

Abstract
Low levels of reactive oxygen species (ROS) can function as redox-active signaling messengers, whereas high levels of ROS induce cellular damage. Menadione generates ROS through redox cycling, and high concentrations trigger cell death. Previous work suggests that menadione triggers cytochrome c release from mitochondria, while other studies implicate activation of the mitochondrial permeability transition poreas the mediator of cell death. We investigated menadione-induced cell death in genetically modified cells lacking specific death-associated proteins. In cardiomyocytes, oxidant stress was assessed using the redox sensor RoGFP, expressed in the cytosol or the mitochondrial matrix. Menadione elicited rapid oxidation in both compartments, while it decreased mitochondrial potential and triggered cytochrome c redistribution to the cytosol. Cell death was attenuated by N-acetyl cysteine and exogenous glutathione (GSH), or by over-expression of cytosolic or mitochondria-targeted catalase. By contrast, no protection was observed in cells over-expressing Cu, Zn-SOD or MnSOD. Over-expression of antiapoptotic Bcl-XLprotected against staurosporine-induced cell death, but it failed to confer protection against menadione. Genetic deletion of Bax and Bak, cytochrome c, cyclophilin D or caspase-9 conferred no protection against menadione-induced cell death. However, cells lacking PARP-1 showed a significant decrease in menadione-induced cell death. Thus, menadione induces cell death through the generation of oxidant stress in multiple subcellular compartments, yet cytochromec, Bax/Bak, caspase-9 and cyclophilin D are dispensable for cell death in this model. These studies suggest that multiple redundant cell death pathways are activated by menadione, but that PARP plays an essential role in mediating each of them.
Index Terms: Reactive oxygen species, apoptosis, mitochondria, redox cycling agents, RoGFP
Introduction
At low levels, intracellular oxidants function as redox-active messengers in signal transduction pathways involving the responses to growth factors, hypoxia, and other receptor-ligand systems [1–7]. However, higher levels of oxidant stress can produce oxidative damage to lipids, proteins, RNA and DNA, which can trigger cell death by apoptosis and/or necrosis [8–17]. A large number of studies have used exogenously applied reactive oxygen species (ROS) to investigate both the signaling and cytotoxic responses to oxidant stress.
Menadione is a polycyclic aromatic ketone that can function as a precursor in the synthesis of Vitamin K. This compound generates intracellular ROS at multiple cellular sites through futile redox cycling. At low concentrations (e.g., 2 μM), menadione-mediated oxidants trigger redox-dependent gene expression responses [18]. For example, low levels of menadione-induced oxidant stress have been shown to mimic endogenous oxidant signals that trigger protection against ischemic injury in the heart [19].
However, higher concentrations of menadione induce toxic oxidant stress associated with tissue injury, mitochondrial DNA damage and cell death [18,20–22]. Previous studies suggest that menadione induces lethality by activating programmed cell death. For example, apoptosis in pancreatic acinar cells treated with menadione was inferred from the observed increases in propidium iodide uptake and Annexin V staining that were inhibited by the caspase inhibitor Z-VADfmk [23]. Those responses were associated with decreases in mitochondrial potential and with a redistribution of cytochrome c from the mitochondria to the cytosol. Mitochondrial depolarization in response to menadione was blocked by pretreatment with bonkrekic acid, an inhibitor of the adenine nucleotide translocator in the mitochondrial inner membrane and a putative component of the mitochondrial permeability transition pore (mPTP). These observations led to the conclusion that apoptosis, induced by ROS-mediated activation of the mPTP, was responsible for the cell death response to menadione treatment [23]. In a relatedstudy, Criddle et al. concluded that menadione activates apoptosis by increasing ROS production through a redox-cycling mechanism [24].
While multiple studies suggest the involvement of apoptosis in the response to menadione, important inconsistencies arise. For example, in neonatal rat cardiomyocytes and H9c2 cardiomyoblasts, Hou and Hsu found that menadione and staurosporine each caused translocation of the proapoptotic protein Bax from the cytosol to the mitochondria. This response triggered the release of cytochrome c to the cytoplasm, which then initiated apoptosis[25]. However, Bax translocation to the outer mitochondrial membrane does not require activation of the mitochondrial permeability transition pore[26], and its oligomerization in the outer membrane causes cytochrome c release without inducing depolarization of the inner membrane [27]. In fact, cells from mice carrying a genetic knockout of cyclophilin D, a principal regulator of the mPTP, are fully capable of undergoing apoptosis in response to staurosporine or other Bax/Bak-dependent activators [28,29]. Therefore, although the studies of Gerasimenko et al. and of Hou and Hsu both invoked apoptosisin the cell death induced by menadione, the mechanisms of apoptotic induction in their findings are at odds. Importantly, neitherstudy demonstratedthat protection can be conferred by genetic interventions that inactivate apoptosis or inhibit mPTP opening.
The present study therefore sought to clarify the mechanisms of cell death induced by menadione. As the subcellular distribution of oxidant stress has never been documented, we began by comparing the cytosolic and mitochondrial distribution of oxidant stresses induced by menadione, and by examining the role of superoxide versus H2O2in the oxidant-mediated cell death. We then explored the effects of menadione on mitochondrial function, in terms of morphology, membrane potential and cytochrome c release. We then used genetic approaches to explore the requirement of specific death-related genes in the cellularresponse to menadione. Our data reveal that menadione induces oxidant stress in both the cytosol and the mitochondrial matrix. The oxidant stress activates redundant cell death pathways, all of which are linked by the common involvement of poly (ADP-ribose) polymerase-1 (PARP-1).
