

Abstract
Autosomal-dominant polycystic kidney disease (ADPKD) is the most common hereditary and systemic disorder associated with various cardiovascular complications. It has been implicated with dysfunction in primary cilia. We and others have shown that the immediate function of endothelial cilia is to sense extracellular signal. The long-term function of cilia is hypothesized to regulate cell cycle. Here, we show that ciliary function (polycystins) and structure (polaris) are required for proper cellular division. Cilia mutant cells undergo abnormal cell division with apparent defects in mitotic spindle formation, cellular spindle assembly checkpoint and centrosome amplification. Down-regulation of the chromosomal passenger survivin contributes to these abnormalities, which further result in cell polyploidy. Re-expression of survivin restores a competent spindle assembly checkpoint and reduces polyploidy. Aged animals show a more severe phenotype in cellular division, consistent with progression of cardiovascular complications seen in older ADPKD patients. For the first time, we show that structure and function of mechanosensory cilia are crucial in maintaining proper cellular proliferation. Furthermore, developmental aging plays a crucial role in the progression of these abnormal cellular phenotypes. We propose that abnormal function or structure of primary cilia not only causes failure to transmit extracellular signals, but also is associated with cytokinesis defects in both mice and humans with polycystic kidney disease.
INTRODUCTION
Autosomal-dominant polycystic kidney disease (ADPKD), a genetic disorder characterized by fluid-filled cysts in the kidney nephrons, is caused by a mutation in PKD1 or PKD2, the coding genes for polycystin-1 and -2, respectively. ADPKD is not only a kidney disease, but also a systemic disorder associated with cardiovascular complications such as cerebral intracranial and aortic aneurysms as well as cardiac valvular defects. These complications represent a continuous concern, particularly in older ADPKD patients (1). In particular, the molecular mechanism in ADPKD has been associated with dysfunction in primary cilia.
A primary cilium is a mechanosensory organelle to which sensory polycystin-1 and polycystin-2 complex is localized. A number of human diseases have been linked to defective cilium structure and function. These diseases have been grouped together as so-called ciliopathies, ranging from hypertension (2–4) to cystic diseases (5–8) and from obesity (9–11) to mental retardation (12). More importantly, aberrant proliferation rates have been observed in many cells with abnormal cilia. For example, abnormal structure and function of cilia results in cystic kidney, of which phenotypes have been associated with polyploidy and hyperplasia (13). It is, therefore, not surprising that primary cilia have been proposed as regulators of the cell cycle, in addition to their role as sensory organelles.
A cilium is projected from a basal body, which is one of the two centrioles possessed by a cell in normal resting state. The two centrioles form a body known as a centrosome. The centrosome duplicates during cell division, and the two centrosomes associate with anchoring points for mitotic spindles during mitosis. As such, resorption of the cilia and liberation of the centriole are crucial for centrosomal duplication and for cell division to proceed properly. Abnormal ciliary function or structure has thus been associated with the aberrant cell cycle due to high proliferation rate (14,15).
Despite the fact that primary cilia have been indicated in various diseases, including ADPKD, the fundamental understanding of primary cilia in cellular proliferation is still lacking. To study the role of cilia in cell division, we used Pkd1−/− and Tg737Orpk/Orpk endothelial cells that have been previously confirmed to have abnormal ciliary function and structure, respectively (3). In addition, we used primary endothelial cells from Pkd2 mice and samples from ADPKD patients to further verify our findings. Using various cells and experimental approaches, our data consistently show that proper function and structure of primary cilia are important and necessary to regulate the cell cycle. The similarity of cellular phenotypes (i.e. mitotic spindle defect and polyploidy) between cilium mutants and chromosomal passenger mutants (such as survivin knockout cells) further revealed a dramatic down-regulation of survivin in cells with abnormal function and structure of primary cilia. We propose that sensory cilia play an important role during mitotic events via regulation of the chromosomal passenger protein, survivin.
RESULTS
Cilia mutant cells are characterized by multipolar spindle formation, mitotic abnormality and centrosomal amplification
Working with the primary endothelial cells or cell lines from various cilia mutant mouse models and ADPKD patient cells, we consistently observed abnormal cellular division in these cells. To examine this phenomenon further, we undertook a simple analysis of mitotic events during different stages of the cell cycle. Our immunostaining studies with acetylated α-tubulin and actin confirm that in contrast to wild-type cells, Pkd1−/− and Tg737Orpk/Orpk endothelial cells are characterized by oversized or abnormal nuclei during interphase (Fig. 1A). Furthermore, tri- and multi-polar spindle formation, as well as micronucleation during the different stages of mitosis, can be observed in cells with abnormal cilia. The number of abnormal dividing cells is significantly greater in Pkd1−/− and Tg737Orpk/Orpk cells than in wild-type cells (Fig. 1B).
Figure 1.

Cell division in cilia mutant cells is characterized by mitotic abnormalities and multipolar spindle formation. (A) Endothelial wild-type, Pkd1−/− and Tg737Orpk/Orpk cells were immunostained with DAPI (blue), acetylated-α-tubulin (green; acet-α-tubulin) and phalloidin (red; actin) to visualize nucleus, cilium, mitotic spindle and actin cytoskeleton. Images were captured at different cell-cycle stages of interphase (I), prophase (II), metaphase (III), anaphase (IV), telophase (V) and cytokinesis (VI). (B) Abnormal dividing cells, indicated as percentages (bar graph) and number of dividing cells (table), are significantly greater in cells with abnormal cilia function (Pkd1−/−) and structure (Tg737Orpk/Orpk) than in wild-type cells. Original magnification, ×100. Asterisks denote significant difference toward corresponding wild-type groups.
It is generally known that improper regulation of centrosome duplication could result in multipolar spindle formation, asymmetric chromosome segregation and genomic instability (16). To study the involvement of abnormal cilia cells in centrosome duplication, we performed immunofluorescence analysis on wild-type, Pkd1−/− and Tg737Orpk/Orpk cells. Antibody to pericentrin is used as a specific marker to confirm the presence of centrosomes (Fig. 2A). We observe that unlike wild-type cells, Pkd1−/− and Tg737Orpk/Orpk cells are associated with multiple centrosomes and oversized nuclei during G0 (interphase). In addition to the multiple centrosomes, multi-polar spindle formation and micronucleation are also observed in cells with abnormal cilia during different stages of mitosis. To confirm the specificity of the centrosomal localization of pericentrin, wild-type, Pkd1−/− and Tg737Orpk/Orpk cells were co-stained with antibodies specific for γ-tubulin, a common centrosomal protein marker (Fig. 2B). This further verifies our observation that abnormal chromosomal segregation is associated with overduplication of centrosomes. Centrosome amplification is significantly greater in Pkd1−/− (15.9 and 22.1%) and Tg737Orpk/Orpk (20.4 and 33.7%) cells than in wild-type cells (1.4 and 2.4%) that are at resting and dividing stages, respectively (Fig. 2C).
