specificity determinant for TEV protease than TVMV protease. Consistent with this notion, the P6 position in all of the naturally occurring TEV polyprotein processing sites is occupied by glutamic acid, whereas variations occur at the P6 sites in the TVMV polyprotein.10
Figure 3.
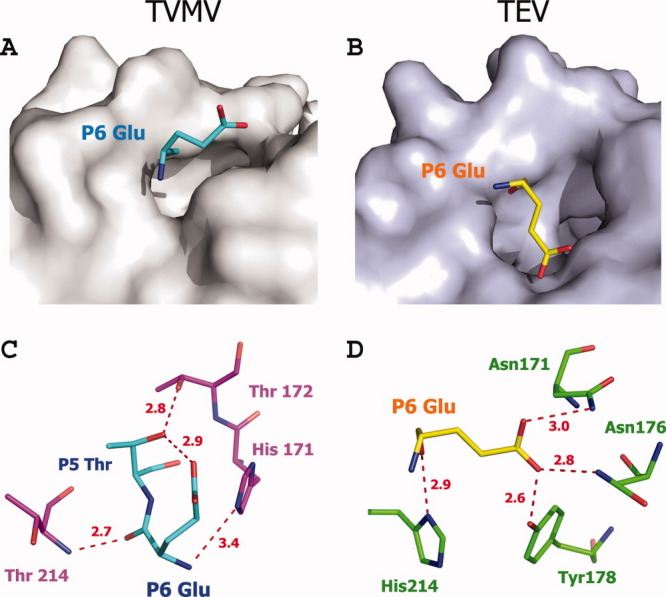
Comparison between the P6 positions of TVMV and TEV protease (PDB ID: 1LVB, Chain A) substrates. (A, B) P6 Glu binds to the surface of its corresponding protease, viewed at the same angle. (C, D) Hydrogen-bond interactions between P6 Glu and TVMV protease (C) and TEV protease (D). Residues are shown in ball-and-stick representation. The hydrogen bonds are shown as dashed lines colored red. Residues from TVMV and TEV protease are colored in purple and green, respectively. The P6 substrate residues for TVMV and TEV proteases are colored cyan and yellow, respectively.
The side chains of P5 Thr in the TVMV peptide and P5 Asn in TEV peptide both project away from the proteins into solvent. Consequently, consistent with the high diversity of residue types that occur at the P5 positions of the natural polyprotein processing sites,8 this position should not be a significant specificity determinant for either enzyme. Furthermore, biochemical studies have shown that almost any residue can occupy the P5 position with little or no impact on the cleavage efficiency of TEV protease.19
The S4 pockets are hydrophobic in both TVMV and TEV proteases. However, the S4 pocket of TVMV protease is shallower than that of TEV protease. The Van der Waals cavity volumes of the S4 pockets in TVMV protease and TEV protease are 137 and 241 Å3, respectively, calculated using VOIDOO20 with a probe radius of 1.4 Å. This may explain why the P4 position is invariably occupied by valine in the natural TVMV processing sites whereas, although leucine is the residue most frequently found at this position in TEV processing sites, valine and isoleucine also occur in some cases.21 Additionally, experiments conducted with oligopeptide substrates in vitro demonstrated that when the P4 valine in an otherwise optimal substrate for TVMV protease was replaced by leucine, it can no longer be cleaved by the enzyme.10 This is understandable because the much smaller S4 pocket of TVMV protease cannot accommodate the larger side chain of leucine. On the other hand, when the P4 leucine is replaced with valine in the TEV protease substrate, the cleavage efficiency is dramatically decreased to only 2% of the canonical substrate,10 indicating TEV protease strongly prefers leucine instead of valine in the P4 position because of its larger S4 pocket. Smaller residues in this position, like alanine, result in even less efficient cleavage.10
Unlike TEV protease, TVMV protease has no S3 pocket [Fig. 4(A–D)]. Rather, the side chain of P3 Arg in the TVMV protease substrate is fully exposed to solvent. Nevertheless, it forms two salt bridges with the side chain of Asp148. The precise geometry required to achieve these strong ionic interactions may be the reason why arginine is found in the P3 positions of all natural TVMV polyprotein processing sites except one, which contains a lysine residue instead. Recognition of the P3 Tyr by TEV protease is totally different and has been described in detail.14 Consequently, the S3/P3 interactions appear to be major discriminators of specificity by the two proteases.
