

1. Introduction
Electron transfer (ET) reactions are fundamental steps in biological redox processes. Respiration is a case in point: at least 15 ET reactions are required to take reducing equivalents from NADH, deposit them in O2, and generate the electrochemical proton gradient that drives ATP synthesis.1–10 Most of these reactions involve quantum tunneling between weakly coupled redox cofactors (ET distances >10 Å) embedded in the interiors of folded proteins. Here we review experimental findings that have shed light on the factors controlling these distant ET events. We also review work on a Re-modified copper protein photosystem in which multistep electron tunneling (hopping) through an intervening tryptophan is orders of magnitude faster than the corresponding single-step ET reaction.
If proton transfers are coupled to ET events, we refer to the processes as proton coupled ET, or PCET, a term introduced by Huynh and Meyer in 1981. 11 Here we focus on two protein redox machines, photosystem II and ribonucleotide reductase, where PCET processes involving tyrosines are believed to be critical for function. Relevant tyrosine model systems also will be discussed.
2. Electron Transfer in Proteins
A great many biological energy transduction pathways depend upon the rapid movement of electrons or holes over long distances (> 30 Å) through proteins. Many redox enzymes require the transfer of holes at high potentials, where sidechains of redox active amino acids, like tyrosine and tryptophan, can become involved. Protein structures are designed to facilitate rapid and efficient charge transport along specific pathways and prevent off-path diffusion of redox equivalents; mutations, denaturants and other disruptions of the redox pathway can hinder or curtail electron transfer.
2.1 Flash-Quench Experiments
Semiclassical ET theory provides a basic framework for understanding the specific rates of reaction between an electron donor (D) and acceptor (A) held at fixed distance and orientation (kET, Eq. 1).12–14 These rates depend on three critical parameters: (1) the driving force for the electron transfer (−ΔG°); (2) the extent of nuclear reorientation in D, A, and the solvent that accompanies formation of D+ and A− (λ); and (3) the electronic coupling between the reactants [D,A] and the products [D+, A−] at
| (1) |
the transition state (HAB). The first two parameters depend largely on the chemical composition and environments of the redox centers, whereas the third is a function of the D-A distance and the structure of the intervening medium.12–18
Inter- and intramolecular laser flash-quench methods have been utilized to trigger ET reactions.12,19–29 These methods have been used to study the distance and medium dependences of long-range electron tunneling reactions,12,14,30–45 to trigger redox enzyme catalysis,46 and initiate multistep tunneling processes.47 In a typical reaction sequence, a laser-excited M-diimine sensitizer (*MS) directly donates (accepts) an electron to (from) a redox partner (*ET), generating the oxidized (reduced) diimine complex and the reduced (oxidized) partner (Figure 1, 1→2→3 (6→4→5)).23 The intermediate formed in this excited-state ET reaction decays in a subsequent charge-recombination reaction to regenerate the original D-A complex (3→1 (5→6)). This scheme is viable only when intramolecular ET competes effectively with excited-state deactivation (typically 100 ns — 1 μs). If this were the only technique available, the short lifetimes of the excited M-diimine complexes would limit measurements of ET rates to systems in which the M-diimine and the protein active site were well coupled.
Figure 1.

Flash-quench scheme for measuring intraprotein ET rates, and generating oxidized and reduced metal centers in proteins. MS is a metal-diimine photosensitizer; MP is the protein metal center.
Intermolecular flash-quench methods have been used to study systems in which charge separation does not compete with excited-state decay, or when the lifetime of the charge-separated state must be protracted.22–23,28 This method takes advantage of bimolecular quenching and scavenging reactions to separate photogenerated holes and electrons onto different molecules that diffuse away from one another. Once the charges are separated onto different molecules, their timescale for recombination moves into the millisecond range — a thousand-fold improvement over intramolecular charge separation. In the flash-quench procedure (Figure 1: oxidative 6→4→3→1; reductive 1→2→5→6), a quencher (Q) is added to the solution to react with *M in a bimolecular ET reaction. This quenching process generates the same intermediates as the intramolecular quenching reaction (3 or 5), but with greater efficiency. Once generated, the intermediate will react via intramolecular ET (3→1, 5→6). Then, on a much longer timescale (~ms), the reduced (oxidized) quencher will react with the oxidized (reduced) protein to regenerate the original complex (6 or 1). Even longer time windows can be examined (seconds) if irreversible redox quenchers are employed.
Flash-quench protocols have been used to measure CuI→RuIII ET (−ΔG° = 0.7 eV) in a set of RuII-modified Pseudomonas aeruginosa azurins to establish the distance dependence of ET along β-strands.14,48–49 A timetable for driving-force-optimized electron tunneling in azurin (Figure 2) reveals a nearly perfect exponential distance dependence, with a decay constant (β) of 1.1 Å−1, and an intercept at close contact (ro = 3 Å) of 1013 s−1.12,14 This decay constant is quite similar to that found for superexchange mediated tunneling across saturated alkane bridges (β ~ 1.0 Å−1),50–51 strongly indicating that a similar coupling mechanism is operative in the polypeptide. Studies have shown that Cu I to RuIII or OsIII ET rates in labeled azurin crystals are nearly identical with solution values for each donor acceptor pair.52
Figure 2.

Timetable for driving-force-optimized electron tunneling in RuII-modified proteins: azurin (red); cytochrome c (green); cytochrome b562 (yellow; myoglolbin (orange); high-potential iron protein (cyan); Zn-cytochrome c crystals (magenta). The solid lines illustrate limiting β values of 1.0 and 1.2 Å−1; the dashed line illustrates a 1.1 Å−1 distance decay.
The kinetics of ET reactions have been examined in more than 30 Ru(diimine)2+ metalloproteins.14,24–26,28,36–37,42–43,45,53–55 Driving-force-optimized tunneling times are scattered around the RuII-azurin 1.1-Å−1 exponential distance decay (Figure 2). ET rates at a single distance can differ by as much as a factor of 103, and D-A distances that differ by as much as 5 Å can produce virtually identical rates. Beratan, Onuchic, and coworkers15,56–57 developed a generalization of the McConnell superexchange coupling model58 that accounts for rate scatter attributable to protein structural complexity. In this tunneling-pathway model, the medium between D and A is decomposed to smaller subunits linked by covalent bonds, hydrogen bonds, and through-space jumps. More elaborate computational protocols also have shed light on the factors that determine distant coupling in proteins.18,59–61
2.2 Hole Hopping
Coupling-limited (activationless) ET reactions in Ru-proteins occur by single-step electron tunneling over a wide distance range (10 to 25 Å).14,48,52,62 Electron transport over very long molecular distances (> 25 Å) likely involves multistep tunneling (hopping)63–70 in which redox-active amino acid sidechains act as intermediate donors or acceptors rather than tunneling bridges.12,14,71 Our work has shown that electrons could be transported 30 Å or more in hundreds of nanoseconds if an intervening redox center (Int) with a reduction potential (E(Int+/0)) well above that of the donor (E(D+/0)) but not more than 200 mV above that of the acceptor (E(A0/−)) is placed between D and A.12,14
Employing three ReI(CO)3(dmp)(His124)-azurins (dmp = (4,7-dimethyl-1,10-phenanthroline), we have demonstrated that an intervening tryptophan can facilitate electron transfer between distant metal redox centers.47 In these ReI-azurins, a histidine is at position 124 on the β strand extending from methionine-121 and either tryptophan, tyrosine, or phenylalanine is at position 122. The X-ray crystal structure of the ReI-labeled Trp122 variant (Re(His124)+(Trp122)CuII-azurin) (Figure 3) shows that the dmp ligand and the Trp122 indole group are near van der Waals contact (~4 Å), and the Cu-Re distance is 19.4 Å.47
Figure 3.

Model of the Cu-W-Re electron-tunneling architecture from the 1.5 Å resolution x-ray crystal structure of Re(His124)+(Trp122)CuII-azurin.. the aromatic rings of the phenanthroline ligand and Trp122 slightly overlap with one methyl group projecting over the indole ring and the plane of the respective π-systems making a 20.9° angle. The average separation of atoms on the overlapped six-membered rings is 3.82 Å, whereas 4.1 Å separates the edge of the Trp122 indole and the His124 imidazole. Distances between redox centers: Cu to Trp122 aromatic centroid, 11.1 Å; Trp122 aromatic centroid to Re, 8.9 Å; Cu to Re, 19.4 Å. Reprinted with permission from Reference 47. Copyright 2008 American Association for the Advancement of Science.
Transient absorption measurements reveal rapid (<50 ns) formation of CuII following 355-nm laser excitation of Re(His124)+(Trp122)CuI-azurin (E°[Re(His124)+*/Re(His124)0] = 1.4 V vs. NHE)72 with concomitant formation of Re(His124)0; charge recombination to regenerate CuI occurs in ~3 μs (Figure 4). Importantly, CuII was not produced following excitation of either Re(His124)+(Phe122)CuI-azurin or Re(His124)+(Tyr122)CuI-azurin. Time-resolved infrared absorption (TRIR) spectra reveal bleaches at 1920 and 2030 cm−1 (ground-state C≡O absorptions) and new features at ~1960, 2012, and ~2040 cm−1, characteristic of the 3MLCT excited state; 73 in addition, peaks attributable to Re(His124)0 appear at 1888 and 2004 cm−1.74 The reduced complex also forms following excitation of Re(His124)+(Trp122)CuII-azurin and Re(His124)+(Trp122)ZnII-azurin, but not after excitation of Re(His124)+(Tyr122)ZnII-azurin. Nanosecond visible transient absorption experiments confirm that no CuII is formed upon excitation of Re(His124)+(Phe122)CuI-azurin or Re(His124)+ (Tyr122)CuI-azurin at 355 nm.
Figure 4.

Transient kinetics of Re(His124)+(Trp122)CuI-azurin. (a) Time resolved luminescence (red: λobs > 450 nm; λex = 355 nm, 10 ps pulsewidth; pH 7.2), instrument response function (blue), and fit to a three exponential kinetics model (black: τ1 = 35 ps (growth); τ2 = 363 ps (decay); τ3 = 25 ns (decay)). (b) Visible transient absorption (λobs = 632.8 (red), 500 nm (blue); λex = 355 nm, 1.5 mJ, 8 ns pulsewidth; pH 7.2). Black lines are fits to a biexponential kinetics model (τ1 = 25 ns (growth); τ2 = 3.1 μs (decay)). (c, d) TRIR spectra measured (λex = 400 nm, ~ 150 fs pulsewidth; D2O, pD = 7.0, phosphate buffer) at selected time delays after femtosecond laser excitation. Reprinted with permission from Reference 47. Copyright 2008 American Association for the Advancement of Science.
The transient spectroscopic data have been interpreted in terms of the kinetics model shown in Figure 5. Optical excitation of Re(His124)+ creates a 1MLCT excited state, which undergoes ~150 fs intersystem crossing75 to a vibrationally excited 3MLCT (*3MLCT) state. Subpicosecond generation of Re(His124)0 is attributable to ET from Trp122 to 1MLCT Re(His124)+. The source of reducing equivalents in these fast ET reactions is the indole side chain of Trp122, as Re(His124)0 was not produced in any protein containing Phe122 or Tyr122.
Figure 5.

Kinetics model of photoinduced electron transfer in Re(His124)+(Trp122)CuI-azurin. Photoexcitation produces electron (red) and hole (blue) separation in the MLCT-excited ReI complex. Hole transfer to CuI via (Trp122) •+ is complete in less than 50 ns. Charge recombination occurs on the microsecond timescale. Rate constants for elementary steps were obtained from fitting time-resolved luminescence, visible absorption, and infrared spectroscopic data. Reprinted with permission from Reference 47. Copyright 2008 American Association for the Advancement of Science.
It is striking that the oxidation of CuI in Re(His124)+(Trp122)CuI-azurin is more than two orders of magnitude faster than expected for electron tunneling over 19 Å.47 Analysis of the reaction kinetics reveals that the reduction potential of Re(His124)+* is just 28 mV greater than that of (Trp122)•+/0, but that is sufficient for very rapid (~ns) ET between closely spaced redox sites. The Trp cation radical is a relatively weak acid (pKa = 4.5(2));76–77 its deprotonation, which is energetically favorable at pH 7, likely would take a few hundred nanoseconds or longer.64 The rate of deprotonation is critical, as (Trp122)•+ can rapidly oxidize CuI in the azurin active site only if it remains protonated in the hopping intermediate.
Semiclassical ET theory was employed to generate a hopping map of driving-force effects on two-step (CuI→Int→*ML) and single-step (CuI→*ML) tunneling rates for a molecular framework analogous to that of Re(His124)+(Trp122)CuI-azurin (Figure 6).47 The map shows that the rate advantage of the multistep process is lost if the first tunneling step is too endergonic (ΔG° (Int→*ML) >200 meV).12,14 Consistent with this prediction, replacement of Trp122 by Tyr or Phe inhibits the initial ET event because the reduction potentials of their cation radicals are more than 200 mV above E°(Re(His124) +*/(Re(His124)0). Concerted oxidation and deprotonation of Tyr122 by Re(His124)+* likely would be accompanied by a significant activation barrier.
Figure 6.