Figure 2.

Cilia mutant cells are characterized by centrosome overduplication and abnormal cell division. (A) Endothelial wild-type, Pkd1−/− and Tg737Orpk/Orpk cells were immunostained with DAPI (blue), acetylated-α-tubulin (green; acet-α-tubulin) and pericentrin (red; centrin) to visualize nucleus, cilium, mitotic spindle and centrosome. Images were captured at different cell-cycle stages of interphase (I), prophase (II), metaphase (III), anaphase (IV), telophase (V) and cytokinesis (VI). (B) To confirm the specificity of centrosome localization, immunostaining was carried out with γ-tubulin (green) and pericentrin (red) in wild-type, Pkd1−/− and Tg737Orpk/Orpk non-dividing (interphase) and dividing (metaphase) cells. (C) Compared with wild-type cells, Pkd1−/− and Tg737Orpk/Orpk cells have significantly more centrosomes in both dividing and non-dividing stages. The number of dividing and non-dividing cells containing ≤2 or >2 centrosomes is indicated as percentages (bar graph) or total cell counts (table). Original magnification, ×100. Asterisks denote significant difference toward corresponding wild-type groups.
Centrosome overduplication is also associated with multiple cilia formation in Pkd1−/− endothelial cells. In contrast to wild-type endothelial cells which have one or no cilia at any time during their life cycles, Pkd1−/− cells showed significantly higher numbers of multiple cilia formation at resting stage (Fig. 3A). At resting stage, we observed about 11% of Pkd1−/− cells with more than one primary cilium (Fig. 3B). To confirm the occurrence of centrosome amplification in vivo, we analyzed aorta sections taken from the whole aorta of wild-type, Pkd2−/− and Tg737Orpk/Orpk mice. Note that we have previously shown that similar to Pkd1−/− cells, Pkd2−/− endothelial cells also have abnormal cilia function (2). Aorta sections from embryonic Pkd2−/− mice showed multiple cilia formation and centrosome amplification compared with sections from wild-type embryos (Fig. 3C). Moreover, we noticed an increased thickening of the intima layer in Pkd2−/− aortas when compared with wild-type aortas. Likewise, aorta sections from Tg737Orpk/Orpk but not wild-type mice showed a significant centrosome amplification phenotype (Fig. 3D).
Figure 3.

Centrosome overduplication results in multiple cilia formation in vitro and in vivo. (A) Endothelial wild-type (WT) and Pkd1−/− cells were immunostained with DAPI (blue), acetylated-α-tubulin (green; acet-α-tubulin) and pericentrin (red; centrin) to visualize nucleus, cilium and centrosome. (B) Compared with wild-type cells, Pkd1−/− cells have more cilia. Scores of randomly chosen non-dividing cells containing one or more cilia are indicated as percentages (bar graph) or total cell counts (table). (C) Aorta sections from wild-type and Pkd2−/− 15.5-day embryos were stained with acetylated α-tubulin (green), pericentrin (red) and counterstained with DAPI (blue) to visualize cilia, centrosome and nucleus, respectively. The boxes indicate areas of arteries that were further magnified, as shown in the insets, which reveal individual cells with multiple cilia and centrosomes in Pkd2−/−, but not in wild-type, aortas. Arrows indicate thickening of intima tissues. (D) Aorta sections from wild-type and Tg737Orpk/Orpk adult mice were stained with pericentrin (red) and counterstained with DAPI. The boxes indicate areas of arteries that were further magnified, as shown in the insets, which reveal individual cells with multiple centrosomes in Tg737Orpk/Orpk, but not in wild-type, aortas. Original magnification, ×100, except for those in phase contrast, ×20. Asterisks denote significant difference toward corresponding wild-type groups. Arrows indicate multiple primary cilia and/or centrosomes.
Cilia mutant cells are characterized by polyploidy and genomic instability
Aberrant mitotic spindle formation and centrosome amplification are known to be the primary causes of polyploidy followed by a possible genomic instability (16). We thus hypothesized that abnormal centrosome amplification would result in cell polyploidy. Polyploidity profiles of wild-type, Pkd1−/− and Tg737Orpk/Orpk cells were analyzed with propidium iodide (PI) and 5-bromodeoxyuridine (BrdU) staining. Although PI is generally used in quantifying DNA content, BrdU is used as a marker for cell division. Our flow cytometry analysis reveals the presence of abnormal polyploidy peaks in both Pkd1−/− and Tg737Orpk/Orpk cells but not in wild-type cells (Fig. 4A). More importantly, the polyploidy cells seem to be able to undergo cell division (Fig. 4B). Regardless, the abnormal polyploidy peaks (>4N) in both Pkd1−/− and Tg737Orpk/Orpk cells represent a significantly higher cell population than in wild-type cells (Fig. 4C). This indicates that the polyploidy levels in ciliary mutant cells are associated with centrosome over-amplification.
Figure 4.

Cells with abnormal cilia structure or function are characterized by cell polyploidy. (A) Endothelial wild-type, Pkd1−/− and Tg737Orpk/Orpk cells were analyzed by flow cytometry by PI and BrdU labeling. Representative BrdU and PI labeling profiles present an apparent polyploidy in Pkd1−/− and Tg737Orpk/Orpk cells, but not in wild-type cells. (B) Further analysis indicates that polyploidy Pkd1−/− and Tg737Orpk/Orpk cells are able to undergo mitosis. (C) Evaluation of the percentage of cells with normal DNA content (2N and 4N) shows that Pkd1−/− and Tg737Orpk/Orpk cells contain a greater DNA content (>4N), compared with wild-type cells. The number of dividing and non-dividing cells containing 2N, 4N and >4N DNA content is presented as percentages in the table. Asterisks denote significant difference toward corresponding wild-type groups. N > 5 for each genotype in flow cytometry experiments.