Materials and Methods
Cell culture
Embryonic chick cardiomyocytes were prepared as previously described and grown at a density of 1 × 105 on 25 mm glass coverslips or 6 well plates in a humidified incubator[30]. Experiments were performed on spontaneously contracting cells at 3–5 days after isolation. Primary mouse embryonic fibroblasts (MEFs) derived from wild type and Bax-Bak double knockout mice, developed by a collaboration between the laboratories of Craig Thompson and Stanley Korsmeyer [27], were immortalized with a plasmid containing SV40 genomic DNA. Immortalized wild-type MEFs were also used to develop a stable cell line over-expressing Bcl-XL. MEFs derived from mice with targeted deletion of Caspase-9, along with wild-type controls, were obtained from Dr. Richard Flavell [31]. MEFs with genetic deletion of Ppif, the gene encoding cyclophilin D, were derived from Ppif−/− mice developed in the laboratory of Stanley Korsmeyer [28]and bred into the C57BL/6 background, along with isogenic wild type control mice. MEFs were also derived from mice carrying a targeted deletion of the PARP-1 gene, strain 129S-PARP1tm1Zqw, obtained from The Jackson Laboratory and developed by Wang et al. [32]. Embryonic cells lacking cytochrome c were obtained from Dr. Celeste Simon at the University of Pennsylvania[33], who derived these along with wild type controls from embryos obtained by matings of mice heterozygous with respect to the cytochrome c gene [34] obtained from the Jackson Laboratory. MEFculture s were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum, L-glutamine (200 mmol/L) and antibiotics (pen/strep).
Redox Measurements
Cardiomyocytes plated on 25 mm coverslips were transduced with an adenovirus to express the ratiometric redox sensor RoGFP in the cytosol or targeted to the mitochondrial matrix [35,36]. After allowing 24–48 hr for expression, cells were studied in a flow through chamber on an inverted microscope as previously described [37]. The chamber was comprised of a stainless steel spacer ring separating two coverslips, one of which carried the cultured cells. The space between the cover slips was perfused with buffered salt solutions at a flow rate of 0.5 ml/min. All solutions were pre-equilibrated in a heated, water jacketed reservoir (Radnoti Glass Co.). The chamber was mounted on a heated stage (37º C) on an inverted microscope (Nikon) fitted with filter wheels (Sutter) and a cooled CCD detector (Cascade 650, Photometrics). Cells were superfused with balanced salt solution (BSS) (NaCl 117 mmol/L, KCl 4 mmol/L, NaHCO3 18 mmol/L, MgSO4 0.76 mmol/L, NaH2PO4 1 mmol/L, CaCl2 1.21 mmol/L and glucose 5.6 mmol/L) bubbled with a normoxic gas mixture (5% CO2/21% O2/74% N2). Cells were studied with a 40X objective using excitation (405 nm and 484 nm) and emission (535 nm) filters. Images were acquired every 60 sec, corrected for background, and ratiometric images were calculated as the ratio of 484/405. Individual cell responses were obtained by selecting regions of interest encompassing single cells; these were averaged to provide a description of the overall response. After studying the effects of menadione (25 μM) on ratiometric behavior, the cells were superfused with dithiothreitol (DTT, 1 mM) and then t-butyl hydroperoxide (TBH, 1 μM) to obtain the maximum reduced and oxidized ratios, respectively. Baseline and menadione-induced ratios were expressed as percent oxidation as previously described [35].
Cell Viability
Cell death was assessed using propidium iodide (PI, 5 μmol/L) in BSS buffer. The dye is excluded from live cells, but enters upon disruption of the plasma membrane integrity. Cells were grown in 6-well plates and PI was added to the media along with menadione. Cells were studied on a fluorescence microscope 10X objective (excitation: 555 nm, emission: 605 nm) equipped with a computer-controlled stage. Plates were kept in the cell culture incubator except during brief periods when they were mounted on the microscope stage for sequential assessment of cell morphology and viability over time. The plate was mounted on the stage, and multiple X–Y locations were programmed to allow repeated measurements of the same cell regions during the experiment. Images were obtained after 0, 1, 3, 4 and 6 hrs of treatment with menadione at various doses. Cells were kept in a 37° C incubator (5% CO2, 21% O2) except for the brief periods required to obtain images. Metamorph imaging software was used to count nuclei stained by PI. After 6 hrs, cells were permeablized by adding digitonin (300 mmol/L) to the perfusate for 45 min. Cell death during the experiment was calculated as the percent of the post-digitonin value.
A lactate dehydrogenase (LDH) cytotoxicity assay (BioVision) was also used to assess cell viability in response to menadione, staurosporine treatment and serum withdrawal. LDH is localized within lysosomes of live cells, and is released into the extracellular supernatant upon disruption of plasma membrane integrity. The LDH assay was used according to the manufacturer’s specifications with minor modifications. Supernatants were collected after treatment with staurosporine (1 μmol/L in 1.5 ml of serum-free media), menadione (25 μM) or prolonged serum withdrawal. Cells were incubated in Triton-X (1.5 ml of 2% in serum-free media) for 30 min. Lysates were collected and centrifuged at 11,000 RPM for 10 min. Samples were mixed with the reaction mixture and incubated for 30 min in a 96 well plate, at which point the absorbance was measured at 490 nm. Cytotoxicity was calculated as the absorbance in the supernatant divided by the sum of absorbance in the supernatant and lysates.
Mitochondrial Fragmentation Assay
Cells expressing the mitochondrial RoGFP protein were exposed to staurosporine, menadione or BSS. Fluorescence microscopy was evaluated using a 60× objective lens to evaluate the mitochondrial morphology. Under baseline conditions, all mitochondria displayed a characteristic irregular curvilinear pattern. Subsequent mitochondrial fragmentation was defined as a distinct change in the morphology of the mitochondria from the normal reticular pattern to one resembling numerous small vacuoles with complete loss of the curvilinear morphology (see figure 3). This appeared to be an all-or-nothing response as there were no cells that displayed partial fragmentation. The total population of cells displaying this pattern was divided by the total number of cells in the field to arrive at a percentage of cells displaying mitochondrial fragmentation. Multiple experiments were performed for each condition to obtain an average and standard error of the mean.
Figure 3.

Mitochondrial morphology in cardiomyocytes treated with menadione. (A) Control cells demonstrate a normal reticular mitochondrial morphology. Cells exposed to menadione (25 μM, 4 hrs) demonstrated significant mitochondrial fragmentation. (B) Analysis of cells exhibiting fragmented mitochondrial morphology after treatment with menadione, or the apoptosis-inducing agent staurosporine (STS, 1 μM, 6 hrs). (n=3; *STS vs. controls p<0.05; **menadione vs. STS and controls p<0.05)
Mitochondrial Membrane Potential
ΔΨm was assessed with tetramethylrhodamine methyl ester (TMRE) under non-quenching conditions as previously described [37]. Cells were loaded with TMRE (100 nmol/L) at 37° C for 30 min. The media was then replaced with a maintenance concentration of TMRE (25 nmol/L). Mitochondrial fluorescence intensity was assessed using excitation at 555 nm and emission at 605 nm. Images were obtained during baseline and at periodic intervals thereafter (10X objective). Intensity values were corrected for background. Stage positions were programmed using data acquisition software (Metamorph, Universal Imaging) allowing repeated images to be obtained from the same fields of cells throughout each experiment. Data are reported as percentage of baseline fluorescence intensity.