Two-step hopping map for electron tunneling through ReI-modified azurin. Colored contours reflect electron-transport time scales as functions of the driving force for the first tunneling step (ordinate, Int→*ML) and the overall electron-transfer process (abscissa, CuI→*ML). The heavy black lines enclose the region in which two-step hopping is faster than single-step tunneling. The dashed black line indicates the driving force for Re(His124)+*(Trp122)CuI-azurin → Re(His124)0(Trp122) •+CuI-azurin ET; the black dot corresponds to Re(His124)+*(Trp122)CuI-azurin → Re(His124)0(Trp122) •+CuI-azurin→Re(His124)0(Trp122)CuII-azurin hopping. Reprinted with permission from Reference 47. Copyright 2008 American Association for the Advancement of Science.
Multistep electron tunneling in Re(His124)+(Trp122)CuI-azurin is reminiscent of the radical transfer reaction involved in the photactivation of DNA photolyase.64 Brettel has shown that a hole migrates over 13 Å in <10 ns from an electronically excited flavin radical cofactor (FADH•) to a solvent-exposed Trp306 residue in the E. coli enzyme. Photochemically generated (Trp306)• + deprotonates with a time constant of ~300 ns, with both water and buffer serving as proton acceptors. The mechanism of electron transport likely involves multistep tunneling via intervening Trp residues (Trp382, Trp359) separated by 4–5 Å. A key requirement for rapid hole migration to Trp306 is that Trp382 and Trp359 are protected from buffer and solvent so that hole transfer along the chain is not interrupted by deprotonation of the transient radical cation.
The effectiveness of Trp residues in mediating multistep tunneling diminishes considerably when the indole side chain is solvent exposed.78–79 Investigations by Giese and coworkers of hole transport along polyproline (PP II) helices report the extent of 20-Å radical migration in 40 ns from a photochemically generated alkoxy-aryl radical cation to a terminal Tyr residue.80–81 The introduction of potential hole-relaying amino acids near the center of the peptide was shown to affect the Tyr radical formation yield. Trp was found to be an inefficient relay residue: the 40-ns transient spectrum revealed little Tyr•, but a substantial population of deprotonated Trp radical. Prior investigations have shown that Tyr to Trp• electron transfer along polyproline peptides is sluggish (kobs ~ 2 × 104 s−1 with a single intervening proline residue).82 As expected, the rates increase by about an order of magnitude when the N-Me-Trp•+ cation radical is used in place of Trp•. Hence, it is not surprising that little Tyr radical formation is observed in 40 ns with three prolines separating Trp and Tyr. The hopping map in Figure 6 illustrates that reduced driving force for the second ET step severely limits the overall timescale for multistep tunneling. By making measurements of a single 40-ns snapshot, it is extremely difficult to evaluate the overall multistep tunneling kinetics.
3. Proton Coupled Electron Transfer (PCET)
Many electron transfer reactions are coupled to proton transfer events. Complete mechanistic descriptions of PCET processes in both proteins and model complexes are research goals in many laboratories. Our review will cover a selected set of investigations in this currently very active area at the interface of chemistry and biology.
3.1 Kinetics Modeling
Square schemes (Figure 7) help visualize different mechanisms for the transfer of an electron and a proton from one chemical species to another.83–85 In the first scheme, often referred to as collinear PCET, a proton and an electron from a single donor transfer along the same direction to a single acceptor, AH + B → A + BH.84 The two limiting stepwise mechanisms are illustrated by the perimeter of the square: either proton transfer to form an intermediate state A− + HB+, followed by electron transfer (PT-ET); or electron transfer to form AH+ + B− followed by proton transfer (ET-PT).86 Alternatively, a concerted pathway,87 referred to here as CPET, where there is no kinetic intermediate, is on the square’s diagonal. The second scheme shows pathways for proton and electron transfer to separate proton and electron acceptors, AH—B + C+ → A—HB+ + C, often referred to as orthogonal or bidirectional PCET or multiple site electron-proton transfer (MS-EPT).11,88–89 Here, similar stepwise mechanisms of proton transfer followed by electron transfer and electron transfer followed by proton transfer proceed through intermediates A−—HB+ + C+ and AH+—B + C, respectively. The CPET diagonal pathway features simultaneous electron and proton transfers to their respective acceptors. It is important to note that in the PCET descriptions above, the electrons and protons originate from different sites on the donor. In contrast, the proton and electron in a hydrogen atom transfer (HAT) reaction come from the same chemical bond.11
Figure 7.
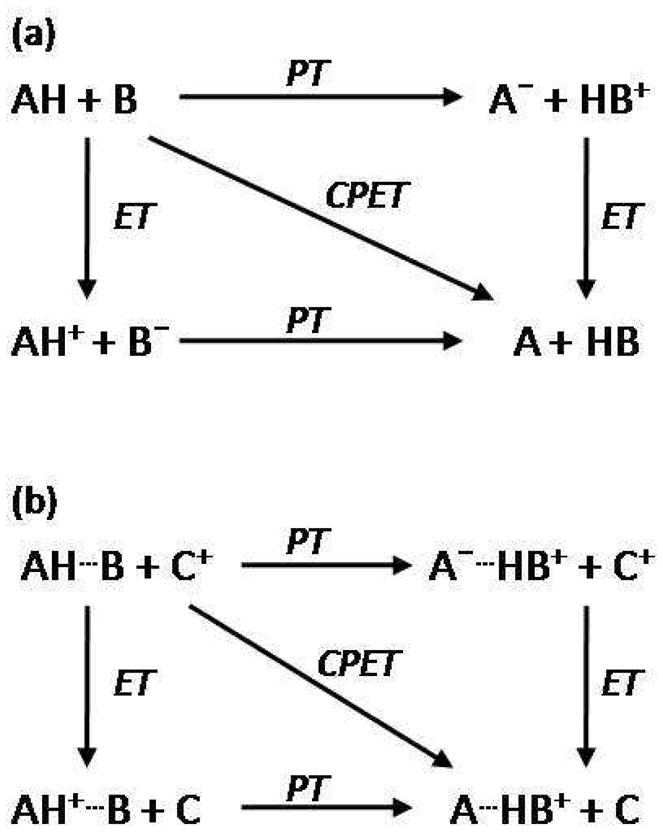
Perimeters of the square illustrate the stepwise, limiting mechanisms of sequential proton and electron transfer steps. The concerted pathway, CPET, is illustrated by the diagonal of the square. (a) Unidirectional or collinear PCET: proton and electron transfer from a single donor along the same direction to a single acceptor. (b) Orthogonal or bidirectional PCET: proton and electron transfer to separate proton and electron acceptors. Adapted from Reference 83. With kind permission from Springer Science+Business Media: Photosynthesis Research, Models for proton-coupled electron transfer in Photosystem II, 87, 2006, 1, Mayer, J. M.; Rhile, I. J.; Larsen, F. B.; Mader, E. A.; Markle, T. F.; Dipasquale, A. G., Scheme 1.
In simultaneous electron and proton transfer processes, the lifetimes of discrete proton or electron transfer intermediates must be much shorter than those for coupled vibrations (~100 fs) and solvent modes (~1 ps).11 While CPET reactions have not been fully explored experimentally, theoretical investigations by Cukier, Hammes-Schiffer, and Savéant have shed light on the coupling of electron and proton transfer events.90–103 (We will not elaborate on this work, as it is discussed in another review in this issue.) CPET is advantageous in many situations, as high energy intermediates are avoided, but proton movement is typically limited to hydrogen-bond distances, whereas electrons are able to tunnel more than 10 Å at reasonable rates.12,88,104 Longer-range proton transfer can be accomplished in CPET reactions by the introduction of a hydrogen-bond relay between the acid and the base. 104
3.2 Tyrosine PCET
The cation radical formed upon oxidation of tyrosine, here denoted TyrOH•+, is a strong acid (pKa = −2) capable of transferring a proton to a water molecule.105–106 For Tyr residues buried in protein interiors, however, solvent water molecules normally are excluded and amino-acid sidechains must serve as proton acceptors. In this context, it is of interest to note that several Tyr residues known to form radicals during enzymatic turnover are hydrogen bonded to potential proton acceptors such as histidine, aspartic acid, glutamic acid, and lysine. Examples include TyrZ in Photosystem II, Tyr122 in E. coli ribonucleotide reductase,63,107–108 Tyr385 in prostaglandin-H synthase-2,109–110 and the Tyr75/Tyr96 pair in cytochrome P450cam.111–112 Tyrosine CPET oxidation likely would be favored if there were a proximal proton acceptor;76,113–115 a sequential electron and proton transfer mechanism also could operate, but only with an oxidant with a reduction potential (>1.34 V vs. NHE to generate TyrOH•+) higher than that (0.93 V vs. NHE, pH 7) of the TyrO•/TyrOH couple.11,76,113–115 In an enzyme active site, the TyrO•/TyrOH formal potential is predicted to vary from that associated with the same reaction in bulk solution, owing to a number of factors—hydrogen bonding to nearby residues, electrostatic effects of charged residues, and the effective dielectric of the protein matrix.83
The redox properties of model complexes containing phenols as tyrosine mimics have been investigated extensively over the last twenty years, with attention focused on systems in which phenols are near proton acceptors.87,116–133 Although unsubstituted phenol is basic (pKa = 10.0), oxidation to PhOH•+ produces a strong acid, with a pKa shift of fully 12 units to a value of −2. Electrochemical studies of phenol show irreversible oxidation waves, as the loss of the acidic phenolic proton to aqueous solvent is highly favorable. Phenols with tert-butyl substituents often are employed to probe oxidation mechanisms, as such sterically bulky groups disfavor radical self-reactivity.
In an investigation of the reactions of triplet-excited states of C 60 and tetracene with phenols, Linschitz and coworkers observed that luminescence quenching was greatly enhanced upon addition of pyridines.87,116,134 Flash-photolysis experiments showed that the products of the quenching reaction are C60•− anion radical, neutral phenoxy radicals, and protonated pyridines.117 A deuterium kinetic isotope effect was observed (kH/kD up to 1.65 ± 0.10), indicating the importance of both proton transfer and hydrogen bonding in promoting these reactions, which in turn were attributed to electron transfer from the phenol to 3*C60 with concerted proton transfer to the hydrogen-bonded base.
Experiments by Lucarini and coworkers have shown that intermolecular hydrogen bonds from hexafluoropropanol (HFP) preferentially stabilize phenoxyl radicals (Figure 8).135 EPR equilibration techniques indicated that the OH bond dissociation enthalpy of the phenol OH bond is lowered in the presence of HFP, owing to stabilization of phenoxyl radicals. Solvents functioning as hydrogen-bond acceptors and donors also were shown to affect the stabilization of phenols and phenoxyls by interacting with their –OR, –OH, and –NH2 substituents.
Figure 8.
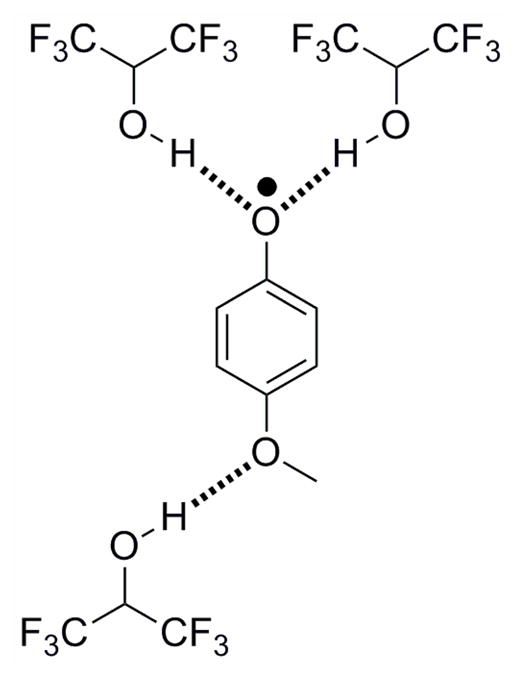
Phenoxyl radicals stabilized via intramolecular hydrogen bonds to hexafluoropropanol. Interactions between substituents and hexafluoropropanol and other hydrogen-bond accepting and donating solvents was shown to affect the stabilization of the phenoxyl radical.
Reactions of phenols with the oxidant trans-[RuVI(L)(O)2]2+ (L=1,12-dimethyl-3,4:9,10-dibenzo-1,2-diaza-5,8-dioxacyclopentadecane) yield phenoxyl radicals and trans-[RuV(L)(O)(OH)]2+.118 Working in aqueous solution, Lau and coworkers observed pH dependent and independent processes, consistent with concurrent oxidation of PhOH and PhO−, in different molar ratios at varying pHs. Based on KIE and other studies of a variety of substituted phenols, the authors concluded that the pH independent pathway was consistent with CPET oxidation of PhOH to form PhO• and trans-[RuV(L)(O)(OH)]2+; and, a similar mechanism was proposed for the reaction in CH3CN solution. In a plot of log(k) vs. bond dissociation energies for a series of substituted phenols, each phenol in the data set fell on one of two lines, with phenols containing substituted 2,6-di-tert-butyl groups falling on the one with lower rates (those without steric crowding of –OH exhibited higher rates). These findings demonstrate that proton transfer distance plays a role in CPET reactions.11
Meyer and coworkers have extracted the kinetics of tyrosine oxidation by M(bpy)33+ complexes (M = Ru, Os) from CV data.136 At pH 7.5, where the basic form of the buffer constitutes a significant percentage of the total, the kinetics of oxidation correlate with the metal complex potential, indicating that electron transfer is not involved in the rate limiting step. However, at lower pH (pH < 6.5) and lower buffer concentrations, the expected relationship between driving force and oxidation rate was found. The authors explained these results by a CPET mechanism that was in competition with PT-ET at high concentrations of base, wherein proton transfer to a hydrogen-bonded phosphate buffer base is the rate limiting step. In a related study, the driving force for oxidation was systematically varied by changing the potential of the oxidant or the pKa of the added base (Figure 9).137 Consistent with previous work, a CPET pathway, with electron transfer to the oxidant and proton transfer to the base could account for the reaction kinetics. Notably, rate constants ranging from 5.0 × 103 to 9.8 × 107 M−1 s−1 correlate well with the driving force for oxidation.
Figure 9.
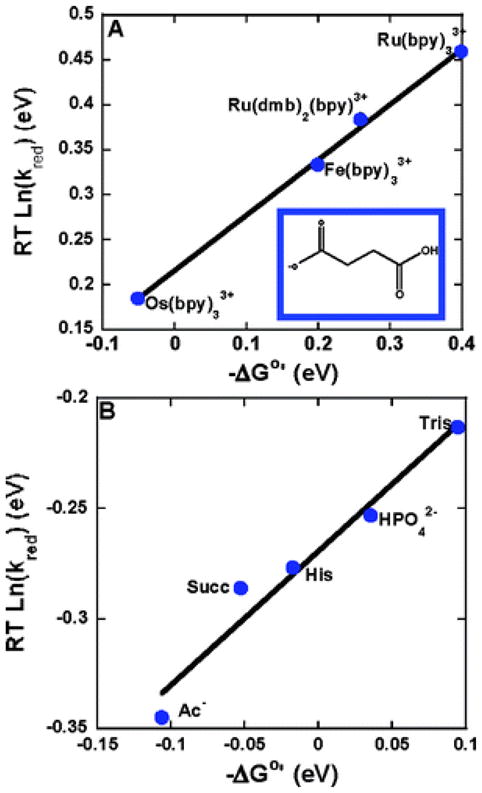
Rates of oxidation (RT lnkred) of tyrosine vs. driving force −ΔG°′ at 298 K. (A) Driving force was varied by utilizing several oxidants (bpy is bipyridine, dmb is 4,4″-dimethyl-2,2′-bipyridine) with different E°′(M3+/2+) with a common base (succinate monoanion) at pH 4.9. (B) Driving force was varied by utilizing several acceptor bases (Ac−: acetate, Succ: succinate monoanion, His: histidine, HPO42−: dibasic phosphate, Tris: tris) with different pKa values with [Os(bpy)3]2+ as the oxidant. 0.050 M buffer solutions with a 10:1 base to acid ratio were utilized. In both figures the slope is 0.61. Reprinted with permission from Reference 137. Copyright 2007 American Chemical Society.
Outer-sphere oxidation of phenol and methyl-substituted phenols by [IrCl6]2− has been examined by Stanbury and coworkers.119 At low pH, the rate is pH independent, but near netural pH, the rate constant is pH dependent and the kinetics are unaffected by buffer concentration. The pH dependence is caused by competing oxidations of PhOH and PhO−, the latter appearing in increasing concentrations at higher pH. CPET with water as the proton acceptor appears to be the dominate pathway for phenol oxidation, as there is a large kinetic isotope effect implicating OH bond cleavage in the rate limiting step. 119
Based on a very thorough investigation of the oxidation of 2,4,6-tri-tert-butylphenol (TTBP) and phenol in nonbuffered aqueous media, Savéant and coworkers proposed that a CPET process forming a phenoxyl radical with proton transfer to water is in competition with a PT-ET mechanism where HO− acts as the proton acceptor.120–122 In electrochemical studies, the cyclic voltammogram shows two separate reversible waves (Figure 10), one corresponding to oxidation of the phenoxide ion, indicating a PT-ET mechanism, which dominates in basic media, and a second wave that becomes more prominent as the pH is decreased, suggesting CPET oxidation. A relatively large H/D kinetic isotope effect was observed for this latter process, supporting the CPET assignment, and successful simulations of the cyclic voltammograms were achieved with this, and only this, model. Based on analyses of a series of laser flash photolysis and stopped-flow experiments with a variety of electron acceptors, the rate constant for the oxidation reaction was obtained as a function of driving force.123 The data from these experiments together with H/D isotope effects rule against a stepwise ET-PT mechanism in favor of a CPET route. Notably, analysis of these data suggests that the concerted process is under activation control albeit with extremely low reorganization energies, a finding that appears to be unique to water as a proton acceptor.
Figure 10.