We next hypothesized that abnormal cell division and cell ploidy would result in genomic instability. Consistent with this view, karyotyping analysis showed abnormal genomic contents in both Pkd1−/− and Tg737Orpk/Orpk endothelial cell lines (data not shown). To confirm this observation in Pkd2−/− mice, we picked and karyotyped endothelia directly from Pkd2 mice. As expected, we consistently observed polyploidy in Pkd2−/− mice (Fig. 5A). A further study on individual chromosomes indicates that this cell is tetraploidy (Fig. 5B). In general, we observe more than 10% of Pkd2−/− endothelial cells with tetraploidy from three separate preparations with a total of 85 cells.
Figure 5.

Cilia mutant cells are characterized by genomic instability. Chromosomal identifications were carried out in mouse and human cells with abnormal function/structure. (A) Freshly isolated endothelial cells from wild-type and Pkd2−/− embryonic E15.5 aortas were studied for their genomic compositions. A simple chromosome count indicates the presence of polyploidy cells in Pkd2−/−. (B) Further characterization of individual chromosomes was performed with fluorescence probes. Identification of individual chromosomes indicates the tetraploidy nature of Pkd2−/− cells, suggesting abnormal segregation of chromosomes in cilia mutant cells. (C) A single endothelium from interlobar arteries of an ADPKD patient was examined for a simple chromosome count. (D) Further characterization of individual chromosomes was performed with fluorescence probes. Genomic instability is further characterized by addition or reduction on a specific chromosome. Abnormal segregation of chromosomes is also apparent in cells from patients with ADPKD.
To further confirm whether our finding is clinically relevant, we utilized samples from our ADPKD patients. Some of these samples have been previously described and partially characterized (2). These vascular tissues from the ADPKD patients have abnormal cilia function. A simple study by counting the chromosomal numbers indicates that non-ADPKD and some ADPKD vascular samples have normal chromosomal numbers of 23 pairs (Fig. 5C). In some ADPKD samples that showed normal chromosomal numbers, we surprisingly observed genomic instability (Fig. 5D). For example, although the ADPKD cell has 23 pairs of chromosomes, this cell had lost one sex chromosome and gained an additional chromosome #7. In general, however, we observed failure of chromosomal segregation, resulting in 46 pairs of chromosomes in ADPKD samples. Overall, we observed 31 individual cells (60%) from three ADPKD samples with normal genomic composition, whereas 7 (13%) and 14 (27%) ADPKD cells show tetraploidy and aneuploidy, respectively. There is no consensus on the chromosomal compositions in aneuploidy cells. This suggests that the genomic instability in ADPKD cells would probably result in tetraploidy, which in turn results in randomized aneuploidy cells.
Cilia mutant cells lack mitotic spindle checkpoint due to aberrant chromosomal passenger
To understand the cause of polyploidy, we performed live cell analysis to examine the mode of chromosome missegregation (17,18). Live imaging allowed us to determine at which mitotic stage the chromosomes fail to segregate and to examine whether over-amplification of the centrosome would allow cells to undergo cytokinesis. Wild-type cells from mouse endothelial cells underwent normal bipolar cell division in which the chromosomes and cytoplasm segregated equally to each side of the poles to generate two diploid daughter cells (Fig. 6A; Supplementary Material, Movie S1). The cells were able to execute all mitotic stages from prophase (chromosome condensation and nuclear membrane dissolution), metaphase (alignment of chromosomes at equatorial plate), anaphase (bipolar segregation of sister chromatids towards opposite poles), telophase (complete chromosome segregation and chromosome decondensation) and cytokinesis (complete separation of cytoplasm to produce two identical progeny cells). These progeny cells are normal and healthy and can continue normal subsequent divisions. A normal mitotic event usually lasts from 15 to 30 min.
Figure 6.

Mouse and human cilia mutant cells are characterized by the loss of mitotic spindle checkpoint. Live-imaging studies were performed to identify the cause of genomic stability in isolated endothelial cells from wild-type (A), Pkd1−/− (B) and Tg737Orpk/Orpk (C) mice. Isolated primary endothelial cells from a patient with ADPKD were also analyzed. (D) In wild-type endothelial cells, normal cell division results in two daughter cells through an even, bipolar chromosomal segregation. In mouse Pkd or primary human ADPKD cells, the DNA was condensed and was able to line up for chromosomal segregations. During anaphase, however, the cells were not able to separate the chromosomes equally. As a result, cytokinesis did not take place, and cells became polyploidy. The movies show that cilia mutant cells could complete the mitotic checkpoint assembly, but failed to maintain spindle tension during anaphase. Numbers indicate time in minutes and seconds as illustrated in the Supplementary Material, Movies S1–S4. Top panels are phase-contrast images to identify numbers of cells; bottom panels are fluorescence images of Hoechst. Arrows indicate cell boundary in phase contrast or cell nucleus in fluorescence images; each color represents one parental cell. Original magnification, ×40.
Mouse Pkd1−/− cells contain significantly larger nuclei and spend a longer period of time in pro-metaphase (Fig. 6B; Supplementary Material, Movie S2). Cells then undergo multiple unequal divisions of the nucleus and cytoplasm to yield several progeny cells that fail to undergo complete cell division and cytokinesis. Due to the failure of chromosomal separation, the daughter cells are re-united in one cell that encloses additional DNA contents. Likewise, Tg737Orpk/Orpk cells showed a very similar mitotic failure (Fig. 6C; Supplementary Material, Movie S3). The cell demonstrated that chromosomes were aligned along the tripolar plate and underwent tripolar cell division. The three progeny daughter cells resulting from this division failed to separate, and hence contracted into one cell that enclosed three nuclei. We further examined primary cells of human ADPKD cells that failed to show cytosolic calcium transient in response to fluid flow (Fig. 6D; Supplementary Material, Movie S4). The cell imaged during prophase spent a long period of time in pro-metaphase. The cell eventually divided into three multipolar cells with abnormal and unequal chromosomal (DNA) divisions in anaphase, which resulted in apparent abnormality during early telophase. Furthermore, the cell was unable to undergo cytokinesis, and the progeny cells failed to develop. As a result, the cell formed a gigantic body with polyploidy DNA content. These multi-nucleated cells, when imaged over a period of 1 week, underwent apoptotic cell death, probably due to the inability to execute further cell divisions (data not shown).