DNA fragmentation assay
Cardiomyocytes (2.1 ×106) were disrupted using lysis buffer, and the DNA was extracted using phenol:chloroform after 2 hrs of proteinase K digestion at 50 C. Genomic DNA (1.5 μg per lane) was treated with 100 μg/ml of RNase A and run on a 2.0% agarose gel containing 0.06 mg/ml of ethidium bromide. Apoptotic DNA fragments consist of multimers of 180–200 base pairs and appear as a DNA ladder. DNA of non-apoptotic populations of cells has a high molecular weight and does not migrate far into the gel.
Immunofluorescence labeling of cytochrome c
To determine subcellular localization of cytochrome c, both treated and untreated cells were fixed in 3% paraformaldehyde with 0.02% glutaraldehyde in PBS for 15 min and subsequently permeablized with ice-cold methanol. Cells were incubated in 0.5 mg/ml sodium borohydride (Sigma–Aldrich) solution, blocked for 30 min in PBS containing 1% goat serum, and incubated overnight at 4 C with 1:100 dilution of mouse anti-cytochrome c antibody (BD Bioscience). Cells were then incubated with a fluorescent secondary antibody (Alexa Fluor 488-labeled anti-mouse IgG, 1:100) for 1 hr at room temperature and visualized under laser scanning confocal microscopy using a 40X objective. Cells with a normal mitochondrial localization of cytochrome c exhibit a fluorescence pattern that is punctuate in appearance, while cells that have undergone redistribution of cytochrome c to the cytosol exhibit a more diffuse pattern of fluorescence. Diffuse cells were enumerated independently by two blinded investigators who examined three separate visual fields each containing 50–60 cells. The percentage of positive cells obtained by the two evaluators was averaged, and mean values across groups were then calculated. Values were expressed as percent of total cells. In separate experiments, cardiomyocytes were infected for 48 hrs with an adenovirus expressing Green Fluorescent Protein (GFP) targeted to the mitochondrial matrix using the targeting sequence from cytochrome oxidase subunit IV, in order to label the mitochondria. Cells were then subjected to menadione or staurosporine. They were then fixed, immunostained for cytochrome c, and were imaged using a laser scanning confocal microscope (Zeiss, 60X) to assess the extent of co-localization.
Reagents
Adenoviruses expressing catalase or mitochondria-targeted catalase were obtained as a gift from Drs. Bai and Cederbaum [38]. Adenoviruses expressing Cu, Zn-SOD (SOD 1) or Mn-SOD (SOD 2) were obtained from the Gene Transfer Vector Core Facility at the University of Iowa [39,40]. Digitonin was obtained from Aldrich. Other chemicals were obtained from Sigma.
Statistical Analysis
Data were analyzed by ANOVA. When a statistically significant effect was detected, individual differences were explored using a Newman-Keuls post hoc analysis. Statistical significance was determined at the 0.05 level. Data are expressed as mean values ± S.E.M..
Results
Detection of oxidant stress during menadione treatment using RoGFP
Cardiomyocytes were transduced with an adenovirus to express the ratiometric redox sensor RoGFP in the cytosol. Under baseline conditions, reducing conditions predominated in the cytosol where the RoGFP oxidation averaged between 20 and 30% (Fig. 1A). Menadione (25 μM) caused progressive oxidation of the RoGFP, which reached 74% oxidation within 15 min. Thus, menadione produces a rapid oxidative stress to cysteine thiols in the cytosol.
Figure 1.
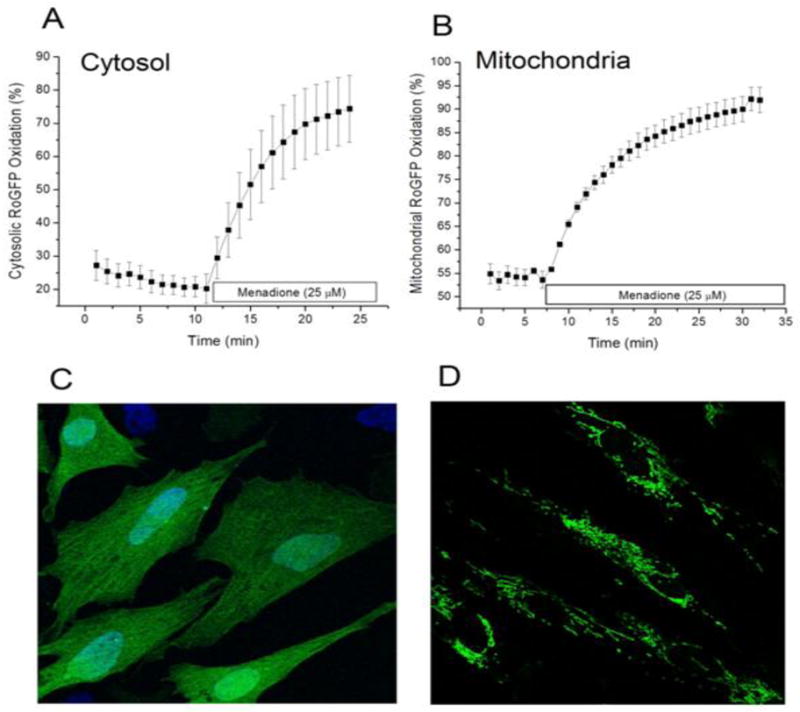
Oxidant stress generated by menadione in cultured cardiomyocytes. Cells expressing the redox-sensitive ratiometric sensor RoGFP were superfused with media while ratiometric images were obtained on an inverted fluorescence microscope. (A) Percent oxidation of the RoGFP sensor under baseline conditions and during menadione treatment (n=4). (B) Percent oxidation of RoGFP targeted to the mitochondrial matrix during menadione treatment (n=5). (C) Representative fluorescence image of cells expressing RoGFP. (D) Representative fluorescence image of cells expressing RoGFP targeted to the mitochondrial matrix.