(a) Cyclic voltammetry of phenol in water at 0.2 V/s in unbuffered water. (b) Peak potentials of cyclic voltammetry plotted as a function of pH. The black stars are the peak potentials in D2O. The blue line is the simulated variation of peak potential for a CPET mechanism. The color code of the voltammograms correspond to the color code of the peak potentials. Reprinted with permission from Reference 121. Copyright 2010 American Chemical Society.
Tyrosines linked to ruthenium polypyridyl photosensitizers, RuII-Tyr, have been employed by Hammarström and coworkers to model the P680-TyrZ system (Figure 11a).105,124–125,138–142 Photoexcitation of the RuII center in the presence of an external electron acceptor, such as methyl viologen, leads to oxidation of the ruthenium center to form RuIII-Tyr. Electron transfer from the tethered tyrosine to the oxidized ruthenium center ensues, and the recovery of RuII is monitored by transient absorption spectroscopy. Ru II-Tyr-O• was generated on the microsecond time scale in neutral water. When the solution pH is lower than the pKa value for tyrosine (~10), the observed rate constant corresponding to this process is pH dependent.105,124,139 Above this value, however, a faster pH independent reaction was observed, consistent with the participation of the TyrO•/TyrO− couple. Near the pKa value, biexponential fitting produced rate constants corresponding to both slow and fast reactions. These data were used to discriminate between possible stepwise and concerted mechanisms. If the deprotonation rate were less than 10 s−1 at pH values below the pKa of tyrosine, a stepwise PT-ET mechanism would be limited by this rate. The steady state approximation for ET-PT gave no pH dependent rate, ruling out this mechanism. The observed pH dependent rate was initially interpreted to be characteristic of a concerted mechanism with proton transfer to water, based on analysis involving the incorrect assumption of a pH dependent driving force.123,143–144 The pH dependence of Tyr oxidation in these model systems, however, could be explained by PCET reactions with one of two proton acceptors, HO− or the basic form of a buffer, in solution, not a pH dependent driving force.11,85,121 Later studies examined the oxidation as a function of buffer concentrations—the rate is first order in the concentration of the basic form of the buffer at high buffer concentrations—indicating a CPET reaction with the buffer acting as a proton acceptor.145 However, the rate of Tyr oxidation by RuIII also was pH dependent at low concentrations and in the absence of buffer, with a plot of log(kobs) vs. pH exhibiting a slope of ca. 0.5. The results were attributed to a CPET reaction with ET to RuIII and proton transfer to the bulk.145 Similar studies with 4,4′-COOEt substituted bipyridine ligands, which yields a stronger oxidant than the parent bipyridine system, suggests a CPET mechanism for this system at higher pH but an ET-PT mechanism at lower pH in the absence of buffer.
Figure 11.

Photosensitizers with appended tyrosine utilized in studies of photochemical oxidation of tyrosine.. (a) RuII-Tyr complexes. R = H or COOEt (b) ReI(P-Y) complex; P-Y is a diphenylphosphinobenzoic acid with an amide linkage to a tyrosine.
The pH dependence of phenol oxidation rate constants is a curious observation. The apparent slope in log(kobs) vs. pH plots (0.5) at low buffer concentrations is inconsistent with kinetics modeling of ET-PT and PT-ET mechanisms.145 Hence, CPET is chosen by elimination. But, if electron and proton transfer are in fact concerted, the challenge is to explain why the barrier height for this process should depend on the proton (or, equivalently, hydroxide) concentration. The notion of a pH dependent driving force has been definitively ruled out.123,143–144 A physically sound explanation of the experimental facts in these systems remains to be provided.
Electronic excitation of a ReI polypyridyl-Tyr complex triggers ET directly to a triplet-excited MLCT state without the need for external quenchers.146 The triplet MLCT state of Re(phen)(CO)3(P-Tyr)+ (ReI(P-Y) Figure 11b), where phen is phenanthroline and P-Tyr is a diphenylphosinobenzoic acid ligand with an amide linkage to a tyrosine residue, is a strong oxidant, E°(ReI*/0) ~ 1.78 V vs. NHE. The tyrosine unit in the complex is oxidized within microseconds upon photoexcitation, as determined by ReI MLCT luminescence quenching. For pH values below the pKa of Tyr, the rate of TyrO• formation is pH dependent. At pH values above the pKa, the rate constant was found to be invariant with pH changes, similar to observations by Hammarström on related systems.124,139,145 Experiments employing several buffer concentrations at different pHs indicated that, upon Tyr photooxidation (and reduction of the excited ReI complex), the proton is transferred to the basic form of the buffer.145 In the absence of buffer, no pH dependence was observed, as expected, and an ET-PT mechanism presumably operates. Theoretical work supports this interpretation. 147
Several model complexes designed and built to mimic charge transfer in the P 680-Tyr-OEC unit also have been investigated. Among these are ones in which ruthenium polypyridyl photosensitizers have been covalently linked to manganese complexes.140–141,148–154 While long-range charge separation has been achieved upon photoexcitation of these systems, few if any oxidize water efficiently. Of special relevance to the mechanism of water oxidation in the OEC are rigorous CPET analyses of electrochemical experiments on osmium aquo-hydroxo model systems.155–158
Phenol systems have been modified with nitrogen and oxygen bases that can act both as hydrogen-bond donors/acceptors and proton acceptors for the acidic phenol proton. Investigators first noted an enhancement of phenol oxidation reversibility in these complexes: they attributed these electrochemical properties to PCET mechanisms with protons transferred to nearby tethered bases; later work, however, including analyses of kinetic isotope effects, cyclic voltammograms, and oxidation rates as a function of driving force, led to a better understanding of these PCET processes (vide infra).
Matsumura and coworkers examined a phenol with α-alkylamino groups in the ortho position (Figure 12) as a model for hydrogen-bonded phenoxyl radicals that are believed to function in biological systems.159 The reversibility of the redox couples of these modified phenols (12a–c) is enhanced relative to a para substituted control (12d), which exhibits an irreversible CV typical of phenols. The observation that the redox couple of 12c is fully reversible suggests that intramolecular transfer of the phenolic proton to the hydrogen-bonded amine accompanies oxidation.
Figure 12.

(top) Phenol derivatives A–D with α-alkyl amino groups at the ortho or para positions. (bottom) The proposed reversible redox process of C/C•+ with intramolecular migration of the phenolic proton to the hydrogen-bonded amine.
The redox chemistry of a phenol-imidazole complex, 2′-(4′,6′-di-tert-butylhydroxyphenol)-4,5-diphenyl imidazole (Figure 14e, R=H, X=H) has been studied by Garner and coworkers.160 One-electron oxidation of this complex was observed to be reversible, with stabilization of the phenoxyl radical cation attributed to an intramolecular hydrogen bond with the imidazole nitrogen, though later work has indicated that CPET with proton transfer to the tethered imidazole is the more likely pathway.126
Figure 14.

Intramolecular hydrogen bonds between phenols and appended (a) amines, (b, e) imidazoles, and (c, d) pyridines.
In studying the oxidation of tertiary amine modified phenols (Figure 13), Pierre and coworkers observed intramolecular proton transfer to the amine to form phenoxyl-ammonium complexes. 161 Upon oxidation of complexes with ester or pyridine substituents on the tertiary amine, proton transfer occurs from the phenoxyl cation to the amine. The proposed stepwise PT-ET pathway may be kinetically and thermodynamically assisted by formation of a multiple hydrogen-bond network that includes the substituents. But note that formation of this network will destabilize the phenoxyl radical by weakening the phenoxyl-ammonium hydrogen bond.
Figure 13.