Figure 4.
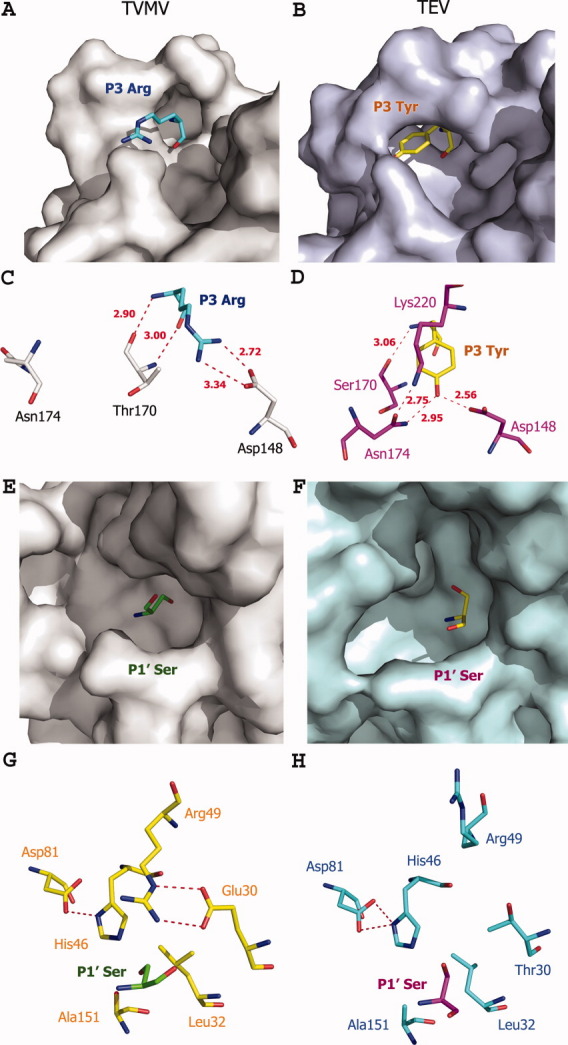
Comparison of the S1′ and S3 subsites in TVMV and TEV proteases. Left and right panels refer to the TVMV and TEV protease, respectively. (A, B) Surface representation of the S3 subsites. P3 residues are colored cyan (TVMV) and yellow (TEV). (C, D) Hydrogen-bond interaction at the S3 subsites. Residues from TVMV and TEV protease are colored gray and magenta, respectively. (E, F) Surface representation of the S1′ pockets. P1′ Ser residues of the peptide substrates are colored green (TVMV) and yellow (TEV). (G, H) Hydrogen-bond interactions in the S1′ pockets. Residues are shown in ball-and-stick representation. The hydrogen bonds are shown as dashed lines colored red. Residues from TVMV and TEV proteases are colored yellow and cyan, respectively. P1′ Ser residues of the peptide substrates are colored in green (TVMV) and magenta (TEV).
The S2 pockets in both TVMV and TEV protease are very hydrophobic, except that in TEV protease, it is more closed from the exposure to solvent. In TEV protease this pocket is formed by four hydrophobic residues (Val 209, Trp 211, Val 216, and Met218) and a face of His46. However, in TVMV protease, because of the different conformation of its C-terminus, the S2 pocket is not covered by a β-strand (β16) as it is in TEV protease, leaving the pocket partially open. In addition to three conserved residues (Trp210, Leu215, and His46), Leu169 from β12 and Phe203 from the loop between α2 and 310-helix C form the S2 pocket in TVMV protease.