To examine the consequences menadione on redox conditions in the mitochondrial matrix, cardiomyocytes were transduced with an adenovirus expressing RoGFP containing the mitochondrial targeting sequence for cytochrome oxidase subunit IV. Unlike the pattern seen with the cytosolic expressing construct (Fig. 1C), cells transduced with this virus expressed a reticular pattern of fluorescence indicative of mitochondrial localization (Fig. 1D). On average, the basal level of RoGFP oxidation was greater in the matrix (55% oxidized) compared with cytosol, reflecting a greater degree of basal oxidant stress in that compartment. With menadione, the percent oxidation in the matrix increased progressively, reaching 90% oxidation after 20 min. These results do not establish the site of ROS generation by menadione, but they do reveal that it produces rapid and significant increases in oxidant stress in multiple subcellular compartments.
Cell death responses to menadione
To assess cell death in response to menadione, cardiomyocytes were treated with different concentrations of menadione (0–25 μmol/L) for 6 hrs. Cell membrane irregularities and evidence of nuclear PI uptake were noted after 4–6 hrs of menadione (25 μmol/L) exposure (Fig. 2A). Using different doses of menadione, a time- and dose-dependent cell death response, evidenced by PI uptake, was observed between 4 and 6 hrs (Fig. 2B), even though the oxidant stress was apparent much earlier (Fig. 1).
Figure 2.

Menadione-induced cell death in cardiomyocytes. (A) Phase contrast images of cardiomyocytes at baseline and after 6 hrs of menadione (25 μmol/L) treatment (upper panel). Loss of plasma membrane integrity was confirmed by propidium iodide uptake (lower panels). (B) Menadione-induced cell death occurred in a time- and dose-dependent manner (Mean values ± SE, [0–3 hrs, n=4] [4–6 hrs, n=6]; * p<.05 compared with 0 hr controls). (C) Mitochondrial potential, as assessed using tetramethylrhodamine ethyl ester (TMRE). Fluorescence intensity was measured in cardiomyocytes loaded with TMRE under control conditions and during exposure to menadione (25 μmol/L). A significant decrease in TMRE fluorescence was detected as early as 1 hr after menadione treatment. Values are expressed as percent of baseline intensity (n=4; * p<.05) compared with controls. White bars indicate control cells and dark bars indicate menadione-treated cells.
To assess the effects of menadione treatment on the mitochondrial membrane potential (ΔΨm), cells loaded with TMRE were imaged during 30 min of baseline normoxia, and after 1, 3 and 4 hrs of menadione exposure (Fig. 2C). Compared with control cells studied over an equivalent period, menadione-treated cells showed a progressive decrease in mitochondrial potential, as indicated by a decrease in TMRE fluorescence, which reached 39±3% of the initial intensity after 4 hrs. The dissipation of TMRE fluorescence is consistent with opening of the mitochondrial permeability transition pore (mPTP) which results in mitochondrial depolarization, swelling and cytochrome c release [37,41–45].
Given the extent of oxidant stress induced by menadione in the mitochondrial matrix, and the subsequent decrease in mitochondrial potential, we assessed mitochondrial morphology in cells expressing the mitochondria-targeted RoGFP. After treatment with menadione (25 μM, 4hrs), cells were fixed with paraformaldehyde, washed, and mounted for visualization by laser scanning confocal microscopy (60X objective, excitation 488 nm, emission 530 nm)(Fig 3A). Significant mitochondrial fragmentation/fission was noted in response to menadione, compared with untreated cells. Enumeration of cells displaying mitochondrial fragmentation showed that a greater percentage of cells experienced mitochondrial fragmentation after menadione than after exposure to the apoptosis-inducing agent, staurosporine (1 μmol/L, 6hrs, Fig 3B). This suggests that mitochondrial fission and loss of membrane potential are early and critical events of menadione-induced cell death but not in staurosporine-induced mitochondrial apoptosis.
Protection against menadione-induced cell death by antioxidants
To determine whether pharmacological antioxidants were protective against menadione-induced cell death, cardiomyocytes were incubated with antioxidants prior to and during exposure to menadione (25 μmol/L). The glutathione precursor N-acetyl-L-cysteine (NAC, 500 μmol/L) significantly decreased cell death (4±2%) compared to controls (41±2.5%) after 4 hrs of menadione exposure (Fig. 4A). Similarly, administration of reduced glutathione (GSH, 100 μmol/L) decreased cell death to 6±0.3%. However, no significant protection was conferred by the iron chelator deferoxamine mesylate (DFO, 100 μmol/L), the thiol reductant pyrolidine dithiocarbamate (PDTC, 10 μmol/L), or the glutathione peroxidase mimetic ebselen (25 mol/L).
Figure 4.

Effect of antioxidant therapy on menadione-induced cell death in cardiomyocytes. (A) N-acetyl-L-cysteine (NAC, 500 μmol/L) and reduced glutathione (GSH, 100 μmol/L) decreased cell death after 6 hrs of menadione (25 μmol/L) treatment compared with controls. DFO (100 μmol/L), pyrolidine dithiocarbamate (PDTC, 10 μmol/L), and ebselen (25 μmol/L) had no significant effect ([control, NAC, DFO, n=6], * p<.05 compared with control). (B) Western blot analysis of adenovirally transduced cardiomyocytes showing over-expression of catalase, mitochondria-targeted catalase, Cu, Zn-SOD and Mn-SOD after 48 hrs. (C) Cell death in adenovirally transduced cardiomyocytes treated with menadione (25 μmol/L, 6 hrs). Both cytosolic (1000 pfu/cell) and mitochondria-targeted catalase (500 pfu/cell) conferred protection compared with Y5-empty virus controls (1000 pfu/cell). Cu, Zn-SOD (SOD 1, 500 pfu/cell) and Mn-SOD over-expression (SOD 2, 20 pfu/cell) conferred no protective effect (n=3; * p<.05) compared with controls.
In other experiments, adenoviral vectors were used to express catalase, catalase targeted to the mitochondrial matrix, SOD 1 or SOD2 in cardiomyocytes. In cells exposed to menadione (25 μM) for 6 hrs, over-expression of catalase, or catalase targeted to the mitochondrial matrix (Fig. 4B), decreased cell death from 56±10% to 14±10% and 9±6%, respectively, compared with cells transduced with an empty adenoviral vector, Y5 (Fig. 3C). Cell death was not attenuated by over-expression of either SOD 1 or SOD2. Collectively, these findings implicate oxidant stress induced by hydrogen peroxide in the mitochondria and the cytosol as important contributors to menadione-induced cell death.