Phenol derivatives with tertiary amines and their corresponding hydrogen-bonded phenoxyl radicals. The multiple hydrogen-bond networks seen in the ester or pyridine substituted pyridines affect the PCET process and the stability of the phenoxyl radical.
Ueyama and coworkers observed less positive peak potentials for oxidation of phenols that feature intramolecular hydrogen bonds to carboxyl groups.127 They attributed this behavior to enhanced acidity of the phenolic proton, consistent with CPET.11
PCET reactions of phenols that are hydrogen-bonded to appended base moieties (primary amine, imidazole, or pyridine Figure 14a, c, e X = p-OMe) have been studied by Mayer and coworkers.83,128–129 Oxidation of these tyrosine models with one-electron outer-sphere oxidants in acetonitrile produces in each case a phenoxyl radical in which the phenolic proton is transferred to the amine by a PCET process. The CV-measured redox potentials are lower than those of corresponding phenols without pendant bases, with the shifts a function of the driving force associated with proton transfer to the appended base. Several arguments that seemingly rule out stepwise pathways that would proceed through high energy intermediates lead to the conclusion that the reaction mechanism is concerted PCET; the primary KIE, kH/kD = 1.6–2.8 (depending on oxidant and phenol), cannot be accounted for by either stepwise pathway, and the rate constants are higher than would be expected for any route that involves a high energy intermediate. What is more, the driving force dependence of reaction rate constants is consistent with Marcus theory predictions for concerted PCET.83 It was suggested from a Marcus-type analysis that PCET in these systems is adiabatic, with the relative sluggishness of the reactions attributable to large reorganization energies.128 Additional experiments, however, led to reinterpretation, as the combined results are more consistent with a nonadiabatic process.129 The proposed nonadiabatic PCET mechanism was further supported by determinations of the temperature dependences of reaction driving forces, which indicated reorganization energies that are much lower than originally reported.130
Interestingly, the CPET reaction of pyridine modified phenol is about 102 times faster than the amine modified phenol, despite similar driving forces and the fact that pyridine is a weaker base than a primary amine.129 Comparison of the rate for one-electron oxidation of the phenol-pyridine complex with that of a related species featuring a methylene linker between the two rings (Figure 14c, d) showed that the latter complex reacts 25–150 times slower than the one with a phenol-pyridine unit, even though the PCET driving forces are similar.131 It was suggested that resonance-assisted hydrogen bonds in the phenol-pyridine complex account for the difference in PCET reaction rates.
Experiments based on a series of phenol-imidazole compounds (Figure 14b, e) have shed some light on various parameters affecting CPET.126 The rate constants for one-electron oxidation of these complexes are well-correlated with the driving forces for these reactions. Structural and electronic factors have much smaller effects on the rate constants, suggesting that CPET tunneling probabilities and intrinsic barriers do not vary significantly in this series of complexes. The specific effects of geometrical and electronic structures in principle could be elucidated by comparing reactions with very similar driving forces.131
Savéant and coworkers have examined several phenol-base model complexes using electrochemical techniques. 132 Careful analysis of the CV responses and H/D kinetic isotope effects of an ortho-substituted phenol with an intramolecular hydrogen bond to the appended amine indicated that oxidation occurs by CPET (Figure 14a, Figure 12a, c).132 Electrochemical oxidation is very nearly reversible, though this reversibility is lost at very slow scan rates and in the presence of an external base, like pyridine, indicating deprotonation of the radical cation. An H/D KIE of 1.8 along with other data rule out stepwise PT-ET and ET-PT mechanisms. In contrast to related oxidations in homogenous solutions, the electrochemical reaction is adiabatic.130 Interestingly, electric fields affect oxidation rates, decreasing proton tunneling barriers that in turn lead to exceptionally large preexponential factors for CPET.
Intermolecular flash-quench techniques have been employed by Hammarström and coworkers to study the oxidation of substituted phenols by photogenerated [Ru(bpy)3]3+.125 Upon oxidation, the phenols, which contain intramolecular hydrogen bonds to carboxylates, exhibit pH dependent rate constants (Figure 15). At low pH, the carboxylate group is protonated and the oxidation rate is pH dependent, which was attributed to concerted CPET with proton transfer to water (or buffer). At intermediate pH, with the carboxylate group deprotonated and the phenol protonated, the rate of phenol oxidation is pH independent and the phenolic proton is transferred intramolecularly to the carboxylate base (CPET mechanism). With a stronger photogenerated oxidant, ET-PT occurs. At higher pH, the phenol also is deprotonated and oxidation occurs only by ET.
Figure 15.

Ortho- and para- carboxylate-substituted phenols with and without intramolecular hydrogen bonds.
This work was extended to include intramolecular flash-quench experiments involving carboxylate substituted phenols covalently linked to ruthenium polypyridyl photosensitizers (Figure 16a, b).133 Analysis of transient absorption data from photoinduced intramolecular oxidation indicated bidirectional CPET with proton transfer to the appended carboxylate. Temperature and H/D-isotope dependences of CPET rates were interpreted with the aid of DFT calculations and MD simulations.
Figure 16.

Ruthenium polypyridyl complexes with covalently linked phenols containing intramolecular hydrogen bonds to (a, b) carboxylates and (c) imidazoles and the (d, e) corresponding control complexes.
Aukauloo and coworkers have investigated a RuII polypyridyl photosensitizer linked to a phenol modified with a hydrogen-bonded imidazole group (Figure 16c).162 Electrochemical measurements produced a quasi-reversible wave that was attributed to formation of the phenoxyl radical, where the potentials are similar to those of other hydrogen-bonded phenols. Photolysis of the Ru II complex in the presence of an irreversible electron acceptor, [Co(NH3)5Cl]2+, produces an oxidized complex with an EPR spectrum attributable to a hydrogen-bonded phenoxyl radical.
Moore and coworkers elucidated the redox chemistry of a hydrogen-bonded tyrosine-histidine complex, BiP (Figure 17a), a model system that serves as a functional mimic of PSII TyrZ-His190.163 The phenoxyl/phenol couple of BiP164 is reversible in the presence of the attached base, which allows the proton to shuttle between the phenol oxygen and the benzimidazole nitrogen lone pair, thereby localizing the proton at the site of electrochemical activity. Additional electrochemical experiments, along with optical and NMR spectroscopic measurements in both acidic and basic solutions, demonstrated that the BiP phenoxyl/phenol couple is capable of water oxidation but the corresponding phenoxyl/phenoxide pair is not.165 The products formed following photoexcitation of BiP linked to a mesityl substituted porphyrin (BiP-PMes, Figure 17b) also were investigated: consistent with expectation, the porphyrin singlet excited state could oxidize the phenoxide but not the phenol, owing to the higher potential of the protonated species. A related BiP-porphyrin (BiP-PF10, Figure 17c) was adsorbed onto the surface of colloidal TiO2 nanoparticles with the goal of mimicking the photosynthetic chlorophyll-Tyr-His complex.163 Photoexcitation of the porphyrin moiety in BiP-PF10:TiO2 triggers electron injection into the TiO2 conduction band, followed by hole transfer from the porphyrin radical cation (PF10•+) to the hydrogen-bonded phenol (BiP) to yield primarily a charge separated state, BiP•+-PF10-TiO2• −. It is likely that the proton of the oxidized BiP phenol is transferred to the appended base, producing the phenoxyl radical that was observed by D-band EPR at low temperature. All of these experiments suggest that concerted PCET events play prominent roles during the oxidation of water in PSII.
Figure 17.

(a) A tyrosine-histidine model complex, BiP, with intermolecular phenol-imidazole hydrogen bonds. (b) BiP-PMes and (c) BiP-PF10, BiP modified porphyrins.
4. Protein Redox Machines
Studies of radical transport in proteins have provided insight into the critical role of proton coupled electron transfer events in biological processes. Redox active amino acids, like tyrosine and tryptophan, are thought to play a key role in radical transport in a number of different enzymatic reactions. Crystal structures of several proteins indicate that these redox active amino acid residues function in radical transport pathways, and proton accepting residues are often positioned nearby. Mutagenesis studies have emphasized that both the redox active amino acid residues and the nearby proton accepting residues are critical for efficient radical transport. Below we discuss studies that illustrate the intimate coupling of proton and electron transfer in two select biological systems, photosystem II and ribonucleotide reductase.
4.1 Photosystem II
Photosystem II (PSII) is a miraculous molecular redox machine that uses solar photons to drive the oxidation of water to dioxygen, thereby producing electrons and protons to reduce carbon dioxide (Figure 18).166–167 Upon illumination, chlorophylls of the primary electron donor P680 are photoexcited and an electron is transferred through pheophytin a (PheoD1) to reduce a bound plastiquinone QA, which in turn reduces plastiquinone QB. Once QB is reduced by two electrons and protonated to form QBH2, with the second reduction/protonation believed to be a PCET event,168 the quinol QBH2 is released to the membrane matrix and transfers reductive equivalents to photosystem I, where CO2 is reduced in the Calvin cycle.169
Figure 18.

Molecular structure of the cofactors involved in electron transfer in Photosystem-II. Image is visualized perpendicular to the internal pseudo-two-fold axis. The electron transfer pathway is indicated by red arrows, and distances are given in angstroms. Reprinted with permission from Reference 204. Copyright 2008 American Chemical Society.
The highly oxidizing P680•+ (E° = +1.26 V vs. NHE)11,170 is reduced by a tyrosine residue, TyrZ (Tyr161 of the D1 subunit)69,171–173 on the nanosecond time scale, generating a strongly oxidizing (1.1–1.2 V vs. NHE)11,174 neutral tyrosine radical TyrZ-O• upon the loss of the phenolic proton. An electron cascade follows, as TyrZ-O• then oxidizes (within 30 microseconds to 1.2 milliseconds) the oxygen evolving complex (OEC), which consists of one calcium and four manganese ions.175 This manganese cluster cycles through four successive photoinduced oxidations (the Kok S-state cycle),176–177 extracting electrons from two associated water molecules and ultimately releasing molecular oxygen and returning to its reduced state.169,175,178–186
The mechanism by which the tyrosyl/tyrosine redox couple mediates charge transport between P680•+ and the OEC is under intense investigation. Upon oxidation of TyrZ, EPR data show a signal consistent with the netural radical form, TyrZ-O•, indicating that transfer of the phenolic proton accompanyies electron transfer.175,187–188 The proton is believed to be transferred to a nearby base, likely the hydrogen bonded histidine residue His190 in the D1 subunit, 182,189–192 as oxidation of tyrosine drops the phenol pKa from 10 to −2 (Figure 18).105–106 Addition of imidazole or other small organic bases has been shown to accelerate the oxidation of TyrZ by P680•+.191 Further, site-directed mutagenesis studies have shown that His190 facilitates the rate of TyrZ oxidation by at least a factor of 200.189,193–196
Since the Tyr-O•/Tyr-OH0 potential (0.93 V vs. NHE at pH 7) is much lower than that for Tyr-OH•+//Tyr-OH0 (1.34 V vs. NHE), it is very likely that CPET oxidation formulated as Tyr-O•…+H-His190/Tyr-OH…His190 occurs, thereby avoiding high energy intermediates.174 As noted above, the potential of this couple has been estimated to be approximately 1.1–1.2 V in the photosynthetic membrane, an increase from the solution value. 11 The shift has been attributed to destabilization in a nonpolar membrane environment or loss of effective protonic contacts between aromatic residues and the bulk solvent. 11,197
The driving forces for electron transfer, CPET, and proton transfer (Figure 19) have been estimated based on the redox couple potentials for P680+/0 (1.26 V vs. NHE)170 and Tyr-OH+•/0 (1.34 V vs. NHE), and pKa values for Tyr-OH+• (−2), Tyr-OH (10), and +H-His (5.5). 11,174 Both the ET and PT reactions are endergonic, +0.08 eV and +0.26 eV uphill, respectively. However, the CPET reaction has a ΔG° = −0.36 eV. While the calculations are solution based values and do not include the difference in ΔG° for forming initial and final H-bonded adducts, they underscore the energetic advantages that exist for CPET reactions over stepwise pathways beginning with ET or PT steps.
Figure 19.