The P1 sites in both TVMV and TEV protease substrates are occupied by glutamine. The hydrogen bonds between atoms N-ɛ2 and O-ɛ1 of P1 Gln and the side chains of His167 and Thr146 are conserved in the two proteases. However, there are also some differences between the conformations of the S1 pockets. As shown in Figure 2(C), the S1 pocket in TVMV protease is partially open, whereas in TEV protease this pocket is fully closed with the P1 residue of the substrate buried inside. The hydrogen-bond network among the main-chain and side-chains atoms of Asp148, Ser170, Asn174, Lys220, and P3 Tyr covers the side chain of P1 Gln in TEV protease. This also explains why in the TEV NIa-Pro carboxy cleavage site, the aliphatic P1 Met is also acceptable. In TVMV protease, this hydrogen-bonding interaction no longer exists because of the movement of Asn174 from the loop between strands β12/β13 and the missing counterpart of Lys220 from its flexible C-terminus.
Unlike the S1′ pocket of TEV protease, which is a shallow and narrow groove on its surface,14 the S1′ pocket of TVMV protease is round and small with the side chain of the P1′ serine residue pointing into its inner surface [Fig. 4(E–H)]. The P1′ positions of both TEV and TVMV polyprotein processing sites are typically occupied by Ser, Gly, or Ala. However, experimental data indicated that TEV protease exhibits a wide tolerance for variation in this position, including Met and Cys residues.22 The fact that TVMV protease has a completely different S1′ pocket raises the question of its sensitivity to variation in the P1′ position. As shown in Figure 4(C), it is the side chains of Arg49 and Glu30 that form the lid of the S1′ pocket of TVMV protease. Further, this conformation is maintained through hydrogen bonds involving N-η1 of Arg49 and O-ɛ2 of Glu30, N-ɛ of Arg49, and O-ɛ1 of Glu30. Although Arg49 is conserved between TEV and TVMV proteases, the hydrogen-bonding partner Glu30 in this interaction is not. The equivalent residue in TEV protease is a threonine. Although threonine is also a hydrophilic residue with a hydroxyl group that might contribute to a hydrogen-bonding interaction, its comparatively shorter side chain eliminates the possibility in this case. Instead, Arg49 in TVMV protease points away from the molecule into the solvent and forms a hydrogen bond with the side chain of Asp207 from the loop between 310-helix C and β-15 [Fig. 2(B)]. It is interesting to note that the conserved equivalent residue to Asp207 in TVMV protease and Asp206 in TEV protease actually is located in the 310-helix C with its Cα atom located 11.3 Å away from that of Asp207 in TVMV protease. The different conformations of their C-termini are the main reason that TVMV and TEV proteases possess such distinct S1′ pockets.
Experimental tests of insights derived from the structure of TVMV protease
Comparison of the structures of the two enzyme/substrate complexes led to a number of insights into the structural basis for their distinct sequence specificities, as described above. Among these, two conjectures were selected to be tested experimentally: (1) that the P6 specificity of TEV protease should be more stringent than that of TVMV protease, and (2) that the P1′ specificity of TVMV protease should be more stringent than that of TEV protease. To this end, a series of oligopeptide substrates with variations in the P6 or P1′ sites were prepared, and the kinetic parameters Km and kcat were determined for reactions with TVMV and TEV proteases. The results are presented in Table II. As predicted, the kinetic data confirm that TVMV protease is much less tolerant than TEV protease of amino acid substitutions in the P1′ position of its substrates. Hence, relaxed specificity in the S1′pocket is not a general feature of potyvirus proteases, but a fortuitous property of TEV protease. Also as predicted, substitutions in the P6 position are more readily tolerated by TVMV than TEV protease. Hence, these results are consistent with the cocrystal structure of the enzyme/substrate complex.
Table II.