The phenotype of genomic instability and evidence from live-imaging analysis generally suggest that such phenotypic characteristics are associated with abnormality in various chromosomal passenger proteins such as survivin (19–22). In addition, live-imaging studies demonstrate that the cilia mutant cells failed to maintain an arrest in the presence of unattached chromosomes, which is another characteristic of abnormal survivin function (23). To confirm whether these abnormalities involve chromosomal passenger proteins, we performed mitotic-stress tests on these cells with taxol to stabilize spindle tubules, nocodazole to depolymerize mitotic tubules or Zm447435 to inhibit the function of chromosomal passenger protein. As expected, wild-type cells underwent mitotic arrest when treated with taxol, nocodazole or Zm447435 (Fig. 7A). The mitotic-stress tests also indicate that nocodazole, but not taxol, induced mitotic arrest in both Pkd1−/− (Fig. 7B) and Tg737Orpk/Orpk (Fig. 7C) cells, when compared with their corresponding control groups. These data suggest that chromosomal passenger protein is involved in the abnormalities seen in our Pkd1−/− and Tg737Orpk/Orpk cells. This further confirms that taxol-induced mitotic arrest involves a mechanism that requires chromosomal passenger protein (23). Consistent with this view, Zm447435 did not show any significant effects in Pkd1−/− or Tg737Orpk/Orpk cells (Fig. 7D). This indicates that the cilia mutant cells might have already had a repressed chromosomal passenger protein, which cannot be further depressed.
Figure 7.

Cilia mutant cells are characterized by aberrant chromosomal passenger protein. Mitotic-stress tests were performed with taxol to stabilize spindle tubules, nocodazole to depolymerize spindle tubules or Zm447435 to inhibit the function of chromosomal passenger protein. Vehicle treatments are indicated as control. Phosphorylated histone 3b (phospho H3) is used as a marker for nuclear division, and PI is to indicate DNA content. In wild-type cells (A), treatments with taxol, nocodazole or Zm447435 resulted in mitotic arrest. In Pkd1−/− (B) and Tg737Orpk/Orpk (C) cells, only nocodazole promoted mitotic arrest when compared with their corresponding controls. Consistent with the results, further inhibition of chromosomal passenger protein with Zm447435 could not promote mitotic arrest and that chromosomal passenger protein was required for mitotic arrest by taxol. Mitotic-stress test was measured by analyzing changes in resting cells (2N), which are PI and phospho-histone-negative, as indicated by the brackets. (D) These changes are reflected in % non-arrested cells from the total cell population. Asterisks denote significant difference toward corresponding control groups.
Survivin has an important role in the genomic instability observed in cilia mutant cells in vitro and in vivo
To examine the possibility of survivin as the chromosomal passenger protein responsible for the phenotypes observed in the cilia mutant cells, we performed western blot analysis on actively dividing wild-type, Pkd1−/− and Tg737Orpk/Orpk cells. Compared with wild-type cells, survivin expression was markedly down-regulated in Pkd1−/− and Tg737Orpk/Orpk cells (Fig. 8A). To verify this finding in vivo, total proteins were isolated from wild-type, Pkd2+/− and Pkd2−/− 15.5-day mouse embryos. Consistent with the data from the cell cultures, survivin expression was down-regulated in Pkd2−/− embryos, compared with wild-type or heterozygous embryos. A more detailed analysis suggests that survivin expression level is regulated at the transcriptional level in Pkd1−/− and Tg737Orpk/Orpk cells (Fig. 8B). Aurora B kinase, on the other hand, does not seem to provide any regulated pattern in cilia mutant cells.
Figure 8.

Survivin plays an important role in genomic instability of cilia mutant cells in vitro and in vivo. (A) Survivin expression was down-regulated in Pkd1−/− and Tg737Orpk/Orpk cells compared with wild-type cells. Likewise, survivin was down-regulated in Pkd2−/− embryos compared with wild-type embryos. Actin expression was used as a loading control. (B) Survivin transcripts were repressed in both Pkd1−/− and Tg737Orpk/Orpk cells. (C) Counterstaining with acetylated-α-tubulin (red) and DAPI (blue) was used to examine subcellular localization of survivin-GFP (green)-transfected cells at metaphase and telophase. (D) Flow cytometry studies show a decrease in polyploidy index in cilia mutant cells transfected with survivin-GFP, but not in those transfected with vector-GFP (empty vector, GFP). Because of the low transfection efficiency in the cells, only transfected (GFP-positive) cells were analyzed. Representative BrdU and PI labeling profiles show an apparent polyploidy in vector-only transfected Pkd1−/− and Tg737Orpk/Orpk cells. Polyploidy Pkd1−/− and Tg737Orpk/Orpk cells are substantially lower in those transfected with survivin-GFP. (E) The number of dividing and non-dividing cells containing 2N, 4N and >4N DNA content is presented as percentages in the table. Asterisks denote significant difference toward corresponding wild-type groups and significant difference between corresponding GFP and survivin-GFP groups.
To further confirm the role of survivin in mutant cells with abnormal cilia structure or function, we transfected the cells with survivin construct. Because the transfection efficiency of these cells is highly variable, we used green fluorescent protein (GFP)-containing construct to measure only those cells that were transfected successfully with either survivin construct or empty vector (control). Under resting state, survivin-GFP was mainly localized in the cytosol (data not shown). Survivin-GFP was co-localized at the mitotic plate and/or kinetochore in metaphase, whereas it was localized at the mid-body at late telophase (Fig. 8C). We also observed that transfected mutant cells still had abnormal division. In some cases, this was probably due to a lower expression level of survivin-GFP in that particular single cell. To obtain a more robust statistics, only survivin-GFP cells were analyzed for BrdU and PI uptakes using flow cytometry (Fig. 8D). The BrdU and PI data were tabulated with proper control groups (Fig. 8E). Our data show that although survivin did not completely rescue the cilia mutant cells, expression of survivin significantly corrected the genomic instability observed in cilia mutant cells.