Markers of mitochondrial apoptosis in response to menadione
The release of cytochrome c from the intermembrane space to the cytosol represents a threshold event in the activation of mitochondrial apoptosis. Cardiomyocytes immunostained for cytochrome c under baseline conditions exhibited a punctuate pattern of fluorescence in confocal images, consistent with mitochondrial localization (Fig. 5A). This pattern was disrupted in cells treated with staurosporine, which increased the number of cells demonstrating a diffuse cytosolic pattern of fluorescence, indicative of cytochrome c release to the cytosol. A significant increase in the number of cells exhibiting a diffuse pattern of fluorescence was also detected after menadione (Fig. 5B), although cytochrome c redistribution was clearly a more predominant feature in staurosporine-treated cells. Moreover, staurosporine-treated cardiomyocytes developed a condensed cellular morphology and a significant redistribution of cytochrome c from the mitochondria to the cytosol after 6 hrs compared with control cells (Fig. 5A, B), although this was not accompanied by a simultaneous dissipation of the mitochondrial membrane potential, as assessed by TMRE fluorescence (Fig. 5C). This is consistent with studies suggesting that release of cytochrome c from the outer mitochondrial membrane is not accompanied by an early dissipation of inner membrane potential [42,46]. However, although evidence of DNA laddering was apparent in response to staurosporine, virtually no DNA laddering was apparent in the cells treated with menadione (Fig. 5D). These results indicate that, although staurosporine and menadione both induce cytochrome c release to the cytosol, important differences exist regarding the mechanisms of cell death induced by the two stimuli. These findings could be explained if ATP levels became depleted during menadione treatment, which could inhibit ATP-dependent caspase activity and DNA cleavage.
Figure 5.

Apoptotic markers in response to menadione or staurosporine (STS). (A) Immunofluorescence analysis of cytochrome c distribution in cardiomyocytes revealed a normal mitochondrial reticular pattern that co-localized with mitochondria-targeted GFP expression under control conditions. A diffuse cytosolic staining pattern developed after menadione treatment, and after staurosporine treatment. (B) A significant increase in the percentage of cells demonstrating cytochrome c release was observed after menadione treatment or staurosporine treatment (n=10; *p<0.05 compared with controls, **p<0.05 compared with menadione). (C) TMRE fluorescence measurements of mitochondrial potential at 6 and 18 hr after STS treatment (dark bars), compared with control cells (white bars). Although a small trend was noted, this did not reach statistical significance. (D) Left: Representative gel electrophoresis showing an absence of DNA fragmentation in cells exposed to menadione (25 μM) for 6 hrs compared with controls. (lanes: 1 - ladder, 2 - control, 3 - menadione). Right: Representative gel showing the presence of DNA fragmentation after staurosporine for 6 hrs. (lanes: 1 - ladder, 2 - control, 3 - staurosporine).
To clarify further the requirement for apoptosis in the response to menadione, we examined the role of BH domain-containing proteins in the regulation of cell death in these models. We induced over-expression of the antiapoptotic protein Bcl-XL using a recombinant adenovirus in cardiomyocytes (Fig. 6B), and found that it conferred protection against staurosporine (Fig. 6A) but not against menadione-induced cell death (Fig. 6C). Over-expression of Bcl-XL is known to inhibit cell death induced by BH domain-containing proteins that regulate outer mitochondrial membrane permeability to cytochrome c [25,34,47,48]. Of note, staurosporine treatment required 18 hrs to induce death in cells treated with the empty viral vector (Y5), while only 4 hrs were required to induce cell death with menadione (25 μmol/L). These findings indicate that menadione-induced cell death does not require activation of the mitochondrial apoptotic pathway, and inhibition of that pathway does not attenuate the cell death response to menadione.
Figure 6.

Role of mitochondrial apoptosis in staurosporine (STS)- and menadione-induced cell death in cardiomyocytes. (A) Cardiomyocytes infected with Y5 (empty vector) or Bcl-XL-expressing adenovirus (1000 MOI) for 36 hrs, then subjected to STS treatment (18 hrs) in serum-free media. LDH release into the supernatant was used to quantify cell death. Bcl-XL over-expression significantly attenuated STS-induced cell death in cardiomyocytes (n=4; * p<.05 compared with controls). (B) Bcl-XL protein over-expression levels with increasing viral titers, after 36 hr. (C) Bcl-XL over-expression (1000 MOI) in cardiomyocytes was not protective against menadione challenge compared with control cells infected with the Y5 virus (1000 MOI) (n=4). (D) Cell death in wild-type (WT) murine embryonic fibroblasts (MEFs), Bax/Bak double knockout (KO) MEFs, and Bcl-XL over-expressing MEFs in response to menadione challenge (25 μmol/L for 6 hrs) or serum withdrawal (72 hrs), as assessed by LDH release. Significant protection against serum-withdrawal-induced cell death was observed in Bax/Bak KO and Bcl-XL over-expressing cells, but these cells were not protected against menadione-induced cell death. (n=4, * p<.05)
To further explore the role of apoptosis in response to menadione, immortalized mouse embryonic fibroblast cells (MEFs) with genetic deletion of the pro-apoptotic proteins Bax and Bak (Bax−/−/Bak−/− double knockout) were compared with immortalized MEFs from isogenic wild-type animals, and with MEFs stably transfected to over-express Bcl-XL (Fig. 6D). Cells were subjected to menadione (25 μmol/L for 4 hrs) or serum withdrawal (72 hrs). Cell death was assessed by LDH cytotoxicity assays rather than by PI because the latter measurement was confounded by detachment of dying MEFs. Significant cell death was observed in response to menadione, but no protection was conferred either by loss of Bax and Bak or by Bcl-XL over-expression. By contrast, Bax/Bak deletion and Bcl-XL over-expression each conferred significant protection against serum removal from the growth media over 72 hrs, consistent with the involvement of mitochondrial apoptosis in response to that stimulus. These findings support the conclusion that, although markers of apoptosis are evident in cells exposed to menadione, neither Bax nor Bak are required for the cell death in response to menadione.