Comparison of the P6 and P1′ Specificity of TEV and TVMV Proteases
| Enzyme | Substrate | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | rel. kcat/Km |
|---|---|---|---|---|---|
| TVMV protease | TETVRFQ↓SGTRR | 0.082 ± 0.020 | 0.092 ± 0.010 | 1.12 ± 0.30 | 1.00 |
| TETVRFQ↓AGTRR | 0.460 ± 0.087 | 0.112 ± 0.015 | 0.24 ± 0.06 | 0.22 | |
| TETVRFQ↓MGTRR | 0.671 ± 0.100 | 0.037 ± 0.003 | 0.06 ± 0.01 | 0.05 | |
| TETVRFQ↓YGTRR | 0.425 ± 0.087 | 0.062 ± 0.008 | 0.15 ± 0.04 | 0.13 | |
| TETVRFQ↓HGTRR | 0.372 ± 0.074 | 0.012 ± 0.001 | 0.03 ± 0.01 | 0.03 | |
| TATVRFQ↓SGTRR | 0.244 ± 0.038 | 0.032 ± 0.002 | 0.13 ± 0.02 | 0.12 | |
| TNTVRFQ↓SGTRR | 0.142 ± 0.035 | 0.074 ± 0.008 | 0.52 ± 0.14 | 0.46 | |
| TQTVRFQ↓SGTRR | 0.169 ± 0.034 | 0.076 ± 0.008 | 0.45 ± 0.10 | 0.40 | |
| TEV protease (S219V) | TENLYFQ↓SGTRR | 0.093 ± 0.009 | 0.345 ± 0.009 | 3.71 ± 0.37 | 1.00 |
| TENLYFQ↓AGTRR | 0.109 ± 0.011 | 0.367 ± 0.016 | 3.36 ± 0.38 | 0.91 | |
| TENLYFQ↓MGTRR | 0.088 ± 0.008 | 0.198 ± 0.006 | 2.25 ± 0.22 | 0.61 | |
| TENLYFQ↓YGTRR | 0.060 ± 0.009 | 0.062 ± 0.004 | 1.03 ± 0.17 | 0.28 | |
| TENLYFQ↓HGTRR | 0.215 ± 0.056 | 0.300 ± 0.030 | 1.40 ± 0.38 | 0.31 | |
| TANLYFQ↓SGTRR | >1.0 | N.D. | 0.15 ± 0.01 | 0.04 | |
| TNNLYFQ↓SGTRR | 0.648 ± 0.130 | 0.244 ± 0.023 | 0.38 ± 0.08 | 0.10 | |
| TQNLYFQ↓SGTRR | 0.535 ± 0.090 | 0.109 ± 0.011 | 0.20 ± 0.04 | 0.05 |
ND, not determined.
The relative specificity constants are given as values relative to that obtained with the respective unmodified substrate of the proteases. Substituted residues in the respective TEV and TVMV cleavage sites are in bold. Cleavage site is indicated in arrow.
Conclusions
Although it is a common practice to analyze and compare substrate specificity determinants of related proteases in a pocket-by-pocket and residue-by-residue manner, one must bear in mind that the substrate-binding site of TVMV protease is not, strictly speaking, composed of independent binding pockets for each residue. Rather, the properties of one pocket may very well influence those of the adjacent ones. Nevertheless, the structure of TVMV protease in complex with its canonical substrate peptide has provided considerable insight into the structural basis of its substrate specificity, leading us to conclude that the S3 and S4 pockets appear to function together as the main specificity determinants. Although structurally similar to TEV protease, TVMV protease has a distinct sequence specificity. In some cases, it may be advantageous to use more than one protease during the production of a recombinant protein or domain. For example, the Midwest Center for Structural Genomics has developed a strategy for protein expression and purification in E. coli that uses both TEV and TVMV proteases.23 Using this approach, they successfully “rescued” several targets that proved very difficult to purify with the conventional single protease method.