Driving forces for (a) ET, (b) CPET, and (c) PT reactions at TyrZ (Tyr161). Adapted from Reference 11.
However, recent work by Rappaport and coworkers has suggested that reduction of P680•+ may be controlled by a stepwise PT-ET mechanism. In this work, the driving force for electron transfer was altered via site-directed mutagenesis of the axial ligand of P680 and for proton transfer by substituting 3-fluorotyrosine (3F-Tyr) for all tyrosines.198 It was concluded that when TyrZ acts as a hydrogen-bond donor, i.e., in the pH range where the proton acceptor is not protonated, reduction of P680•+ by tyrosine is thus controlled by the proton transfer to the nearby base, His190, in a stepwise PT-ET mechanism. The salt bridge formed between the tyrosinate and the protonated base was assumed to affect the tyrosyl/tyrosinate redox couple.
Mechanisms for OEC water oxidation mediated by TyrZ have been extensively reviewed.11,174 We refer the interested reader to these detailed accounts, as here we will only discuss the role of TyrZ. Based on estimates for the successive transitions of the Kok cycle, the average potential for each of the S state transitions is approximately 0.9 V vs. NHE, so it could be oxidized by TyrZ-O• if the estimated membrane potential of 1.1–1.2 V vs. NHE is correct. Babcock and coworkers initially proposed that TyrZ• abstracts H• from water bound to the manganese atoms in OEC.69,169,199–202 Protons were thought to be shuttled from TyrZ to His190 then on to the lumen via an exit channel,69,169 where they appeared in the bulk phase on timescales similar to that for electron removal from TyrZ.203
Not so fast! Recent structural evidence indicates that the nearest Mn ion in the OEC cluster is over 6 Å away from TyrZ182,190,204 and the TyrZ-His190 pair is relatively isolated by α-helices88 that would preclude rapid proton transfer.11,174 A PCET pathway must play an important role in the oxidation of water, however, to avoid charge buildup associated with the 4 protons lost during O2 production from water. TyrZ-O• is believed to oxidize the OEC through a PCET process with electron transfer from Mn orbitals to the tyrosyl radical while water based protons are transferred to the nearby hydrogen-bonded aspartic acid residue (Asp61) and a proton from the protonated His190 is transferred back to the tyrosyl radical upon its reduction. Asp61 is believed to be the entryway to a hydrophilic proton exit channel, and upon protonation, the proton is shuttled to the lumen by a series of conserved titratable residues.205–208 Asp170 also is thought to be the internal base required for PCET. Mechanisms of this sort, which feature strong coupling between electron transfer and proton transfer, maintain charge neutrality in going from reactants to products, thereby bypassing barriers attributable to high energy intermediates.
4.2 Ribonucleotide Reductase
Ribonucleotide reductase (RNR), the enzyme responsible for the production of deoxyribonucleic acids, utilizes the oxidizing power of molecular oxygen to carry out hydrogen atom abstraction chemistry.65–66,107–108,209–210 In E. coli ribonucleotide reductase, a hole originating on the Tyr122 radical (Tyr122•) in the β2 subunit is transferred some 35 Å to the active site in the α2 subunit, retaining sufficient oxidizing power to generate the Cys439 radical that initiates conversion of nucleotides to deoxynucleotides. 63,65–67,211–213 Electron tunneling across the 35 Å that separates Tyr122• and Cys439 would be much slower14,28,45 than the observed kcat of ~2 to 10 s−1.85 Multistep electron tunneling architectures in this enzyme facilitate the movement of charges rapidly over long distances with only a small loss of free energy. Because the enzyme operates at very high potentials, the sidechains of aromatic amino acids (e.g., tryptophan, tyrosine) are believed to participate in the charge migration process.63 A hopping mechanism involving the conserved amino acids Tyr122• →Trp48→Tyr356→Tyr731→Tyr730→Cys439 has been proposed,66,212 supported by site direct mutagenesis studies indicating that activity is inhibited in these absence of these residues.214–217
The electron transport mechanism through RNR is thought to be closely coupled to proton motion along and orthogonal to the participating amino acid residues in the hopping chain. The PCET pathways, which have been elucidated after many years of work, include both orthogonal CPET reactions and unidirectional H• propagation. The synchronization of protons and electrons during transport through the enzyme is favored thermodynamically, and experiments based on site-directed mutagenesis214–221 and photoinitiated radical transport have pointed to a critical role for redox active amino acids in charge migration; and amino acid radical intermediates have been observed in certain cases.
Experiments involving site-directed mutagenesis as well as the incorporation of unnatural amino acids to perturb pKas and redox potentials have provided information about the putative PCET pathway of the enzyme. Further, “photoRNRs” have been created, allowing radical initiation by phototriggering, permitting spectroscopic examination of transient radical intermediates. These investigations are detailed below. Note that the conserved amino acids in the consensus ET pathway in non E. coli RNRs (e.g., mouse RNR) are at positions in the polypeptide sequence different from those in the E. coli enzyme.
The radical hopping mechanism is initiated upon the formation of a diferric tyrosyl radical cofactor Tyr122•, the assembly of which requires Fe2+ binding and the four-electron reduction of O2 to H2O.66 In β2 subunits with a Tyr122Phe mutation, EPR active intermediates attributed to iron cluster cofactor based radicals exhibit extended lifetimes as compared to those of the wild type subunit, suggesting that these radical intermediates are responsible for Tyr122 oxidation.107 In addition, Trp48 has been postulated to help modulate cofactor assembly prior to oxidation of Tyr122.107,222–223
Reversible electron transfer between Tyr122 and Trp48 is proposed to play a key role in radical transport. Structural work on E. coli RNR has identified a conserved tryptophan (Trp48) that could participate in the radical transport pathway via direct charge transfer with Tyr122. Trp48 is hydrogen bonded to Asp237 and His118, residues that also are conserved in all species.224–225 In the β2 subunit of mouse RNR, the conserved tryptophan analogous to Trp48 (Trp103) was replaced by phenylalanine and tyrosine.216 Upon initiation, the tyrosyl radical Tyr122• formed in the Trp103Tyr mutant, but not in the Trp103Phe protein. Neither of the mutants was active in an enzymatic assay, however, suggesting that the conserved tryptophan is a required intermediate in the multistep electron transfer pathway. Transient kinetics studies of Trp-Tyr dipeptides have shown that radical transfer between these two amino acids can be controlled by pH.226–227 TyrO• has a lower reduction potential than Trp• at physiological pHs, and Trp• was found to oxidize Tyr, whereas charge transfer at higher pHs occurs in the reverse direction, Trp-TyrO• →Trp•-TyrO−. Coupled with findings from structural and biochemical work, these results emphasize that Trp48 and its cation radical Trp48•+ play key roles both in initiation of nucleotide reduction and cofactor assembly.107,222–223
Asp237, which is hydrogen bonded to Trp48 (2.9 Å), is the most probable site for orthogonal PCET to or from Trp48.228 E. coli RNR β2 mutants with glutamic acid substituted at the 237 position (Asp237Glu-β2) exhibit enzymatic activity at 7% of the rate of wild type β2, suggesting the importance of an acidic residue at the 237 site.214 Mutation of aspartic acid to asparagine (Asp237Asn- β2) knocks out catalytic activity. In mouse RNR, mutants with the conserved aspartic acid replaced by alanine are able to form the tyrosyl radical, but do not exhibit any enzymatic activity.216 It has been postulated that the position of a proton between Trp48 and Asp237 modulates the reduction potential of Trp48.66 Further, proton transfer between Trp48H•+ and Asp237 may couple with ET between Tyr122• and Trp48, such that oxidation of Trp48 is triggered by proton transfer to Asp237.229
Tyr356 modulates electron transfer between Trp48 in the β2 subunit and Tyr731 in the α2 subunits. Though its location has not been specifically located in α2 or β2 crystal structures,228 sequence conservation and mutagenesis studies221,230 have confirmed its role in radical transport. Work by Nocera and Stubbe has elucidated the role of Tyr356 through a series of mutant β2 subunits incorporating unnatural amino acids.67,231–233 In one such experiment, β2 subunits containing a series of fluorinated tyrosine derivatives234 with reduction potentials that ranged from −50 to +270 mV vs. the tyrosine potential and pKas that ranged from 5.6 to 9.9 were used to map the pH rate profiles of deoxynucleotide production.231 The results of this study suggested that the rate-determining step of the natural protein, attributed to a physical or conformational step, could be switched to radical propagation by varying the reduction potential of Tyr356•, emphasizing the role of Tyr356 as a redox-active amino acid in multistep, long-range ET between Tyr122• and Cys439. Efficient nucleotide reduction was observed even with the fluorinated amino acids deprotonated at the pHs studied, suggesting that a hydrogen-bonding pathway between Trp48, Tyr356, and Tyr731 is not necessary for radical hopping nor is hydrogen-atom transfer compulsory. Upon oxidation of Tyr356, the proton is believed to be transferred to bulk solution, directly or possibly assisted by amino acid residues.
Preparation of Tyr730 and Tyr731 mutants showed that activity is curtailed when these tyrosines are replaced by phenylalanine.215 The absence of enzymatic activity in the mutants indicates that these residues play a critical role in radical initiation, with hydrogen atom transfer the most likely mechanism.66
Roles for Tyr730 and Tyr731 as redox-active residues in the radical transport pathway of RNR also is supported from work in which 3-aminotyrosine (NH2Tyr) was incorporated at these two positions.235 The lower reduction potential of NH2TyrO• as compared to TyrO• provides a thermodynamic trap for the radical transport pathway in the mutated protein, which is capable of turnover. Freeze-quenching of an initiated reaction allowed observation of an organic radical assigned as NH2Tyr730/731• by X-band EPR. This study was the first to identify a radical intermediate in a radical propagation pathway.
Work in the Nocera and Stubbe research groups also has focused on methods to photoinitiate and monitor RNR radical intermediates by transient spectroscopy. In these experiments, the β2 subunit is replaced by a 20-mer C-terminal peptide tail, which contains the critical Tyr356 as well as amino acids required for subunit binding to the α2 subunit.218,236 A photooxidant is appended nearby the Tyr356 amino acid and laser excitation produces Tyr356•. This photochemical radical generation method effects turnover in the presence of CDP substrate and ADP effectors when the sensitizer-modified peptide tail is docked to the α2 subunit.227,237 In one case, [Ru(bpy)3]2+ was employed in conjunction with a quencher-oxidant [Co(NH3)5Cl]2+ to generate a tyrosyl radical at position 356; the low observed activities were attributed to inefficiency of [Ru(bpy)3]3+ oxidation of tyrosine.238 Enhanced activities were obtained when photoionization of tryptophan initiated oxidation of Tyr, though “inner-filter” optical effects and protein stability with the deep UV wavelengths (<290 nm) required for photoionization greatly limited the system.226–227 Benzophenone and anthraquinone also were utilized as photooxidants with excitation wavelengths up to 365 nm.237 ReI polypyridyl complexes are particularly attractive, as their excited states are powerful oxidants (Re(phen)(CO)3(PPh3)*+ can oxidize TyrOH).89,237
Photochemical investigations of mutants with amino acid substitutions along the proposed radical transport pathway have shed light on details of the RNR mechanism. In the presence of CDP substrate and ATP effector, turnover can be photoinitiated. Interruption of the hydrogen bond network in α2 by mutation of Tyr730 to phenylalanine239 leads to curtailment of photoinduced nucleotide reduction activity with benzophenone or anthraquinone photooxidants.237 From analysis of potential mechanisms for radical transport in α2, it was concluded that the proton-transfer pathway is critical for turnover and it was further suggested that a proton-dependent hopping mechanism is responsible for Tyr731• →Tyr730→Cys439 charge transport (PCET in which both an electron and proton transfer unidirectionally).
High turnovers were observed in photoiniated experiments incorporating Re(bpy)(CO)3CN as a photochemical Tyr• generator and incorporating 3,5-difluorotyrosine (3,5-F2Tyr) in “position 356” of the β20-mer C-terminal peptide tail.240–241 When coupled to an α2 subunit with a Tyr731Phe mutation, radical transport into α2 is prevented. Employing transient absorption spectroscopy, a tyrosyl radical intermediate, 3,5-F2Tyr• was observed.
A model has been developed to account for the observations of radical transport in RNR (Figure 20). In the β2 subunit, the diiron oxo/hydroxo cofactor accepts a proton from Tyr122 upon oxidation. Oxidation of Tyr356 requires coupling proton release to electron transfer, and a mechanism with PT orthogonal to ET is invoked. The orthogonal PCET upon oxidation of Tyr122 and Tyr356 allows short distance PT steps to be coupled with longer-distance ET steps. In the α2 subunit, the studies discussed above have suggested that the radical transport pathway through Tyr731→Tyr730→Cys439 involves collinear PCET, where the electron and proton are transported together.
Figure 20.