To further explore the requirement for apoptosis in the response to menadione, murine cells obtained from cyt c−/− embryos or from wild type littermate embryos were subjected to menadione (25 μmol/L) for 6 hr. Compared with untreated cells, menadione induced significant cell death, as indicated by LDH release. However, genetic deletion of cytochrome c did not confer any protection from death induced by this treatment (Fig. 7A). Absence of cyt c protein was confirmed by Western blotting. These findings reveal that menadione-induced cell death does not require cytochrome c release to the cytosol.
Figure 7.

(A) Cell death (LDH Assay) in wild type (n=3) and cytochrome c−/− (n=3) murine embryonic cells in response to menadione treatment (25 μM for 6 hrs). Genetic deletion of cytochrome c did not lessen cell death in response to menadione. Inset: Immunoblot of cytochrome c protein expression. (B) Cell death (LDH Assay) in wild type (n=3) and cyclophilin-D−/− murine embryonic fibroblasts (n=3) in response to menadione treatment (25 μM for 6 hrs). Genetic deletion of Cyp-D did not lessen cell death in response to menadione.
Role of the mitochondrial permeability transition pore
The mitochondrial depolarization observed in menadione-treated cells suggested that activation of the mitochondrial permeability transition pore may contribute to cell death in this model. Indeed, previous studies had implicated this pathway in pancreatic acinar celldeath after challenge with menadione [23]. To examine this mechanism more directly, we generated murine embryonic fibroblasts (MEFs) from mice lacking the Ppif gene [28] which encodes cyclophilin D, along with wild type control MEFs in the same genetic background. Menadione treatment induced significant cell deathin wild type cells, but this was not decreased in cells lacking cyclophilin D (Cyp-D−/−) (Fig. 7B). Mouse genotyping was confirmed by RT-PCR analysis (data not shown). These data indicate that the mPTP is not required for cell death in response to menadione.
To assess the requirement for caspase-9 in the cell death pathway, we compared menadione responsiveness in immortalized MEFs from wild type and caspase-9−/− mouse embryos. However, no decrease in menadione-induced cell death was observed in cells lacking caspase-9 (Fig. 8A). As caspase-9 activation occurs in response to cytochrome c release from mitochondria, these results are consistent with our earlier results showing a lack of protection by cytochrome c deletion.
Figure 8.

(A) Cell death (LDH Assay) in wild type (n=5) and immortalized caspase-9−/− murine embryonic fibroblasts (n=5) in response to menadione treatment (25 μM for 6 hrs). Genetic deletion of caspase-9 did not lessen cell death in response to menadione. (B) Cell death (LDH Assay) in wild type (n=4) and PARP-1−/− murine embryonic fibroblasts (n=5) in response to menadione treatment (25 μM for 6 hrs). Genetic deletion of PARP-1 significantly decreased cell death in response to menadione. (** p<.05 in comparison to wild type cells). (C) Immunoblot showing poly-(ADP-ribosylation) of cellular proteins in response to menadione (25 μM for 60 min).
Conceivably, oxidant stress-induced DNA damage could trigger activation of poly-(ADP ribose) polymerase (PARP), whose activation has been linked to mitochondrial disruption, cytochrome c release, and cell death associated with bioenergetic deficiency [49]. Although multiple members of the PARP superfamily have been described, PARP-1 represents an important component of that system [50]. Accordingly, we generated MEFs from wild type and PARP-1−/− murine embryos and compared their responsiveness to menadione challenge. Wild type MEFs treated with menadione for 60 min showed a significant increase in poly-(ADP ribosylation) (Fig 8C). While wild type MEFs exhibited cell death of approximately 40% on average, PARP-1−/− cells showed significantly less cell death (Fig. 8B). These findings indicate that, although multiple redundant cell death pathways are activated in response to menadione, PARP activation represents an important common component in the cell death pathways induced by menadione.
Discussion
Menadione has been used at low concentrations to mimic oxidant signaling in cells [18], and at higher concentrations to induce lethal oxidant stress [10,12,14,16,22–24]. The present study advances this previous work in four respects. First, we provide a quantitative comparison of the effects of menadione on oxidant stress in the cytosol and the mitochondrial matrix, using a ratiometric redox sensor. Second, we demonstrate that the cell death arising from menadione challenge requires hydrogen peroxide generation, but not superoxide. Third, we describe a genetic and functional dissection of the apoptotic and necrotic pathways activated by menadione, we find that both pathways become activated in response to menadione, and we show that each is sufficient to cause cell death. Finally, we demonstrate that genetic deletion of PARP-1 confers significant protection, indicating that PARP is a common element in the redundant death pathways activated during menadione challenge.
RoGFP measurements and oxidant stress
The ratiometric characteristics of the RoGFP sensor reveal that, under baseline conditions, proteins in the mitochondrial matrix are more highly oxidized than are proteins in the cytosol. Previous studies have shown that menadione induces oxidant stress in cells, but the question of whether menadione generates oxidant stress in subcellular compartments has not been addressed. Our results reveal that significant protein thiol oxidation begins in the cytosol immediately upon menadione addition, and oxidation continues to increase for more than 20 minutes. This pattern is mirrored in the mitochondrial matrix, where rapid oxidation of RoGFP was also observed. Of course these measurements do not reveal the site of oxidant generation, since H2O2 generated in one compartment could conceivably diffuse into adjacent compartments. Despite the rapid onset of oxidant stress, cell death does not develop for several hours, suggesting that a progressive accumulation of oxidative damage may be necessary to trigger the death response. The oxidant stress during menadione treatment was associated with progressive mitochondrial morphological fragmentation and depolarization, which was accompanied by cytochrome c release to the cytosol.
Protection by antioxidants
The antioxidant compounds NAC and GSH both conferred significant protection against menadione-induced cell death, which is consistent with the involvement of oxidant stress in that response. By contrast, the thiol reductant PDTC, the glutathione peroxidase mimetic ebselen, and the iron chelator DFO failed to protect. The differential responses to these various antioxidant compounds most likely is a reflection of their chemical dissimilarities, which would allow them to work by different mechanisms. When antioxidant enzymes were expressed using adenoviruses, catalase was highly protective when expressed in either the cytosol or the mitochondrial matrix, indicating that hydrogen peroxide is the principal oxidant responsible for the lethal effects of menadione in these cells. By comparison, neither SOD1nor SOD 2 over-expression conferred protection. This finding indicates that superoxide, by itself, is not as important for cell death as is H2O2. Hydrogen peroxide generated in one intracellular compartment can diffuse through membranes to access other compartments, which could explain why catalase expression in either compartment is protective against menadione.