Molecular convergence and developmental aging may also involve in polyploidy
We previously showed that acetylcholine-induced nitric oxide synthesis was intact in cilia mutant endothelial cells (3). In particular, cilia mutant cells seem to ‘behave' better after acetylcholine treatment, as indicated by numbers of centrosomes at resting or dividing states (Fig. 9A). Further analysis indicates that although acetylcholine tends to decrease centrosome amplification in cilia mutant cells, this effect was very minimal at best (Fig. 9B). Because survivin has been shown to be directly downstream of PI3K/Akt(PKB) signaling (24–26), which is also involved in cilia function (2), we examined whether PI3K/Akt(PKB) signaling is altered or can be induced with vascular endothelial growth factor (VEGF) in mutant cells (Fig. 9C). The western blot data indicate that although Akt levels were not altered, basal phosphorylation of Akt was consistently greater in cilia mutant cells than in wild-type cells. More important is that VEGF increases Akt phosphorylation and induces survivin expression, which can be blocked by either PI3K or Akt/PKB inhibitor. To correlate these results with cell ploidy level, we employed flow cytometry to analyze polyploid cells with various pharmacological combinations (Fig. 9D). In particular, VEGF but not acetylcholine tends to reduce polyploid cilia mutant cells (statistically significant in Pkd1−/− cells). This tendency was diminished when VEGF-induced survivin expression was blocked with either PI3K or Akt/PKB inhibitor. In Tg737Orpk/Orpk cells, PI3K or Akt/PKB inhibitor significantly worsened the cell polyploidity. Together, we propose a possible intra-molecular convergence paradigm that links cilia function, cellular growth and polyploidy (Fig. 9E).
Figure 9.

Effect of VEGF on cell ploidy is reversible by PI3K and Akt/PKB inhibitors. (A) Cells with and without 10 μm acetylcholine (ACh) treatment were analyzed with acetylated α-tubulin (green), pericentrin (red) and DAPI (blue) during resting or dividing state. Arrows indicate centrosomes. (B) Their centrosome numbers were calculated and presented in the table. Cells were treated with or without 40 ng/ml of VEGF, 10 μm Akt/PKB inhibitor (Akt inh) or 20 μm PI3K inhibitor (PI3K inh). (C) The cells were analyzed for survivin, phosphorylated-Akt (p-Akt), Akt, PI3K and GADPH. (D) Polyploid cells were studied with flow cytometry and plotted in the graph. (E) A hypothetical pathway is depicted to assess the molecular mechanism of polyploidy.
Biologically speaking, cellular growth is connotated with a typical process of physiological development or aging. More specifically, it has been suggested that aged human endothelial cells may occasionally become polyploidy (27). To examine the relationship between polyploidy and developmental aging in the cilia mutant mouse model, we freshly isolated aortic endothelial cells from E15.5 embryo, 3-week-old pup and 3-month-old adult of wild-type or Tg737Orpk/Orpk mice. Centrosome amplification was observed in resting and dividing Tg737Orpk/Orpk cells isolated from embryo, pup and adult mice (Fig. 10A). Although centrosome amplification was more readily found in cells from adult mice, abnormal centrosome is always significantly greater in Tg737Orpk/Orpk than in wild-type cells, regardless of the age groups (Fig. 10B). Polyploidity was also studied in these cells (Fig. 10C). Of note are the readily dividing embryonic cells compared with cells isolated from pup or adult (BrdU-positive cells). Regardless of the age group, however, the polyploidy is consistently and significantly greater in Tg737Orpk/Orpk than in wild-type mice. This further indicates that a mutation which results in cilia dysfunction will present a more severe phenotype as the disease progresses at an older age.
Figure 10.

Aging compounds the severity of cilia mutant on centrosome number and cell polyploidy. Aortic endothelial cells were freshly isolated from E15.5-day embryos, 3-week-old pup and 3-month-old adult of wild-type or Tg737Orpk/Orpk mice (C3H background). (A) Cells were stained with pericentrin (red), acetylated-α-tubulin (green) and DAPI (blue) to count the centrosome numbers of resting and dividing cells. Arrows indicate centrosomes. (B) Percentage of abnormal centrosome number from three to four isolations was averaged and plotted. (C) Flow cytometry was also used to analyze cell polyploidy for each age group. (D) Percentage of polyploidy cells from three to four isolations was averaged and plotted. Asterisks denote significant difference toward corresponding wild-type groups.
DISCUSSION
We show here for the first time that primary cilia in endothelial cells are important sensory organelles required for proper cell division and chromosomal segregation. Failing to have a normal cilia structure and function, a cell will undergo abnormal cell division through centrosomal overduplication and multipolar spindle formation. Consequently, cilia mutant cells from mice or human patients are characterized by abnormal mitosis and chromosomal separation, resulting in genomic instability. Such instability will result in cell polyploidy and is further exacerbated by developmental aging. Survivin re-expression can partially rescue the cellular phenotypes of centrosome over-amplification and cell polyploidy.
Primary cilia are mechanosensory organelles that transmit extracellular fluid flow to intracellular biochemical signaling. Abnormal transmittance of extracellular signal prevents cells from differentiating properly (data not shown). Failure to differentiate is usually associated with the abnormal cell cycle. Consistent with this view, abnormally dividing cells were significantly greater in cilia mutant cell populations than in wild-type cell populations. Surprisingly, these abnormalities can be observed throughout different cell-cycle stages of interphase, prophase, metaphase, anaphase, telophase and cytokinesis (Fig. 1).
The abnormal mitosis is most likely caused by a significant number of cells with centrosome overduplication because amplification of the centrosome can be seen in both dividing and non-dividing stages of cilia mutant cells (Fig. 2). This observation is consistent with other studies, which suggests that centrosome amplification happens early in cystic kidney disease (28,29). We further show that Pkd1−/− and Pkd2−/− cells with centrosome overduplication have multiple primary cilia in vitro and in vivo (Fig. 3). In Tg737Orpk/Orpk cells or tissues, the multiple cilia formations were less obvious, if present at all, probably due to their short and stubby cilia. Nonetheless, multiple centrosomes are also observed in Tg737Orpk/Orpk cells and tissues.
Centrosome overduplication generally induces the unevenness of chromosomal segregation, which can result in greater DNA content. When cilia mutant cells were further examined for their cell-cycle profiles, they were indeed shown to have greater DNA content. Analysis with BrdU and PI indicates that cilia mutant cells are polyploidy with additional DNA content (Fig. 4). To further verify that such polyploidy and abnormal cell division are not due to cell line artifact, we performed the additional experiments on primary cells isolated from Pkd mice and samples from ADPKD patients (Fig. 5). When chromosomal identifications were examined, abnormal numbers of chromosomes were encountered, verifying our findings from the cell lines. Abnormal chromosomal segregation immediately became apparent in polycystic kidney disease.