Putative PCET pathway for radical transport from Tyr122• to C439 in E. coli RNR, based on conserved residues, crystal structures of subunits β2 and α2, and a docking model. Tyr356 has not been located in either the β2 or α2 crystal structure; other distances are taken from crystal structures. Reprinted with permission from Reference 231. Copyright 2006 American Chemical Society.
5. Concluding Remarks
Proton-coupled electron transfers are key reactions in many biological redox processes. Work on model systems has shown that stepwise pathways often involve high energy intermediates, whereas concerted reactions require synchronous proton and electron motions. Much additional work will be required before we will be able to design redox machines that run efficiently by incorporating low barrier CPET reactions.
Extensive investigations have shown that proton acceptors positioned close to redox active amino acid residues are able to couple distant ET reactions to short-range proton transfer. Notably, model systems with bases appended to phenols have been employed to study PCET involving intramolecular proton transfer in solution.
Research on the roles protons play in electron flow though molecules is expanding at a rapid pace. Work on biological as well as small model systems will continue to advance our understanding of the inner workings of protein redox machines. We urgently need to ramp up both theoretical and experimental investigations of the factors that control the coupling of electron and proton motions to build a firm foundation for the design and construction of artificial photosynthetic machines to produce clean fuel from sunlight and water.
Acknowledgments
Our work is supported by NIH (DK019038, GM068461), NSF Center for Chemical Innovation Grant (CHE-0802907), GCEP (Stanford), CCSER (Gordon and Betty Moore Foundation), and the Arnold and Mabel Beckman Foundation.
Biography
 Jillian L. Dempsey (center) received an S.B. in chemistry from MIT in 2005. She completed her PhD at Caltech in 2010 under the mentorship of Jay Winkler and Harry Gray. Currently, she is beginning a postdoctoral position in the laboratory of Daniel Gamelin at the University of Washington.
Jillian L. Dempsey (center) received an S.B. in chemistry from MIT in 2005. She completed her PhD at Caltech in 2010 under the mentorship of Jay Winkler and Harry Gray. Currently, she is beginning a postdoctoral position in the laboratory of Daniel Gamelin at the University of Washington.
Jay R. Winkler (right) received a B.S. in chemistry from Stanford University in 1978 and a PhD from Caltech in 1984, where he worked with Harry Gray. After carrying out postdoctoral work with Norman Sutin and Thomas Netzel at Brookhaven National Laboratory, he was appointed to the permanent staff in the Brookhaven Chemistry Department. In 1990 he moved to Caltech, where he is currently the Director of the Beckman Institute Laser Resource Center, Member of the Beckman Institute, and a Powering the Planet NSF CCI Principal Investigator.
Harry B. Gray (right) received a B.S. in chemistry from Western Kentucky University in 1957. and a PhD from Northwestern University, where he worked with Fred Basolo and Ralph Pearson. After carrying out postdoctoral work with Carl Ballhausen at the University of Copenhagen (1960–1961), he joined the chemistry faculty of Columbia University. In 1966 he moved to Caltech, where he is the Arnold O. Beckman Professor of Chemistry, Founding Director of the Beckman Institute, and a Powering the Planet NSF CCI Principal Investigator.
References
- 1.Saraste M. Science. 1999;283:1488. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 2.Hinchliffe P, Sazanov LA. Science. 2005;309:771. doi: 10.1126/science.1113988. [DOI] [PubMed] [Google Scholar]
- 3.Sazanov LA, Hinchliffe P. Science. 2006;311:1430. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 4.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawaitoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 5.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 6.Sun F, Huo X, Zhai YJ, Wang AJ, Xu JX, Su D, Bartlam M, Rao ZH. Cell. 2005;121:1043. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 7.Zhang ZL, Huang LS, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Nature. 1998;392:677. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 8.Xia D, Yu CA, Kim H, Xian JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Science. 1997;277:60. doi: 10.1126/science.277.5322.60. [DOI] [PubMed] [Google Scholar]
- 9.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Science. 1998;281:64. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 10.Abrahams JP, Leslie AGW, Lutter R, Walker JE. Nature. 1994;370:621. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 11.Huynh MHV, Meyer TJ. Chem Rev. 2007;107:5004. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray HB, Winkler JR. Proc Nat Acad Sci USA. 2005;102:3534. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265. [Google Scholar]
- 14.Gray HB, Winkler JR. Q Rev Biophys. 2003;36:341. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 15.Beratan DN, Betts JN, Onuchic JN. Science. 1991;252:1285. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 16.Skourtis SS, Beratan DN. In: Electron Transfer - from Isolated Molecules to Biomolecules Part 1. Prigogine I, Rice SA, editors. Vol. 106 John Wiley & Sons, Inc; New York: 1999. [Google Scholar]
- 17.Beratan DN, Skourtis SS. Curr Opin Chem Biol. 1998;2:235. doi: 10.1016/s1367-5931(98)80065-3. [DOI] [PubMed] [Google Scholar]
- 18.Prytkova TR, Kurnikov IV, Beratan DN. Science. 2007;315:622. doi: 10.1126/science.1134862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winkler JR, Nocera DG, Yocom KM, Bordignon E, Gray HB. J Am Chem Soc. 1982;104:5798. [Google Scholar]
- 20.Gray HB. Chem Soc Rev. 1986;15:17. [Google Scholar]
- 21.Mayo SL, Ellis WR, Crutchley RJ, Gray HB. Science. 1986;233:948. doi: 10.1126/science.3016897. [DOI] [PubMed] [Google Scholar]
- 22.Chang IJ, Gray HB, Winkler JR. J Am Chem Soc. 1991;113:7056. [Google Scholar]
- 23.Bjerrum MJ, Casimiro DR, Chang IJ, Di Bilio AJ, Gray HB, Hill MG, Langen R, Mines GA, Skov LK, Winkler JR, Wuttke DS. J Bioenerg Biomembr. 1995;27:295. doi: 10.1007/BF02110099. [DOI] [PubMed] [Google Scholar]
- 24.Mines GA, Bjerrum MJ, Hill MG, Casimiro DR, Chang I-J, Winkler JR, Gray HB. J Am Chem Soc. 1996;118:1961. [Google Scholar]
- 25.Di Bilio AJ, Hill MG, Bonander N, Karlsson BG, Villahermosa RM, Malmström BG, Winkler JR, Gray HB. J Am Chem Soc. 1997;119:9921. [Google Scholar]
- 26.Di Bilio AJ, Dennison C, Gray HB, Ramirez BE, Sykes AG, Winkler JR. J Am Chem Soc. 1998;120:7551. [Google Scholar]
- 27.Tezcan FA, Crane BR, Winkler JR, Gray HB. Proc Nat Acad Sci USA. 2001;98:5002. doi: 10.1073/pnas.081072898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray HB, Winkler JR. Chem Phys Lett. 2009;483:1. doi: 10.1016/j.cplett.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartings MR, Kurnikov IV, Dunn AR, Winkler JR, Gray HB, Ratner MA. Coord Chem Rev. 2010;254:248. doi: 10.1016/j.ccr.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gray HB, Malmström BG. Biochemistry. 1989;28:7499. doi: 10.1021/bi00445a001. [DOI] [PubMed] [Google Scholar]
- 31.Therien MJ, Selman MA, Gray HB, Chang I-J, Winkler JR. J Am Chem Soc. 1990;112:2420. [Google Scholar]
- 32.Bowler BE, Raphael AL, Gray HB. Prog Inorg Chem. 1990;38:259. [Google Scholar]
- 33.Beratan DN, Onuchic JN, Betts JN, Bowler BE, Gray HB. J Am Chem Soc. 1990;112:7915. [Google Scholar]
- 34.Jacobs BA, Mauk MR, Funk WD, MacGillivray RTA, Mauk AG, Gray HB. J Am Chem Soc. 1991;113:4390. [Google Scholar]
- 35.Winkler JR, Gray HB. Chem Rev. 1992;92:369. [Google Scholar]
- 36.Wuttke DS, Bjerrum MJ, Winkler JR, Gray HB. Science. 1992;256:1007. doi: 10.1126/science.256.5059.1007. [DOI] [PubMed] [Google Scholar]
- 37.Wuttke DS, Bjerrum MJ, Chang I-J, Winkler JR, Gray HB. Biochim Biophys Acta. 1992;1101:168. [Google Scholar]
- 38.Casimiro DR, Wong L-L, Colón JL, Zewert TE, Richards JH, Chang I-J, Winkler JR, Gray HB. J Am Chem Soc. 1993;115:1485. [Google Scholar]
- 39.Casimiro DR, Richards JH, Winkler JR, Gray HB. J Phys Chem. 1993;97:13073. [Google Scholar]
- 40.Beratan DN, Onuchic JN, Winkler JR, Gray HB. Science. 1992;258:1740. doi: 10.1126/science.1334572. [DOI] [PubMed] [Google Scholar]
- 41.Karpishin TB, Grinstaff MW, Komar-Panicucci S, McLendon G, Gray HB. Structure. 1994;2:415. doi: 10.1016/s0969-2126(00)00043-5. [DOI] [PubMed] [Google Scholar]
- 42.Langen R, Colón JL, Casimiro DR, Karpishin TB, Winkler JR, Gray HB. J Biol Inorg Chem. 1996;1:221. [Google Scholar]
- 43.Gray HB, Winkler JR. J Electroanal Chem. 1997;438:43. [Google Scholar]
- 44.Ponce A, Gray HB, Winkler JR. J Am Chem Soc. 2000;122:8187. [Google Scholar]
- 45.Gray HB, Winkler JR. Biochim Biophys Acta, Bioenerg. 2010;1797:1563. doi: 10.1016/j.bbabio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Berglund J, Pascher T, Winkler JR, Gray HB. J Am Chem Soc. 1997;119:2464. [Google Scholar]
- 47.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlcek A, Jr, Richards JH, Winkler JR, Gray HB. Science. 2008;320:1760. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 48.Langen R, Chang IJ, Germanas JP, Richards JH, Winkler JR, Gray HB. Science. 1995;268:1733. doi: 10.1126/science.7792598. [DOI] [PubMed] [Google Scholar]
- 49.Regan JJ, Di Bilio AJ, Langen R, Skov LK, Winkler JR, Gray HB, Onuchic JN. Chem Biol. 1995;2:489. doi: 10.1016/1074-5521(95)90266-x. [DOI] [PubMed] [Google Scholar]
- 50.Smalley JF, Finklea HO, Chidsey CE, Linford MR, Creager SE, Ferraris JP, Chalfant K, Zawodzinsk T, Feldberg SW, Newton MD. J Am Chem Soc. 2003;125:2004. doi: 10.1021/ja028458j. [DOI] [PubMed] [Google Scholar]
- 51.Smalley JF, Feldberg SW, Chidsey CED, Linford MR, Newton MD, Liu YP. J Phys Chem. 1995;99:13141. [Google Scholar]
- 52.Crane BR, Di Bilio AJ, Winkler JR, Gray HB. J Am Chem Soc. 2001;123:11623. doi: 10.1021/ja0115870. [DOI] [PubMed] [Google Scholar]
- 53.Gray HB, Winkler JR. Annu Rev Biochem. 1996;65:537. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 54.Winkler JR, Di Bilio AJ, Farrow NA, Richards JH, Gray HB. Pure Appl Chem. 1999;71:1753. [Google Scholar]
- 55.Babini E, Bertini I, Borsari M, Capozzi F, Luchinat C, Zhang XY, Moura GLC, Kurnikov IV, Beratan DN, Ponce A, Di Bilio AJ, Winkler JR, Gray HB. J Am Chem Soc. 2000;122:4532. [Google Scholar]
- 56.Beratan DN, Onuchic JN. Photosynth Res. 1989;22:173. doi: 10.1007/BF00048296. [DOI] [PubMed] [Google Scholar]
- 57.Onuchic JN, Beratan DN, Winkler JR, Gray HB. Annu Rev Biophys Biomol Struct. 1992;21:349. doi: 10.1146/annurev.bb.21.060192.002025. [DOI] [PubMed] [Google Scholar]
- 58.McConnell H. J Chem Phys. 1961;35:508. [Google Scholar]
- 59.Zheng XH, Georgievskii Y, Stuchebrukhov AA. J Chem Phys. 2004;121:8680. doi: 10.1063/1.1796751. [DOI] [PubMed] [Google Scholar]
- 60.Prytkova TR, Kurnikov IV, Beratan DN. J Phys Chem B. 2005;109:1618. doi: 10.1021/jp0457491. [DOI] [PubMed] [Google Scholar]
- 61.Onuchic JN, Kobayashi C, Miyashita O, Jennings P, Baldridge KK. Philos Trans R Soc London, Ser B. 2006;361:1439. doi: 10.1098/rstb.2006.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skov LK, Pascher T, Winkler JR, Gray HB. J Am Chem Soc. 1998;120:1102. [Google Scholar]
- 63.Stubbe J, van der Donk WA. Chem Rev. 1998;98:705. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]
- 64.Aubert C, Vos MH, Mathis P, Eker AP, Brettel K. Nature. 2000;405:586. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 65.Sjoberg BM. Struct Bond. 1997;88:139. [Google Scholar]
- 66.Stubbe J, Nocera DG, Yee CS, Chang MC. Chem Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 67.Chang MC, Yee CS, Nocera DG, Stubbe J. J Am Chem Soc. 2004;126:16702. doi: 10.1021/ja044124d. [DOI] [PubMed] [Google Scholar]