Materials and Methods
Protein expression and purification
The catalytically inactive C151A TVMV protease mutant was constructed by polymerase chain reaction (PCR) amplification of the open reading frame (ORF) from pRK10359 (http://www.addgene.org, plasmid #8832) using the forward primer PE-1449 (5′-GAG AAC CTG TAC TTC CAG TCT AAA GCT TTG CTG AAG GGC GTG-3′), which annealed to the 5′end of the TVMV ORF and added an in-frame tobacco etch virus (TEV) protease recognition site upstream, of the ORF, and the reverse primer PE-639 (5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT AGT CCA TGA TGG CGG CAA CAG-3′), which annealed to the 3′ end of TVMV ORF and added an attB2 Gateway recombination site (Invitrogen, Carlsbad, CA) to the end of the amplicon. An attB1 Gateway recombination site was subsequently added to the 5′ end of the resulting PCR amplicon in a second round of PCR, using the attB1-TEV primer PE-27724 and the same reverse primer. The final PCR product was recombined into pDONR201 via the Gateway BP reaction to generate pKP1485, and the DNA sequence was confirmed. The C151A mutation was subsequently introduced by QuikChange mutagenesis (Stratagene, La Jolla, CA). The C151A mutant TVMV protease ORF, preceded by an in-frame TEV protease recognition site, was moved into the destination vector pDEST-HisMBP25 via the Gateway LR reaction to generate pKP1487. The surface entropy reduction mutations (K65A/K67A) were introduced into pKP1487 by overlap extension PCR26 to create pBA1675.
The expression vector for C-terminally truncated TVMV protease (TVMV1–217) was constructed by (PCR) amplification of the ORF from pRK1035 using the forward primer PE-727 (5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GGA AAC CGT GCG TTT CCA GTC TC-3′), which annealed to the 5' end of the TVMV ORF and the upstream TVMV protease recognition site while adding an attB1 recombination site to this end of the amplicon, and the reverse primer PE-2070 (5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT ATT CAA CCA GGG TAA AGG AAC-3′), which introduced a termination codon after residue 217 of the TVMV ORF while adding an attB2 Gateway recombination site to the other end of the amplicon. The PCR product was recombined into pDONR201 via the Gateway BP reaction to generate pPS2021, and the DNA sequence was confirmed. The truncated TVMV protease ORF, now preceded by an in-frame TVMV protease recognition site, was moved into the destination vector pDEST-HisMBP25 via the Gateway LR reaction to generate pPS2022. The construction of vectors for the production of full-length TEV (pRK793) and TVMV (pRK1035) proteases has been described previously.9,16
All recombinant proteins were expressed in E. coli BL21(DE3) CodonPlus-RIL cells (Stratagene, La Jolla, CA), which were induced at mid-log phase with 1 mM IPTG for 4 h at 30°C. The cells were harvested by centrifugation at 4°C and frozen at −80°C until use. Purification of full-length TEV and TVMV proteases was carried out as described.9,27 Truncated TVMV protease (TVMV1–217) was purified in the same manner as full-length TVMV protease.9 The catalytically inactive mutant TVMV protease (K65A/K67A/C151A) was purified as follows. The cell pellet was resuspended in 50 mM sodium phosphate, pH 8.0, 150 mM NaCl, and 25 mM imidazole, and the cells were disrupted using a APV Model G1000 homogenizer (Invensys, Roholmsvej, Denmark). The lysate was centrifuged at 15,000 rpm at 4°C using an SA-600 rotor, filtered, and then the HisMBP-TVMV fusion protein was purified by immobilized metal affinity chromatography (IMAC) as described.25 Fractions containing the fusion protein were pooled, cleaved overnight with hexahisidine-tagged TEV protease,27 and then subjected to another round of IMAC as described.25 The flow-through fractions were pooled and concentrated to 5 mL, using an Amicon stirred cell with a YM10 membrane (Millipore, Billerica, MA) and applied to a 26/60 Superdex-75 preparative size exclusion column (GE Healthcare, Piscataway, NJ) equilibrated in 25 mM Tris, pH 7.5, and 2 mM tris(2-carboxyethyl) phosphine hydrochloride, and the peak fractions corresponding to TVMV protease were pooled and concentrated to ∼15 mg/mL with an Amicon stirred cell (Millipore) as above.