To identify the defects in chromosomal segregation and/or cytokinesis, we performed live-imaging analysis with phase contrast to identify individual cell body, and fluorescence imaging to identify individual cell nucleus (Fig. 6 and Supplementary Material, Movies S1–S4). The most obvious defect in cilia mutant cells is particularly observed during anaphase. To further eliminate cell line artifact, we also used primary cell culture from ADPKD patients. Both mouse cell lines and primary human cells indeed provided very similar results, further substantiating our hypothesis that proper function and structure of cilia are required in cell division. All abnormalities observed in polyploidy and during chromosomal segregation are characteristics of dysfunction in chromosomal passenger proteins. In particular, the live-imaging analysis reveals that cilia mutant cells could complete the mitotic checkpoint assembly but failed to maintain spindle tension during anaphase, which further supports the involvement of chromosomal passenger proteins (23).
We next performed mitotic-stress tests (Fig. 7). Basically, these tests utilize both taxol and nocodazole to promote mitotic arrest. In particular, chromosomal passenger proteins are not required during early mitotic spindle formation. However, they are required to maintain ‘cell commitment' to complete cell division. Consistent with this view, our data indicate that stabilizing microtubules with taxol failed to arrest cilia mutant cells but not wild-type cells. On the other hand, de-polymerizing microtubules with nocodazole arrested both wild-type and cilia mutant cells. Our data, thus, support the idea that chromosomal passenger proteins play an important role in checkpoint maintenance, such as microtubule tension of mitotic spindles. Chromosomal passenger protein complex is known to phosphorylate their substrate, histone H3. Accordingly, inhibitor for chromosomal passenger complex Zm447439 induces a decrease in histone H3 phosphorylation in wild-type cells. Surprisingly, Zm447439 did not cause a further decrease in histone H3 phosphorylation in cilia mutant cells, presumably because these cells have already had a depressed activity of chromosomal passenger.
To investigate this possibility, we measured expression levels of the chromosomal passenger protein, survivin (Fig. 8). Levels of survivin are consistently and significantly repressed at the transcriptional and translational levels in mutant cilia cells and tissues. Because the downstream effector of survivin is aurora B kinase, we also measured expression levels of aurora B kinase by re-blotting western membrane for survivin. However, we do not see any consistent pattern of changes in the expression levels of aurora B kinase. Because survivin is expressed only during cell division, survivin expression level may thus be regulated after cells are committed to undergo cell division. The results are consistent with our live-imaging observation.
VEGF has been shown to increase survivin expression (26), and re-introducing survivin is sufficient to correct cell polyploidy (Fig. 8). Our data show that VEGF-induced survivin expression can partially rescue the polyploidy phenotype in cilia mutant cells (Fig. 9). This effect depends on the molecular functions of PI3K and Akt/PKB. Acetylcholine, which also involves PI3K and Akt/PKB (30), has a minor effect on centrosome amplification but has no significant role in cell ploidy. This indicates that although acetylcholine prevents apoptosis through PI3K/Akt/PKB pathway, it does not have any role in endothelial ploidy level.
Survivin is a chromosomal passenger protein involved in coordinating proper chromosomal events during mitosis. In addition to inhibiting apoptosis, survivin is also essential for proper cell division during mitosis and is a cell-cycle-regulated protein expressed in G2/M phase (19). It has been shown that altering survivin level can cause abnormal cell division (20–22). An abnormally high survivin expression is observed in many types of cancer but is undetectable in normal, differentiated adult tissues. Consistent with this information, higher polyploidy has been reported in endothelial cells isolated from older humans (27). Our data also support this view (Fig. 10). In particular, the polyploid cells with abnormal numbers of chromosomes were observed at the rate of approximately 10–20% at a time. We would argue that the small rate is significantly relevant, over years and decades, to the progression of human diseases.
Although the exact mechanism is still unknown, the down-regulation of survivin in cilia mutant cells might induce mitotic cell-cycle arrest, which might lead to mitotic slippage and result in polyploid cells. Survivin and aurora B interact with each other, and in the presence of survivin, aurora B kinase activity is increased (31). Thus, survivin binding to aurora B is required for aurora kinase activity to phosphorylate its substrates, such as histone H3. In particular, aurora kinase homolog in Chlamydomonas is believed to be a key regulator for flagella structure (32,33). In fact, aurora kinase could promote ciliary disassembly in mammalian cells (34).
In summary, we propose that the sensory function and structure of cilia are crucial in regulating cell differentiation and cellular division, especially during anaphase of the cell cycle. Abnormalities in such regulation will result in abnormal expression of survivin, which leads to multipolar spindle formation, asymmetric chromosome segregation and genomic instability. Our studies also provide evidence that abnormal cell division will result in cell polyploidy, which might be the hallmark behind the pathophysiological abnormalities associated with ADPKD, including cystogenesis and aneurysms. Furthermore, the severity of these abnormalities may be compounded by developmental aging, which partly explains the long-term pathogenesis event of atherosclerosis, also a cilia-related disorder (35).
MATERIALS AND METHODS
The use of animal tissues was approved by the animal care and use committee of The University of Toledo. Signed and informed consent to collect disposed ADPKD human tissues was obtained from the patients, and tissue collection protocols were approved by the Department for Human Research Protections of the Biomedical Institutional Review Board of The University of Toledo.
Cell cultures
Endothelial cell lines were generated from wild-type, Pkd1−/− and Tg737Orpk/Orpk 15.5-day embryonic (E15.5) mouse aortas, as previously described (3). Prior to the experiments, these cells were grown in Dulbecco's Modified Eagle's Medium (DMEM; Cellgro, Manassas, VA, USA) containing 2% of fetal bovine serum at 39°C. For the primary culture, vascular endothelial cells were obtained from wild-type and Pkd2 E15.5 mouse aortas. Freshly isolated endothelial cells from E15.5 embryos, 3-week-old pups and 3-month-old adults were obtained from wild-type and Tg737Orpk/Orpk mice. These mice were bred in C3H background for 15 generations to study age-related effects on cell division. Human endothelial cells were isolated from interlobar arteries. All primary cells were grown in DMEM with 15% serum (and 1% penicillin/streptomycin as needed) at 37°C with 5% CO2 incubator. Isolation and partial characterization of all primary cultures have been described previously (2). The experimental set-up for the functional study on shear stress-induced cilia activation has also been discussed in detail (2,3). Regardless of cell lines or primary cells, all media supplements were not used or were withdrawn prior to our experiments to prevent unintended cell growth (culture artifact).