- 68.Page CC, Moser CC, Chen XX, Dutton PL. Nature. 1999;402:47. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 69.Tommos C, Babcock GT. Biochim Biophys Acta. 2000;1458:199. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 70.Frey PA. Chem Rev. 1990;90:1343. [Google Scholar]
- 71.Berlin YA, Hutchison GR, Rempala P, Ratner MA, Michl J. J Phys Chem A. 2003;107:3970. [Google Scholar]
- 72.Connick WB, Di Bilio AJ, Hill MG, Winkler JR, Gray HB. Inorg Chim Acta. 1995;240:169. [Google Scholar]
- 73.Blanco-Rodriguez AM, Busby M, Gradinaru C, Crane BR, Di Bilio AJ, Matousek P, Towrie M, Leigh BS, Richards JH, Vlcek A, Jr, Gray HB. J Am Chem Soc. 2006;128:4365. doi: 10.1021/ja057451+. [DOI] [PubMed] [Google Scholar]
- 74.Gabrielsson A, Hartl F, Zhang H, Lindsay Smith JR, Towrie M, Vlcek A, Jr, Perutz RN. J Am Chem Soc. 2006;128:4253. doi: 10.1021/ja0539802. [DOI] [PubMed] [Google Scholar]
- 75.Cannizzo A, Blanco-Rodriguez AM, El Nahhas A, Sebera J, Zalis S, Vlcek A, Jr, Chergui M. J Am Chem Soc. 2008;130:8967. doi: 10.1021/ja710763w. [DOI] [PubMed] [Google Scholar]
- 76.Harriman A. J Phys Chem. 1987;91:6102. [Google Scholar]
- 77.Solar S, Getoff N, Surdhar PS, Armstrong DA, Singh A. J Phys Chem. 1991;95:3639. [Google Scholar]
- 78.Di Bilio AJ, Crane BR, Wehbi WA, Kiser CN, Abu-Omar MM, Carlos RM, Richards JH, Winkler JR, Gray HB. J Am Chem Soc. 2001;123:3181. doi: 10.1021/ja0043183. [DOI] [PubMed] [Google Scholar]
- 79.Miller JE, Gradinaru C, Crane BR, Di Bilio AJ, Wehbi WA, Un S, Winkler JR, Gray HB. J Am Chem Soc. 2003;125:14220. doi: 10.1021/ja037203i. [DOI] [PubMed] [Google Scholar]
- 80.Cordes M, Kottgen A, Jasper C, Jacques O, Boudebous H, Giese B. Angew Chem Int Ed. 2008;47:3461. doi: 10.1002/anie.200705588. [DOI] [PubMed] [Google Scholar]
- 81.Wang M, Gao J, Muller P, Giese B. Angew Chem Int Ed. 2009;48:4232. doi: 10.1002/anie.200900827. [DOI] [PubMed] [Google Scholar]
- 82.Mishra AK, Chandrasekar R, Faraggi M, Klapper MH. J Am Chem Soc. 1994;116:1414. [Google Scholar]
- 83.Mayer JM, Rhile IJ, Larsen FB, Mader EA, Markle TF, Dipasquale AG. Photosynth Res. 2006;87:1. doi: 10.1007/s11120-005-8164-3. [DOI] [PubMed] [Google Scholar]
- 84.Mayer JM, Rhile IJ. Biochim Biophys Acta, Bioenerg. 2004;1655:51. doi: 10.1016/j.bbabio.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 85.Reece SY, Nocera DG. Annu Rev Biochem. 2009;78:673. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Laviron E. J Electroanal Chem. 1981;124:1. [Google Scholar]
- 87.Biczók L, Linschitz H. J Phys Chem. 1995;99:1843. [Google Scholar]
- 88.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Philos Trans R Soc, B. 2006;361:1351. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reece SY, Lutterman DA, Seyedsayamdost MR, Stubbe J, Nocera DG. Biochemistry. 2009;48:5832. doi: 10.1021/bi9005804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cukier RI. J Phys Chem. 1994;98:2377. [Google Scholar]
- 91.Cukier RI, Nocera DG. Annu Rev Phys Chem. 1998;49:337. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- 92.Cukier RI. J Phys Chem. 1996;100:15428. [Google Scholar]
- 93.Cukier RI. J Phys Chem B. 2002;106:1746. [Google Scholar]
- 94.Cukier RI. Biochim Biophys Acta, Bioenerg. 2004;1655:37. doi: 10.1016/j.bbabio.2003.06.011. [DOI] [PubMed] [Google Scholar]
- 95.Decornez H, Hammes-Schiffer S. J Phys Chem A. 2000;104:9370. [Google Scholar]
- 96.Hammes-Schiffer S. Acc Chem Res. 2001;34:273. doi: 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]
- 97.Hammes-Schiffer S. Chem Phys Chem. 2002;3:33. [Google Scholar]
- 98.Hammes-Schiffer S, Soudackov AV. J Phys Chem B. 2008;112:14108. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Venkataraman C, Soudackov AV, Hammes-Schiffer S. J Phys Chem C. 2008;112:12386. [Google Scholar]
- 100.Edwards SJ, Soudackov AV, Hammes-Schiffer S. J Phys Chem B. 2009;113:14545. doi: 10.1021/jp907808t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Navrotskaya I, Hammes-Schiffer S. J Chem Phys. 2009;131:024112. doi: 10.1063/1.3158828. [DOI] [PubMed] [Google Scholar]
- 102.Hammes-Schiffer S. Acc Chem Res. 2009;42:1881. doi: 10.1021/ar9001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Costentin C, Robert M, Savéant JM. J Electroanal Chem. 2006;588:197. [Google Scholar]
- 104.Costentin C, Robert M, Savéant JM, Tard C. Angew Chem Int Ed. 2010;49:3803. doi: 10.1002/anie.200907192. [DOI] [PubMed] [Google Scholar]
- 105.Sjödin M, Styring S, Wolpher H, Xu Y, Sun L, Hammarström L. J Am Chem Soc. 2005;127:3855. doi: 10.1021/ja044395o. [DOI] [PubMed] [Google Scholar]
- 106.Dixon WT, Murphy D. J Chem Soc, Faraday Trans 2. 1976;72:1221. [Google Scholar]
- 107.Bollinger JM, Jr, Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 108.Stubbe J. Curr Opin Chem Biol. 2003;7:183. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 109.Degray JA, Lassmann G, Curtis JF, Kennedy TA, Marnett LJ, Eling TE, Mason RP. J Biol Chem. 1992;267:23583. [PubMed] [Google Scholar]
- 110.Rogge CE, Liu W, Wu G, Wang LH, Kulmacz RJ, Tsai AL. Biochemistry. 2004;43:1560. doi: 10.1021/bi035717o. [DOI] [PubMed] [Google Scholar]
- 111.Schunemann V, Lendzian F, Jung C, Contzen J, Barra AL, Sligar SG, Trautwein AX. J Biol Chem. 2004;279:10919. doi: 10.1074/jbc.M307884200. [DOI] [PubMed] [Google Scholar]
- 112.Spolitak T, Dawson JH, Ballou DP. J Biol Inorg Chem. 2008;13:599. doi: 10.1007/s00775-008-0348-9. [DOI] [PubMed] [Google Scholar]
- 113.Jovanovic SV, Harriman A, Simic MG. J Phys Chem. 1986;90:1935. [Google Scholar]
- 114.Defelippis MR, Murthy CP, Faraggi M, Klapper MH. Biochemistry. 1989;28:4847. doi: 10.1021/bi00437a049. [DOI] [PubMed] [Google Scholar]
- 115.Defelippis MR, Murthy CP, Broitman F, Weinraub D, Faraggi M, Klapper MH. J Phys Chem. 1991;95:3416. [Google Scholar]
- 116.Biczók L, Bérces T, Linschitz H. J Am Chem Soc. 1997;119:11071. [Google Scholar]
- 117.Biczók L, Gupta N, Linschitz H. J Am Chem Soc. 1997;119:12601. [Google Scholar]
- 118.Yiu DTY, Lee MFW, Lam WWY, Lau TC. Inorg Chem. 2003;42:1225. doi: 10.1021/ic026184v. [DOI] [PubMed] [Google Scholar]
- 119.Song N, Stanbury DM. Inorg Chem. 2008;47:11458. doi: 10.1021/ic8015595. [DOI] [PubMed] [Google Scholar]
- 120.Costentin C, Louault C, Robert M, Savéant J-M. J Am Chem Soc. 2008;130:15817. doi: 10.1021/ja8064914. [DOI] [PubMed] [Google Scholar]
- 121.Costentin C, Robert M, Savéant J-M. Acc Chem Res. 2010;43:1019. doi: 10.1021/ar9002812. [DOI] [PubMed] [Google Scholar]
- 122.Costentin C, Louault C, Robert M, Savéant J-M. Proc Natl Acad Sci U S A. 2009;106:18143. doi: 10.1073/pnas.0910065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonin J, Costentin C, Louault C, Robert M, Routier M, Savéant J-M. Proc Natl Acad Sci U S A. 2010;107:3367. doi: 10.1073/pnas.0914693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sjödin M, Ghanem R, Polivka T, Pan J, Styring S, Sun LC, Sundstrom V, Hammarström L. Phys Chem Chem Phys. 2004;6:4851. [Google Scholar]
- 125.Sjödin M, Irebo T, Utas Josefin E, Lind J, Merenyi G, Åkermark B, Hammarström L. J Am Chem Soc. 2006;128:13076. doi: 10.1021/ja063264f. [DOI] [PubMed] [Google Scholar]
- 126.Markle TF, Rhile IJ, Dipasquale AG, Mayer JM. Proc Nat Acad Sci USA. 2008;105:8185. doi: 10.1073/pnas.0708967105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kanamori D, Furukawa A, Okamura T, Yamamoto H, Ueyama N. Org Biomol Chem. 2005;3:1453. doi: 10.1039/b419361j. [DOI] [PubMed] [Google Scholar]
- 128.Rhile IJ, Mayer JM. J Am Chem Soc. 2004;126:12718. doi: 10.1021/ja031583q. [DOI] [PubMed] [Google Scholar]
- 129.Rhile IJ, Markle TF, Nagao H, DiPasquale AG, Lam OP, Lockwood MA, Rotter K, Mayer JM. J Am Chem Soc. 2006;128:6075. doi: 10.1021/ja054167+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Costentin C, Robert M, Savéant J-M. J Am Chem Soc. 2007;129:9953. doi: 10.1021/ja071150d. [DOI] [PubMed] [Google Scholar]
- 131.Markle TF, Mayer JM. Angew Chem Int Ed. 2008;47:738. doi: 10.1002/anie.200702486. [DOI] [PubMed] [Google Scholar]
- 132.Costentin C, Robert M, Savéant J-M. J Am Chem Soc. 2006;128:4552. doi: 10.1021/ja060527x. [DOI] [PubMed] [Google Scholar]
- 133.Johannissen LO, Irebo T, Sjödin M, Johansson O, Hammarström L. J Phys Chem B. 2009;113:16214. doi: 10.1021/jp9048633. [DOI] [PubMed] [Google Scholar]
- 134.Gupta N, Linschitz H, Biczók L. Fullerene Sci Technol. 1997;5:343. [Google Scholar]
- 135.Lucarini M, Mugnaini V, Pedulli GF, Guerra M. J Am Chem Soc. 2003;125:8318. doi: 10.1021/ja034963k. [DOI] [PubMed] [Google Scholar]
- 136.Fecenko CJ, Meyer TJ, Thorp HH. J Am Chem Soc. 2006;128:11020. doi: 10.1021/ja061931z. [DOI] [PubMed] [Google Scholar]
- 137.Fecenko CJ, Thorp HH, Meyer TJ. J Am Chem Soc. 2007;129:15098. doi: 10.1021/ja072558d. [DOI] [PubMed] [Google Scholar]
- 138.Magnuson A, Berglund H, Korall P, Hammarström L, Åkermark B, Styring S, Sun L. J Am Chem Soc. 1997;119:10720. [Google Scholar]
- 139.Sjödin M, Styring S, Åkermark B, Sun L, Hammarström L. J Am Chem Soc. 2000;122:3932. [Google Scholar]
- 140.Sun LC, Hammarström L, Åkermark B, Styring S. Chem Soc Rev. 2001;30:36. [Google Scholar]
- 141.Sun LC, Burkitt M, Tamm M, Raymond MK, Abrahamsson M, LeGourrierec D, Frapart Y, Magnuson A, Kenez PH, Brandt P, Tran A, Hammarström L, Styring S, Åkermark B. J Am Chem Soc. 1999;121:6834. [Google Scholar]
- 142.Lomoth R, Magnuson A, Sjödin M, Huang P, Styring S, Hammarström L. Photosynth Res. 2006;87:25. doi: 10.1007/s11120-005-9005-0. [DOI] [PubMed] [Google Scholar]
- 143.Krishtalik LI. Biochim Biophys Acta, Bioenerg. 2003;1604:13. doi: 10.1016/s0005-2728(03)00020-3. [DOI] [PubMed] [Google Scholar]
- 144.Costentin C, Robert M, Savéant JM. J Am Chem Soc. 2007;129:5870. doi: 10.1021/ja067950q. [DOI] [PubMed] [Google Scholar]
- 145.Irebo T, Reece Steven Y, Sjödin M, Nocera Daniel G, Hammarström L. J Am Chem Soc. 2007;129:15462. doi: 10.1021/ja073012u. [DOI] [PubMed] [Google Scholar]
- 146.Reece SY, Nocera DG. J Am Chem Soc. 2005;127:9448. doi: 10.1021/ja0510360. [DOI] [PubMed] [Google Scholar]
- 147.Ishikita H, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2007;129:11146. doi: 10.1021/ja072708k. [DOI] [PubMed] [Google Scholar]
- 148.Burdinski D, Bothe E, Wieghardt K. Inorg Chem. 2000;39:105. doi: 10.1021/ic990755a. [DOI] [PubMed] [Google Scholar]
- 149.Burdinski D, Wieghardt K, Steenken S. J Am Chem Soc. 1999;121:10781. [Google Scholar]
- 150.Magnuson A, Frapart Y, Abrahamsson M, Horner O, Åkermark B, Sun LC, Girerd JJ, Hammarström L, Styring S. J Am Chem Soc. 1999;121:89. [Google Scholar]
- 151.Sun L, Raymond MK, Magnuson A, LeGourrierec D, Tamm M, Abrahamsson M, Kenez PH, Martensson J, Stenhagen G, Hammarström L, Styring S, Åkermark B. J Inorg Biochem. 2000;78:15. doi: 10.1016/s0162-0134(99)00200-7. [DOI] [PubMed] [Google Scholar]
- 152.Sun LC, Berglund H, Davydov R, Norrby T, Hammarström L, Korall P, Borje A, Philouze C, Berg K, Tran A, Andersson M, Stenhagen G, Martensson J, Almgren M, Styring S, Åkermark B. J Am Chem Soc. 1997;119:6996. [Google Scholar]
- 153.Sun LC, Hammarström L, Norrby T, Berglund H, Davydov R, Åndersson M, Borje A, Korall P, Philouze C, Almgren M, Styring S, Akermark B. Chem Commun. 1997:607. [Google Scholar]
- 154.Magnuson A, Anderlund M, Johansson O, Lindblad P, Lomoth R, Polivka T, Ott S, Stensjo K, Styring S, Sundstrom V, Hammarström L. Acc Chem Res. 2009;42:1899. doi: 10.1021/ar900127h. [DOI] [PubMed] [Google Scholar]
- 155.Costentin C, Robert M, Savéant JM, Teillout AL. Chem Phys Chem. 2009;10:191. doi: 10.1002/cphc.200800361. [DOI] [PubMed] [Google Scholar]
- 156.Costentin C, Robert M, Savéant JM, Teillout AL. Proc Nat Acad Sci USA. 2009;106:11829. doi: 10.1073/pnas.0905020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Haddox RM, Finklea HO. J Phys Chem B. 2004;108:1694. [Google Scholar]