Crystallization and data collection
The inactive enzyme–substrate complex was prepared by mixing the protein solution (10 mg/mL) with a fivefold molar excess of the peptide substrate (Ac-RETVRFQSD). The complex was then subjected to crystallization trials with various kits from Hampton Research, Qiagen, and Emerald Biosystems. The Hydra II Plus One crystallization robot (Matrix Technologies, Hudson, NH) was used to setup the screens in a sitting drop vapor diffusion format at 18°C. The crystal used for data collection was grown from a solution consisting of 0.2M potassium formate, 20% PEG 3350 with a ratio of protein to reservoir solution of 1:3. Crystals of mutant 1 (K65A/K67A/C151A) appeared within 4 days. The crystals belong to the space group P212121 and contain two monomers per asymmetric unit. The solvent content of the crystal was estimated to be ∼43.4% (v/v) with a Matthews coefficient (VM) of 2.13 Å3 Da−1 for two monomers in the asymmetric unit. The unit cell has dimensions of a = 76.5 Å, b = 77.6 Å, and c = 78.5 Å. Crystals were equilibrated in a cryoprotectant composed of reservoir buffer plus 5% (v/v) ethylene glycerol and were flash frozen in a cold nitrogen stream at −170°C. One dataset was collected at the SER-CAT beamline 22-ID (Advanced Photon Source, Argonne National Laboratory). Data were processed and scaled using the HKL2000 program suite.28 Data collection and processing statistics are summarized in Table III.
Table III.
Summary of the TVMV Protease Crystallographic Data
| Parameter | TVMV |
|---|---|
| Data collection | |
| Space group | P212121 |
| Unit cell dimensions | |
| a, b, c (Å) | 76.5, 77.6, 78.5 |
| α, β, γ (°) | 90, 90, 90 |
| Protein molecules/A.U. | 2 |
| Wavelength (Å) | 0.97939 |
| Data processing | |
| Resolution range (Å)a | 35–1.7 (1.76–1.70) |
| No. of reflections | 50064 |
| Rmergea | 0.08 (0.431) |
| I/σIa | 17.4 (2.7) |
| Completeness (%)a | 96.1 (92.7) |
| Redundancya | 5.2 (3.4) |
| Refinement statistics | |
| Resolution range (Å) | 35–1.7 |
| Rwork/Rfree (%) | 17.5/21.0 |
| No. atoms/molecules | |
| Protein | 3591 |
| Water | 553 |
| Formic acid | 1 |
| Average B-factor (Å2) | |
| Protein | 17.4 |
| Water | 32.3 |
| Formic acid | 36.5 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.33 |
| Ramachandran plot statistics | |
| Residues in most favored regions | 351 |
| Residues in additional allowed regions | 38 |
| Residues in generously allowed regions | 1 |
| Residues in disallowed regions | 0 |
A.U., asymmetric unit.
Values in parentheses refer to the highest resolution shell.
Structure determination and refinement
The structure of TVMV protease mutant (K65A/K67A/C151A) in complex with the peptide substrate was solved by molecular replacement method using the monomer of TEV protease (PDB code: 1Q3129) as the search model and the program MOLREP.30 The substrate peptide and the loops were deleted from the search model. Five percent of the reflections were set aside for cross validation (Rfree). After initial rigid body refinement, the molecular replacement solution yielded an Rfactor of 0.45 and Rfree of 0.48. The resulting model was manually corrected and finished in O.31 Refinement was carried out with REFMAC5.32 The final model consists of residues 1–217 of Chain A, 3–216 of Chain B, 2–8 of Chain C, and 2–9 of Chain D. In addition, one molecule of formic acid and 553 water molecules were located during structure refinement and included in the model. Model quality was assessed with PROCHECK33 and MolProbity.34 All nonglycine residues reside either in the most favorable or in the allowed regions of the Ramachandran plot. Model refinement statistics are listed in Table III. The atomic coordinates and structure factors for the TVMV protease/peptide structure have been deposited in the PDB35 with accession code 3MMG. All figures were generated by the graphics program PyMOL.36
Enzyme kinetics
Enzyme assays were initiated by mixing 20 μL of protease (50–5700 nM) in 50 mM sodium phosphate (pH 7.0), 5 mM dithiothreitol, 800 mM NaCl, 10% glycerol, with 20 μL of substrate (0.04–1.1 mM; the actual range was selected on the basis of approximate Km values) in the same buffer. The enzyme concentrations were determined by amino acid analysis. Measurements were performed at six different substrate concentrations. The reaction mixtures were incubated at 30°C for 30 min and then stopped by the addition of 160 μL 4.5M guanidine hydrochloride containing 1% trifluoroacetic acid. Aliquots were injected on to a Nova-Pak C18 reversed-phase chromatography column (3.9 mm × 150 mm; Waters Corporation, Milford, MA) using an automatic injector. Substrates and the cleavage products were separated using an increasing water/acetonitrile gradient (0–100%) in the presence of 0.05% trifluoroacetic acid. To determine the correlation between peak areas of the cleavage products and their amount, fractions were collected and analyzed by amino acid analysis. The kcat values were calculated by assuming 100% activity for the enzyme. Kinetic parameters were determined by fitting the data obtained at less than 20% substrate hydrolysis to the Michaelis–Menten equation by using the FIG P program (Fig. P Software Corp., Durham, NC). The standard deviations for the kcat/Km values were calculated as described.37 If no saturation was obtained in the studied concentration range, the kcat/Km value was determined from the linear part of the rate versus concentration profile. Because of slight variations in the activity of different protease preparations, all measurements were performed with the same batch of enzymes.