Requirement for mitochondrial apoptosis in menadione-induced cell death
Menadione triggered a redistribution of cytochrome c from the intermembrane space to the cytosol. This finding is consistent with previous studies implicating apoptotic activation as the mechanism of death induced by menadione [23–25]. However, our data also reveal that mitochondrial apoptosis is not required for menadione-induced death. In this regard, over-expression of Bcl-XL in cardiomyocytes or MEFs, or genetic deletion of Bax and Bak in MEFs, each conferred protection against staurosporine and serum deprivation, yet these interventions conferred no protection against menadione. Similarly, deletion of caspase-9 and genetic deletion of cytochrome c conferred no protection. These findings indicate that although mitochondrial apoptosis becomes activated, it is dispensable for the cell death response.
Requirement for the mitochondrial permeability transition pores
Studies have suggested that menadione induces mPTP opening by generating excessive oxidant stress [43]. Opening of the mPTP induces necrotic cell death in response to oxidant stress or ischemia, through a process that is independent of Bax and Bak [28,41,51–53]. Subsequent swelling of the mitochondrial matrix can then cause rupture of the outer membrane, release of mitochondrial contents including cytochrome c to the cytosol, ATP depletion, and subsequent necrotic death. Other investigators report that the loss of mitochondrial potential with menadione is inhibited by bonkrekic acid, an inhibitor of the ATP/ADP translocator (ANT) in the inner membrane [23]. Those authors concluded that the mPTP must have mediated the cell death response, based on the putative role of ANT as a component of the pore. However, studies of mice with genetic deletions of ANTs demonstrated that the ATP/ADP translocators are not required for activation of the mPTP [54]. Hence, the significance of the inhibitory effect of bonkrekic acid on membrane depolarization in menadione treated cells is unknown, and the requirement for mPTP in the cell death remains uncertain. We observed a progressive loss in mitochondrial potential (ΔΨm) after menadione treatment, consistent with the activation of mPTP. However, MEFs from Cyp-D-deficient cells showed no protection against cell death, indicating that mPTP opening is not required for the death response. Cyp-D is a known promoter of mPTP opening [55], so the lack of protection with loss of Cyp-D demonstrates that the mPTP is not required for the cell death response.
Redundant cell death pathways induced by menadione
Collectively, our data reveal that multiple, redundant death pathways are activated in response to menadione-induced oxidant stress in cells. Although markers of apoptosis were observed, attempts to lessen cell death using genetic tools that inhibit apoptosis were unsuccessful. Similarly, evidence of mPTP opening was also seen, yet genetic deletion of a known modulator of the pore failed to confer protection. The most reasonable explanation for the lack of protection in these cases is that the alternate pathway remained active, providing a redundant death mechanism. However, our studies using cells carrying a genetic deletion of PARP-1 did demonstrate significant protection. This indicates that PARP activation is a common element in the two death pathways. Conceivably, menadione-induced oxidative damage to DNA could lead to excessive PARP activation, which would deplete cellular NAD+ levels and contribute to bioenergetic deficiency. PARP activation can also trigger cytochrome c and AIF release from mitochondria[56], leading to the early stages of apoptotic activation. Although early markers of apoptosis were observed, DNA laddering was absent, presumably reflecting the lack of ATP. Excessive oxidant stress may also activate mPTP opening, which would contribute further to the bioenergetic deficiency, cytochrome c loss, and further PARP activation.
Acknowledgments
The authors thank Craig B. Thompson for the Bax/Bak double knockout and wild-type MEFs. We also thank Dr. Celeste Simon for the cytochrome c embryonic cells. We are indebted to Bhumika Sharma for general technical assistance, Danijela Dokic for assistance with immunofluorescence studies, and Juan Li for the isolation of the cardiomyocytes.
This work was supported by HL35440, HL32646, HL66315, HL079650 (PTS) and the American Heart Association, Midwest Affiliates(GL and JS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW, Schumacker PT. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res. 2006;99:970–978. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 2.Mansfield KD, Simon MC, Keith B. Hypoxic reduction in cellular glutathione levels requires mitochondrial reactive oxygen species. J Appl Physiol. 2004;97:1358–1366. doi: 10.1152/japplphysiol.00449.2004. [DOI] [PubMed] [Google Scholar]
- 3.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxides-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 5.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 6.Shibata Y, Branicky R, Landaverde IO, Hekimi S. Redox regulation of germline and vulval development in Caenorhabditis elegans. Science. 2003;302:1779–1782. doi: 10.1126/science.1087167. [DOI] [PubMed] [Google Scholar]
- 7.Fallon JF, Lopez A, Ros MA, Savage MP, Olwin BB, Simandl BK. FGF-2: apical ectodermal ridge growth signal for chick limb development. Science. 1994;264:104–107. doi: 10.1126/science.7908145. [DOI] [PubMed] [Google Scholar]
- 8.Ostadal P, Elmoselhi AB, Zdobnicka I, Lukas A, Elimban V, Dhalla NS. Role of oxidative stress in ischemia-reperfusion-induced changes in Na+, K(+)-ATPase isoform expression in rat heart. Antioxid Redox Signal. 2004;6:914–923. doi: 10.1089/ars.2004.6.914. [DOI] [PubMed] [Google Scholar]
- 9.Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem. 2006;281:33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- 10.Morgan WA. DNA single-strand breakage in mammalian cells induced by redox cycling quinones in the absence of oxidative stress. J Biochem Toxicol. 1995;10:227–232. doi: 10.1002/jbt.2570100407. [DOI] [PubMed] [Google Scholar]
- 11.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim KA, Lee JY, Park KS, Kim MJ, Chung JH. Mechanism of menadione-induced cytotoxicity in rat platelets. Toxicol Appl Pharmacol. 1996;138:12–19. doi: 10.1006/taap.1996.0092. [DOI] [PubMed] [Google Scholar]