Immunofluorescence and western blot
Confluent cells grown to full differentiation on glass coverslips were rinsed with 1× phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde containing 2% sucrose for 10 min, and permeabilized with 1% Triton-X in PBS for 5 min. Acetylated-α-tubulin (clone 6–11B-1 from Sigma, St Louis, MO, USA) was used at a dilution of 1:10 000, actin (clone AC-40; Sigma) at 1:20 000, pericentrin (Covance, Princeton, NJ, USA) at 1:500 and γ-tubulin (clone C-20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:500. Texas red and fluorescein-conjugated anti-mouse and anti-rabbit (Pierce, Rockford, IL, USA) were used at 1:500. Cells were counterstained with 4.6-diamidino-2-phenylindol DAPI (Vector Laboratories, Burlingame, CA, USA).
For western blot analysis, cells were lysed with radioimmunoprecipitation assay buffer. Intracellular contents were collected by centrifugation at 100g for 10 min. Total cell lysates were analyzed with a standard 10 or 8–14% gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Survivin and aurora B kinase (ABCam, Cambridge, MA, USA) were used at dilutions of 1:100 and 1:200, respectively. Antibodies against Akt, p-Akt, PI3K, GAPDH (Cell Signaling Technologies, Danvers, MA, USA; 1:1000, 1:500, 1:500 and 1:1500, respectively) and p-survivin (Novus Biologicals, Littleton, CO, USA, 1:500) were also used.
Flow cytometry
Endothelial cells were incubated with 30 μm BrdU (Sigma) for 15 min, rinsed with 1× PBS, detached with trypsin-EDTA and permeabilized with ice-cold 100% ethanol. Total RNA was digested with 0.5 mg/ml of RNase A (Boehringer Mannheim, Ingelheim am Rhein, Germany) at 37°C for 30 min, pelleted at 1100 rpm for 8 min at 4°C and then resuspended in 1 ml of HCl-Triton solution. The DNA was then denatured at 97°C for 15 min and quickly chilled in an ice-water bath for 15 min. Anti-BrdU Alexa Fluor 488 at a dilution of 1:100 (A21303; Invitrogen, Carlsbad, CA, USA) was used. The cells were then pelleted and resuspended in 0.5 ml of 20 μg/ml PI (P-4170; Sigma) for 1 h at room temperature before being analyzed. In some cases, anti-phosphorylated histone 3A antibody (Sigma) at a dilution of 1:50 and PI was used to stain and analyze the cells.
For survivin-rescued experiments, human full-length survivin construct was inserted into pEGFPc1 (Clontech, Mountain View, CA, USA). Endothelial cells were transiently transfected with survivin-GFP or empty-GFP vector with Fugene 6, according to the manufacturer's instructions (Roche Diagnostics, Indianapolis, IN, USA). About 24 h after transfection, cells expressing GFP were randomly selected for analysis with C6 Flow Cytometer (Accuri Cytometers, Ann Arbor, MI, USA).
Quantitative reverse-transcriptase polymerase chain reaction
Total RNA was collected from three different plates, pooled together and isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA). The yield and quality of total RNA was first analyzed by agarose gel electrophoresis. Five micrograms of total RNA were used for reverse transcription (RT) reactions in a 100-μl reaction to synthesize single-strand cDNA using SuperScript II RNase H-Reverse transcriptase RT-PCR system (Invitrogen). Survivin, aurora B kinase and actin RNAs were amplified in separate tubes to avoid competition. Genes were amplified using the following primers. Survivin_F: 5′-ATC GCC ACC TTC AAG AAC TG-3′; Survivin_R: 5′-CAG GGG AGT GCT TTC TAT GC-3′; AuroraB_F: 5′-GCC AGA AGT TGG CTG AGA AC-3′; AuroraB_R: 5′-GAT CTT GAG TGC CAC GAT GA-3′; Actin_F: 5′-TGT TAC CAA CTG GGA CGA CA-3′; Actin_R: 5′-GGG GTG TTG AAG GTC TCA AA-3′. RNA expression profile was analyzed by real-time PCR using iScript™ One-Step RT-PCR Kit with SYBR® green (Bio-Rad, Hercules, CA, USA) and carried in an iCycler iQ™ Real-time PCR detection system. The complete reactions were incubated in the real-time thermal detection system and subjected to the following program of thermal cycling: 10 min at 50°C for cDNA synthesis, 5 min at 95°C for iScript Reverse Transcriptase inactivation and 40 cycles of 10 s at 95°C and 30 s at 62°C for PCR cycling and detection. A melting curve was run after the PCR cycles, followed by a cooling step.
The real-time PCRs were quantitated by selecting the amplification cycle where the PCR product of interest was first detected (CT). The CT value was defined as the cycle in which an increase in reporter signal (fluorescence) crosses the threshold. The CT value was then averaged from the triplicate PCR reactions. Each sample was thus run in triplicate in every experiment, and each experiment was repeated at least three times. The expression level of survivin and aurora B kinase was normalized to the expression level of the endogenous reference gene, actin. The fold change in survivin and aurora B kinase mRNA relative to the endogenous standard (actin) was determined by 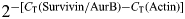 , summarized as
, summarized as 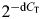 .
.
Spectral karyotyping analysis
For chromosome spreads, cells were isolated and plated for at least 1 day and grown to 80% confluency. Cells were then grown for an additional 2 h after incubating with 0.05 μg/ml colcemid solution for 30 min at 37°C in the dark. Next, cells were detached and incubated with 0.56% KCl solution for 45 min at 37°C, followed by fixing with 3:1 methanol:acetic acid. The cells were dropped onto pre-cleaned glass slides and counterstained with DAPI for microscopy analysis. At least 50 chromosome spreads were counted from each cell analysis.
For spectral karyotyping (SKY) analysis, chromosome spreads were prepared using air-drying methods. After sequential digestion with RNase and pepsin, according to the procedure recommended by Applied Spectral Imaging (ASI, Vista, CA, USA), the chromosomal DNA on slides was denatured in 70% formamide and then hybridized with a cocktail of mouse or human SKY paint probes tagged with various nucleotide analogues (i.e. a mixture of individual chromosome DNA prepared by flow-sorting and PCR amplification). Thirty to fifty mitoses were chosen at random. The images were developed by combinations of five different fluorophores. Rhodamine, Texas-Red, Cy5, FITC and Cy5.5 were captured with a Spectral cube and interferometer module installed on an Olympus microscope. Spectral karyotypes were carried out using SKY View software (Version 1.62).