Acknowledgments
The authors thank Scott Cherry and Kerri Penrose for technical assistance. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31–109-Eng-38. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Ryan MD, Flint M. Virus-encoded proteinases of the picornavirus super-group. J Gen Virol. 1997;78(Part 4):699–723. doi: 10.1099/0022-1317-78-4-699. [DOI] [PubMed] [Google Scholar]
- 2.Stanway G. Structure, function and evolution of picornaviruses. J Gen Virol. 1990;71(Part 11):2483–2501. doi: 10.1099/0022-1317-71-11-2483. [DOI] [PubMed] [Google Scholar]
- 3.Domier LL, Franklin KM, Shahabuddin M, Hellmann GM, Overmeyer JH, Hiremath ST, Siaw MF, Lomonossoff GP, Shaw JG, Rhoads RE. The nucleotide sequence of tobacco vein mottling virus RNA. Nucleic Acids Res. 1986;14:5417–5430. doi: 10.1093/nar/14.13.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellmann GM, Shaw JG, Rhoads RE. In vitro analysis of tobacco vein mottling virus NIa cistron: evidence for a virus-encoded protease. Virology. 1988;163:554–562. doi: 10.1016/0042-6822(88)90296-6. [DOI] [PubMed] [Google Scholar]
- 5.Seipelt J, Guarne A, Bergmann E, James M, Sommergruber W, Fita I, Skern T. The structures of picornaviral proteinases. Virus Res. 1999;62:159–168. doi: 10.1016/s0168-1702(99)00043-x. [DOI] [PubMed] [Google Scholar]
- 6.Hwang DC, Kim DH, Kang BH, Song BD, Choi KY. Molecular cloning, expression, and purification of nuclear inclusion A protease from tobacco vein mottling virus. Mol Cells. 2000;10:148–155. doi: 10.1007/s10059-000-0148-8. [DOI] [PubMed] [Google Scholar]
- 7.Hwang DC, Kim DH, Lee JS, Kang BH, Han J, Kim W, Song BD, Choi KY. Characterization of active-site residues of the NIa protease from tobacco vein mottling virus. Mol Cells. 2000;10:505–511. doi: 10.1007/s10059-000-0505-7. [DOI] [PubMed] [Google Scholar]
- 8.Yoon HY, Hwang DC, Choi KY, Song BD. Proteolytic processing of oligopeptides containing the target sequences by the recombinant tobacco vein mottling virus NIa proteinase. Mol Cells. 2000;10:213–219. doi: 10.1007/s10059-000-0213-3. [DOI] [PubMed] [Google Scholar]
- 9.Nallamsetty S, Kapust RB, Tozser J, Cherry S, Tropea JE, Copeland TD, Waugh DS. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr Purif. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 10.Tozser J, Tropea JE, Cherry S, Bagossi P, Copeland TD, Wlodawer A, Waugh DS. Comparison of the substrate specificity of two potyvirus proteases. FEBS J. 2005;272:514–523. doi: 10.1111/j.1742-4658.2004.04493.x. [DOI] [PubMed] [Google Scholar]
- 11.Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr Purif. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper DR, Boczek T, Grelewska K, Pinkowska M, Sikorska M, Zawadzki M, Derewenda Z. Protein crystallization by surface entropy reduction: optimization of the SER strategy. Acta Crystallogr D Biol Crystallogr. 2007;63:636–645. doi: 10.1107/S0907444907010931. [DOI] [PubMed] [Google Scholar]
- 13.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 14.Phan J, Zdanov A, Evdokimov AG, Tropea JE, Peters HK, III, Kapust RB, Li M, Wlodawer A, Waugh DS. Structural basis for the substrate specificity of tobacco etch virus protease. J Biol Chem. 2002;277:50564–50572. doi: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]