- 13.Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 14.Harrison JF, Hollensworth SB, Spitz DR, Copeland WC, Wilson GL, LeDoux SP. Oxidative stress-induced apoptosis in neurons correlates with mitochondrial DNA base excision repair pathway imbalance. Nucleic Acids Res. 2005;33:4660–4671. doi: 10.1093/nar/gki759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gauduel Y, Duvelleroy MA. Role of oxygen radicals in cardiac injury due to reoxygenation. J Mol Cell Cardiol. 1984;16:459–470. doi: 10.1016/s0022-2828(84)80617-3. [DOI] [PubMed] [Google Scholar]
- 16.Gabai VL, Seilanov AS, Makarova Y, Mosin AF. Oxidative stress, disturbance of energy balance, and death of ascites tumour cells under menadione (vitamin K3) action. Biomed Sci. 1990;1:407–413. [PubMed] [Google Scholar]
- 17.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 18.Chuang YY, Chen Y, Gadisetti, Chandramouli VR, Cook JA, Coffin D, Tsai MH, DeGraff W, Yan H, Zhao S, Russo A, Liu ET, Mitchell JB. Gene expression after treatment with hydrogen peroxide, menadione, or t-butyl hydroperoxide in breast cancer cells. Cancer Res. 2002;62:6246–6254. [PubMed] [Google Scholar]
- 19.Heinzel FR, Luo YK, Li XK, Boengler K, Buechert A, Garcia-Dorado D, Di LF, Schulz R, Heusch G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in Connexin 43 deficient mice. Circ Res. 2005;97:583–586. doi: 10.1161/01.RES.0000181171.65293.65. [DOI] [PubMed] [Google Scholar]
- 20.Luo X, Pitkanen S, Kassovska-Bratinova S, Robinson BH, Lehotay DC. Excessive formation of hydroxyl radicals and aldehydic lipid peroxidation products in cultured skin fibroblasts from patients with complex I deficiency. J Clin Invest. 1997;99:2877–2882. doi: 10.1172/JCI119481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakagami H, Satoh K, Hakeda Y, Kumegawa M. Apoptosis-inducing activity of vitamin C and vitamin K. Cell Mol Biol (Noisy -le-grand) 2000;46:129–143. [PubMed] [Google Scholar]
- 22.Grishko V, Solomon M, Wilson GL, LeDoux SP, Gillespie MN. Oxygen radical-induced mitochondrial DNA damage and repair in pulmonary vascular endothelial cell phenotypes. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1300–L1308. doi: 10.1152/ajplung.2001.280.6.L1300. [DOI] [PubMed] [Google Scholar]
- 23.Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Petersen OH, Watson AJ. Menadione-induced apoptosis: roles of cytosolic Ca(2+) elevations and the mitochondrial permeability transition pore. J Cell Sci. 2002;115:485–497. doi: 10.1242/jcs.115.3.485. [DOI] [PubMed] [Google Scholar]
- 24.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Petersen OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 25.Hou Q, Hsu YT. Bax translocates from cytosol to mitochondria in cardiac cells during apoptosis: development of a GFP-Bax-stable H9c2 cell line for apoptosis analysis. Am J Physiol Heart Circ Physiol. 2005;289:H477–H487. doi: 10.1152/ajpheart.00879.2004. [DOI] [PubMed] [Google Scholar]
- 26.Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(Suppl):S97–S107. S97–S10707. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 27.Wei MC, Zong WX, Cheng EHY, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacCregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schinzel AC, Takeuchi O, Huang ZH, Fisher JK, Zhou ZP, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, II, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 30.Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury in cardiac myocytes after simulated ischemia. Am J Physiol. 1996;270:H1334–H1341. doi: 10.1152/ajpheart.1996.270.4.H1334. [DOI] [PubMed] [Google Scholar]
- 31.Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- 32.Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 33.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- 35.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 36.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 37.Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–H558. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- 38.Bai J, Cederbaum AI. Adenovirus-mediated overexpression of catalase in the cytosolic or mitochondrial compartment protects against cytochrome P450 2E1-dependent toxicity in HepG2 cells. J Biol Chem. 2001;276:4315–4321. doi: 10.1074/jbc.M008895200. [DOI] [PubMed] [Google Scholar]
- 39.Zwacka RM, Dudus L, Epperly MW, Greenberger JS, Engelhardt JF. Redox gene therapy protects human IB-3 lung epithelial cells against ionizing radiation-induced apoptosis. Hum Gene Ther. 1998;9:1381–1386. doi: 10.1089/hum.1998.9.9-1381. [DOI] [PubMed] [Google Scholar]
- 40.Zwacka RM, Zhou W, Zhang Y, Darby CJ, Dudus L, Halldorson J, Oberley L, Engelhardt JF. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat Med. 1998;4:698–704. doi: 10.1038/nm0698-698. [DOI] [PubMed] [Google Scholar]
- 41.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 42.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 43.Kim JS, Jin YG, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH-and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 44.Petronilli V, Penzo D, Scorrano L, Bernardi P, Di LF. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem. 2001;276:12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- 45.Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen AL, Herman B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr. 1999;31:305–319. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]
- 46.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–1191. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 47.Rabkin SW. Prevention of staurosporine-induced cell death in embryonic chick cardiomyocyte is more dependent on caspase-2 than caspase-3 inhibition and is independent of sphingomyelinase activation and ceramide generation. Arch Biochem Biophys. 2001;390:119–127. doi: 10.1006/abbi.2001.2350. [DOI] [PubMed] [Google Scholar]
- 48.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 49.Yu SW, Wang HM, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 50.Gagne JP, Hendzel MJ, Droit A, Poirier GG. The expanding role of poly(ADP-ribose) metabolism: current challenges and new perspectives. Curr Opin Cell Biol. 2006;18:145–151. doi: 10.1016/j.ceb.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 53.Mizuta T, Shimizu S, Matsuoka Y, Nakagawa T, Tsujimoto Y. A Bax/Bak-independent mechanism of cytochrome c release. J Biol Chem. 2007;282:16623–16630. doi: 10.1074/jbc.M611060200. [DOI] [PubMed] [Google Scholar]
- 54.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cal JY, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 56.Chiarugi A, Moskowitz MA. Cell biology. PARP-1--a perpetrator of apoptotic cell death? Science. 2002;297:200–201. doi: 10.1126/science.1074592. [DOI] [PubMed] [Google Scholar]