- 158.Madhiri N, Finklea HO. Langmuir. 2006;22:10643. doi: 10.1021/la061103j. [DOI] [PubMed] [Google Scholar]
- 159.Maki T, Araki Y, Ishida Y, Onomura O, Matsumura Y. J Am Chem Soc. 2001;123:3371. doi: 10.1021/ja002453+. [DOI] [PubMed] [Google Scholar]
- 160.Benisvy L, Blake AJ, Collison D, Davies ES, Garner CD, McInnes EJL, McMaster J, Whittaker G, Wilson C. Dalton Trans. 2003:1975. [Google Scholar]
- 161.Thomas F, Jarjayes O, Jamet M, Hamman S, Saint-Aman E, Duboc C, Pierre JL. Angew Chem Int Ed. 2004;43:594. doi: 10.1002/anie.200352368. [DOI] [PubMed] [Google Scholar]
- 162.Lachaud F, Quaranta A, Pellegrin Y, Dorlet P, Charlot MF, Un S, Leibl W, Aukauloo A. Angew Chem Int Ed. 2005;44:1536. doi: 10.1002/anie.200461948. [DOI] [PubMed] [Google Scholar]
- 163.Moore GF, Hambourger M, Gervaldo M, Poluektov OG, Rajh T, Gust D, Moore TA, Moore AL. J Am Chem Soc. 2008;130:10466. doi: 10.1021/ja803015m. [DOI] [PubMed] [Google Scholar]
- 164.Benisvy L, Bill E, Blake AJ, Collison D, Davies ES, Garner CD, Guindy CI, McInnes EJL, McArdle G, McMaster J, Wilson C, Wolowska J. Dalton Trans. 2004:3647. doi: 10.1039/b410934a. [DOI] [PubMed] [Google Scholar]
- 165.Moore GF, Hambourger M, Kodis G, Michl W, Gust D, Moore TA, Moore AL. J Phys Chem B. 2010 doi: 10.1021/jp101592m. [DOI] [PubMed] [Google Scholar]
- 166.Barber J. Biochim Biophys Acta, Bioenerg. 1998;1365:269. doi: 10.1016/s0005-2728(98)00079-6. [DOI] [PubMed] [Google Scholar]
- 167.Nelson N, Ben-Shem A. Nat Rev Mol Cell Biol. 2004;5:971. doi: 10.1038/nrm1525. [DOI] [PubMed] [Google Scholar]
- 168.Okamura MY, Paddock ML, Graige MS, Feher G. Biochim Biophys Acta. 2000;1458:148. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 169.Tommos C, Babcock GT. Acc Chem Res. 1998;31:18. [Google Scholar]
- 170.Rappaport F, Guergova-Kuras M, Nixon PJ, Diner BA, Lavergne J. Biochemistry. 2002;41:8518. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- 171.Barber J. Q Rev Biophys. 2003;36:71. doi: 10.1017/s0033583502003839. [DOI] [PubMed] [Google Scholar]
- 172.Kim S, Liang J, Barry BA. Proc Natl Acad Sci U S A. 1997;94:14406. doi: 10.1073/pnas.94.26.14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vrettos JS, Limburg J, Brudvig GW. Biochim Biophys Acta. 2001;1503:229. doi: 10.1016/s0005-2728(00)00214-0. [DOI] [PubMed] [Google Scholar]
- 174.Meyer TJ, Huynh MHV, Thorp HH. Angew Chem Int Ed. 2007;46:5284. doi: 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]
- 175.Hoganson CW, Babcock GT. Science. 1997;277:1953. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 176.Kok B, Forbush B, McGloin M. Photochem Photobiol. 1970;11:457. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 177.Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Science. 2005;310:1019. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- 178.Yachandra VK, Sauer K, Klein MP. Chem Rev. 1996;96:2927. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- 179.Ruttinger W, Dismukes GC. Chem Rev. 1997;97:1. doi: 10.1021/cr950201z. [DOI] [PubMed] [Google Scholar]
- 180.Penner-Hahn JE. Metal Sites in Proteins and Models Redox Centres. Vol. 90 Springer-Verlag Berlin; Berlin: 1998. p. 33. [Google Scholar]
- 181.Nugent JHA, Rich AM, Evans MCW. Biochim Biophys Acta, Bioenerg. 2001;1503:138. doi: 10.1016/s0005-2728(00)00223-1. [DOI] [PubMed] [Google Scholar]
- 182.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 183.Goussias C, Boussac A, Rutherford AW. Philos Trans R Soc London, Ser B. 2002;357:1369. doi: 10.1098/rstb.2002.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sauer K, Yachandra VK. Biochim Biophys Acta, Bioenerg. 2004;1655:140. doi: 10.1016/j.bbabio.2003.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rappaport F, Diner BA. Coord Chem Rev. 2008;252:259. [Google Scholar]
- 186.Dau H, Haumann M. Coord Chem Rev. 2008;252:273. [Google Scholar]
- 187.Ahlbrink R, Haumann M, Cherepanov D, Bogershausen O, Mulkidjanian A, Junge W. Biochemistry. 1998;37:1131. doi: 10.1021/bi9719152. [DOI] [PubMed] [Google Scholar]
- 188.Diner BA, Force DA, Randall DW, Britt RD. Biochemistry. 1998;37:17931. doi: 10.1021/bi981894r. [DOI] [PubMed] [Google Scholar]
- 189.Chu H-A, Nguyen AP, Debus RJ. Biochemistry. 1995;34:5839. doi: 10.1021/bi00017a016. [DOI] [PubMed] [Google Scholar]
- 190.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Nature. 2005;438:1040. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 191.Hays AMA, Vassiliev IR, Golbeck JH, Debus RJ. Biochemistry. 1999;38:11851. doi: 10.1021/bi990716a. [DOI] [PubMed] [Google Scholar]
- 192.Rappaport F, Lavergne J. Biochemistry. 1997;36:15294. doi: 10.1021/bi971287o. [DOI] [PubMed] [Google Scholar]
- 193.Diner BA, Nixon PJ, Farchaus JW. Curr Opin Struct Biol. 1991;1:546. [Google Scholar]
- 194.Debus RJ. Biochim Biophys Acta, Bioenerg. 2001;1503:164. doi: 10.1016/s0005-2728(00)00221-8. [DOI] [PubMed] [Google Scholar]
- 195.Mamedov F, Sayre RT, Styring S. Biochemistry. 1998;37:14245. doi: 10.1021/bi980194j. [DOI] [PubMed] [Google Scholar]
- 196.Hays AM, Vassiliev IR, Golbeck JH, Debus RJ. Biochemistry. 1998;37:11352. doi: 10.1021/bi980510u. [DOI] [PubMed] [Google Scholar]
- 197.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. Biochemistry. 1999;38:9495. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 198.Rappaport F, Boussac A, Force DA, Peloquin J, Brynda M, Sugiura M, Un S, Britt RD, Diner BA. J Am Chem Soc. 2009;131:4425. doi: 10.1021/ja808604h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Babcock GT, Espe M, Hoganson C, LydakisSimantiris N, McCracken J, Shi WJ, Styring S, Tommos C, Warncke K. Acta Chem Scand. 1997;51:533. doi: 10.3891/acta.chem.scand.51-0533. [DOI] [PubMed] [Google Scholar]
- 200.Tommos C, Hoganson CW, Di Valentin M, Lydakis-Simantiris N, Dorlet P, Westphal K, Chu HA, McCracken J, Babcock GT. Curr Opin Chem Biol. 1998;2:244. doi: 10.1016/s1367-5931(98)80066-5. [DOI] [PubMed] [Google Scholar]
- 201.Hoganson CW, LydakisSimantiris N, Tang XS, Tommos C, Warncke K, Babcock GT, Diner BA, McCracken J, Styring S. Photosynth Res. 1995;46:177. doi: 10.1007/BF00020428. [DOI] [PubMed] [Google Scholar]
- 202.Westphal KL, Tommos C, Cukier RI, Babcock GT. Curr Opin Plant Biol. 2000;3:236. [PubMed] [Google Scholar]
- 203.Lavergne J, Junge W. Photosynth Res. 1993;38:279. doi: 10.1007/BF00046752. [DOI] [PubMed] [Google Scholar]
- 204.Barber J. Inorg Chem. 2008;47:1700. doi: 10.1021/ic701835r. [DOI] [PubMed] [Google Scholar]
- 205.Iwata S, Barber J. Curr Opin Struct Biol. 2004;14:447. doi: 10.1016/j.sbi.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 206.Barber J, Ferreira K, Maghlaoui K, Iwata S. Phys Chem Chem Phys. 2004;6:4737. [Google Scholar]
- 207.De Las Rivas J, Barber J. Photosynth Res. 2004;81:329. doi: 10.1023/B:PRES.0000036889.44048.e4. [DOI] [PubMed] [Google Scholar]
- 208.Ishikita H, Saenger W, Loll B, Biesiadka J, Knapp EW. Biochemistry. 2006;45:2063. doi: 10.1021/bi051615h. [DOI] [PubMed] [Google Scholar]
- 209.Bollinger JM., Jr Science. 2008;320:1730. doi: 10.1126/science.1160001. [DOI] [PubMed] [Google Scholar]
- 210.Jordan A, Reichard P. Annu Rev Biochem. 1998;67:71. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 211.Stubbe J, Riggs-Gelasco P. Trends Biochem Sci. 1998;23:438. doi: 10.1016/s0968-0004(98)01296-1. [DOI] [PubMed] [Google Scholar]
- 212.Uhlin U, Eklund H. Nature. 1994;370:533. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 213.Sjoberg BM. Structure. 1994;2:793. doi: 10.1016/s0969-2126(94)00080-8. [DOI] [PubMed] [Google Scholar]
- 214.Ekberg M, Potsch S, Sandin E, Thunnissen M, Nordlund P, Sahlin M, Sjoberg BM. J Biol Chem. 1998;273:21003. doi: 10.1074/jbc.273.33.21003. [DOI] [PubMed] [Google Scholar]
- 215.Ekberg M, Sahlin M, Eriksson M, Sjoberg BM. J Biol Chem. 1996;271:20655. doi: 10.1074/jbc.271.34.20655. [DOI] [PubMed] [Google Scholar]
- 216.Rova U, Goodtzova K, Ingemarson R, Behravan G, Graslund A, Thelander L. Biochemistry. 1995;34:4267. doi: 10.1021/bi00013a016. [DOI] [PubMed] [Google Scholar]
- 217.Rova U, Adrait A, Potsch S, Graslund A, Thelander L. J Biol Chem. 1999;274:23746. doi: 10.1074/jbc.274.34.23746. [DOI] [PubMed] [Google Scholar]
- 218.Climent I, Sjoberg BM, Huang CY. Biochemistry. 1992;31:4801. doi: 10.1021/bi00135a009. [DOI] [PubMed] [Google Scholar]
- 219.Sahlin M, Lassmann G, Potsch S, Sjoberg BM, Graslund A. J Biol Chem. 1995;270:12361. doi: 10.1074/jbc.270.21.12361. [DOI] [PubMed] [Google Scholar]
- 220.Katterle B, Sahlin M, Schmidt PP, Potsch S, Logan DT, Graslund A, Sjoberg BM. J Biol Chem. 1997;272:10414. doi: 10.1074/jbc.272.16.10414. [DOI] [PubMed] [Google Scholar]
- 221.Ekberg M, Birgander P, Sjoberg BM. J Bacteriol. 2003;185:1167. doi: 10.1128/JB.185.4.1167-1173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JH. J Am Chem Soc. 2000;122:12195. [Google Scholar]
- 223.Krebs C, Chen SX, Baldwin J, Ley BA, Patel U, Edmondson DE, Huynh BH, Bollinger JM. J Am Chem Soc. 2000;122:12207. [Google Scholar]
- 224.Nordlund P, Sjoberg BM, Eklund H. Nature. 1990;345:593. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 225.Nordlund P, Eklund H. J Mol Biol. 1993;232:123. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 226.Reece SY, Stubbe J, Nocera DG. Biochim Biophys Acta, Bioenerg. 2005;1706:232. doi: 10.1016/j.bbabio.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 227.Chang MC, Yee CS, Stubbe J, Nocera DG. Proc Nat Acad Sci USA. 2004;101:6882. doi: 10.1073/pnas.0401718101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hogbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzian F. Proc Natl Acad Sci U S A. 2003;100:3209. doi: 10.1073/pnas.0536684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Reece SY, Nocera DG. In: Quantum Tunnelling Enzyme-Catal React. Allermann RK, Scrutton NS, editors. RSC Publishing; Cambridge, U.K: 2009. [Google Scholar]
- 230.Tong W, Burdi D, Riggs-Gelasco P, Chen S, Edmondson D, Huynh BH, Stubbe J, Han S, Arvai A, Tainer J. Biochemistry. 1998;37:5840. doi: 10.1021/bi9728811. [DOI] [PubMed] [Google Scholar]
- 231.Seyedsayamdost MR, Yee CS, Reece SY, Nocera DG, Stubbe J. J Am Chem Soc. 2006;128:1562. doi: 10.1021/ja055927j. [DOI] [PubMed] [Google Scholar]
- 232.Yee CS, Chang MC, Ge J, Nocera DG, Stubbe J. J Am Chem Soc. 2003;125:10506. doi: 10.1021/ja036242r. [DOI] [PubMed] [Google Scholar]
- 233.Yee CS, Seyedsayamdost MR, Chang MC, Nocera DG, Stubbe J. Biochemistry. 2003;42:14541. doi: 10.1021/bi0352365. [DOI] [PubMed] [Google Scholar]
- 234.Seyedsayamdost MR, Reece SY, Nocera DG, Stubbe J. J Am Chem Soc. 2006;128:1569. doi: 10.1021/ja055926r. [DOI] [PubMed] [Google Scholar]
- 235.Seyedsayamdost MR, Xie J, Chan CTY, Schultz PG, Stubbe J. J Am Chem Soc. 2007;129:15060. doi: 10.1021/ja076043y. [DOI] [PubMed] [Google Scholar]
- 236.Climent I, Sjoberg BM, Huang CY. Biochemistry. 1991;30:5164. doi: 10.1021/bi00235a008. [DOI] [PubMed] [Google Scholar]
- 237.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. J Am Chem Soc. 2007;129:8500. doi: 10.1021/ja0704434. [DOI] [PubMed] [Google Scholar]
- 238.Chang CJ, Chang MCY, Damrauer NH, Nocera DG. Biochim Biophys Acta, Bioenerg. 2004;1655:13. doi: 10.1016/j.bbabio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 239.Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnstrom K, Sjoberg BM, Eklund H. Structure. 1997;5:1077. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 240.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. J Am Chem Soc. 2006;128:13654. doi: 10.1021/ja0636688. [DOI] [PubMed] [Google Scholar]
- 241.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. J Am Chem Soc. 2007;129:13828. doi: 10.1021/ja074452o. [DOI] [PMC free article] [PubMed] [Google Scholar]