- 15.Parks TD, Howard ED, Wolpert TJ, Arp DJ, Dougherty WG. Expression and purification of a recombinant tobacco etch virus NIa proteinase: biochemical analyses of the full-length and a naturally occurring truncated proteinase form. Virology. 1995;210:194–201. doi: 10.1006/viro.1995.1331. [DOI] [PubMed] [Google Scholar]
- 16.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 17.Dougherty WG, Carrington JC, Cary SM, Parks TD. Biochemical and mutational analysis of a plant virus polyprotein cleavage site. EMBO J. 1988;7:1281–1287. doi: 10.1002/j.1460-2075.1988.tb02942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrington JC, Haldeman R, Dolja VV, Restrepo-Hartwig MA. Internal cleavage and trans-proteolytic activities of the VPg-proteinase (NIa) of tobacco etch potyvirus in vivo. J Virol. 1993;67:6995–7000. doi: 10.1128/jvi.67.12.6995-7000.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dougherty WG, Cary SM, Parks TD. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology. 1989;171:356–364. doi: 10.1016/0042-6822(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 20.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 21.Carrington JC, Dougherty WG. Small nuclear inclusion protein encoded by a plant potyvirus genome is a protease. J Virol. 1987;61:2540–2548. doi: 10.1128/jvi.61.8.2540-2548.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapust RB, Tozser J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease. Biochem Biophys Res Commun. 2002;294:949–955. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 23.Eschenfeldt WH, Maltseva N, Stols L, Donnelly MI, Gu M, Nocek B, Tan K, Kim Y, Joachimiak A. Cleavable C-terminal His-tag vectors for structure determination. J Struct Funct Genomics. 2010;11:31–39. doi: 10.1007/s10969-010-9082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evdokimov AG, Tropea JE, Routzahn KM, Waugh DS. Three-dimensional structure of the type III secretion chaperone SycE from Yersinia pestis. Acta Crystallogr D Biol Crystallogr. 2002;58:398–406. doi: 10.1107/s090744490200015x. [DOI] [PubMed] [Google Scholar]
- 25.Tropea JE, Cherry S, Nallamsetty S, Bignon C, Waugh DS. A generic method for the production of recombinant proteins in Escherichia coli using a dual hexahistidine-maltose-binding protein affinity tag. Methods Mol Biol. 2007;363:1–19. doi: 10.1007/978-1-59745-209-0_1. [DOI] [PubMed] [Google Scholar]
- 26.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 27.Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Nunn CM, Jeeves M, Cliff MJ, Urquhart GT, George RR, Chao LH, Tscuchia Y, Djordjevic S. Crystal structure of tobacco etch virus protease shows the protein C terminus bound within the active site. J Mol Biol. 2005;350:145–155. doi: 10.1016/j.jmb.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Vagin AA, Isupov MN. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr D Biol Crystallogr. 2001;57:1451–1456. doi: 10.1107/s0907444901012409. [DOI] [PubMed] [Google Scholar]
- 31.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Part 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 34.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 37.Boross P, Bagossi P, Copeland TD, Oroszlan S, Louis JM, Tozser J. Effect of substrate residues on the P2' preference of retroviral proteinases. Eur J Biochem. 1999;264:921–929. doi: 10.1046/j.1432-1327.1999.00687.x. [DOI] [PubMed] [Google Scholar]