Live-imaging study
Because of the low transfection efficiency of fluorescence constructs (GFP or DsRed), we incubated the cells with membrane-permeable and DNA-specific dye, Hoechst. After 20 min and being rinsed of the excess dye, cells were randomly selected for live-imaging analysis for 24 h with a Nikon TE2000 microscope equipped with an environmental chamber. For better focusing, the microscope was equipped with XY-axis motorized flat top inverted stage, Nikon automatic focusing RFA Z-axis drive and custom-designed vibration isolation platform. For a better controlled environment, the body of the microscope was enclosed inside a custom-built chamber to control CO2, humidity, heat and light. Images were obtained every 5 min with an exposure time of ∼20 ms. Phase-contrast images were captured simultaneously at the same interval, but with an exposure time of 2 ms. Multiple focal planes were also taken to obtain the best focus for the dividing cell.
Pharmacological treatments
For mitotic-stress tests, cells were first incubated with 2 μm Zm447435 for 1 h to inhibit aurora B kinase activity, 0.1 μg/ml of nocodazole for 12–16 h to depolymerize microtubules or 33.3 nm taxol for 12–16 h to stabilize microtubules. In some cases, cells were treated with 10 μm acetylcholine. In other cases, cells were incubated with either 40 ng/ml of VEGF in the presence or absence of 10 μm Akt/PKB inhibitor or 20 μm PI3K inhibitor (LY294-002). All chemicals were purchased from Sigma, except Akt/PKB inhibitor (Calbiochem, Gibbstown, NJ, USA) and Zm447435 (Tocris Bioscience, Ellisville, MO, USA).
Data analysis
Experiments were repeated on different sets of cell populations at least three times. When primary mouse or human cells were used, they were also isolated from at least three different subjects (N ≥ 3 for each group). For immunostaining analysis, a minimum of six coverslips were used for each experiment. All quantifiable data were reported as mean ± SEM. Comparison between two groups was carried out using Student's t-test. Data comparisons for more than two groups were done using analysis of variance test, followed by Tukey's post-test analysis. Unless otherwise indicated, the difference between groups was statistically significant at P < 0.05. All statistical analyses were done with GraphPad Prism, version 5.0.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by awards from the NIH (DK080640) and The University of Toledo research program (to S.M.N.). The additional funding from the NIH through American Recovery and Reinvestment Act (to S.M.N.) allows us to accelerate the completion of this study. This work was also supported partially by grant GM077238 (to J.V.S.).
ACKNOWLEDGEMENTS
We thank Charisse Montgomery for her editing service and Maki Takahashi and Blair Mell for their technical support. We also like to extend our gratitude for resources obtained from the University of Alabama at Birmingham, Recessive PKD core center (http://www.rpkdcc.uab.edu) and the Yale Polycystic Kidney Disease Research Center.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Harris P.C., Torres V.E. Polycystic kidney disease. Annual Rev. Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. doi:10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.AbouAlaiwi W.A., Takahashi M., Mell B.R., Jones T.J., Ratnam S., Kolb R.J., Nauli S.M. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. doi:10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nauli S.M., Kawanabe Y., Kaminski J.J., Pearce W.J., Ingber D.E., Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. doi:10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poelmann R.E., Van der Heiden K., Gittenberger-de Groot A., Hierck B.P. Deciphering the endothelial shear stress sensor. Circulation. 2008;117:1124–1126. doi: 10.1161/CIRCULATIONAHA.107.753889. doi:10.1161/CIRCULATIONAHA.107.753889. [DOI] [PubMed] [Google Scholar]
- 5.Nauli S.M., Alenghat F.J., Luo Y., Williams E., Vassilev P., Li X., Elia A.E., Lu W., Brown E.M., Quinn S.J., et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003;33:129–137. doi: 10.1038/ng1076. doi:10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 6.Nauli S.M., Rossetti S., Kolb R.J., Alenghat F.J., Consugar M.B., Harris P.C., Ingber D.E., Loghman-Adham M., Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. doi:10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- 7.Siroky B.J., Ferguson W.B., Fuson A.L., Xie Y., Fintha A., Komlosi P., Yoder B.K., Schwiebert E.M., Guay-Woodford L.M., Bell P.D. Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am. J. Physiol. 2006;290:F1320–F1328. doi: 10.1152/ajprenal.00463.2005. doi:10.1152/ajprenal.00463.2005. [DOI] [PubMed] [Google Scholar]
- 8.Xu C., Rossetti S., Jiang L., Harris P.C., Brown-Glaberman U., Wandinger-Ness A., Bacallao R., Alper S.L. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am. J. Physiol. 2007;292:F930–F945. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davenport J.R., Watts A.J., Roper V.C., Croyle M.J., van Groen T., Wyss J.M., Nagy T.R., Kesterson R.A., Yoder B.K. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. doi:10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo S., Guo D.F., Bugge K., Morgan D.A., Rahmouni K., Sheffield V.C. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet. 2009;18:1323–1331. doi: 10.1093/hmg/ddp031. doi:10.1093/hmg/ddp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z., Li V., Chan G.C., Phan T., Nudelman A.S., Xia Z., Storm D.R. Adult type 3 adenylyl cyclase-deficient mice are obese. PloS ONE. 2009;4:e6979. doi: 10.1371/journal.pone.0006979. doi:10.1371/journal.pone.0006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacoby M., Cox J.J., Gayral S., Hampshire D.J., Ayub M., Blockmans M., Pernot E., Kisseleva M.V., Compere P., Schiffmann S.N., et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. doi:10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- 13.Evan A.P., Gardner K.D., Jr, Bernstein J. Polyploid and papillary epithelial hyperplasia: a potential cause of ductal obstruction in adult polycystic disease. Kidney Int. 1979;16:743–750. doi: 10.1038/ki.1979.191. doi:10.1038/ki.1979.191. [DOI] [PubMed] [Google Scholar]
- 14.Jonassen J.A., San Agustin J., Follit J.A., Pazour G.J. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J. Cell Biol. 2008;183:377–384. doi: 10.1083/jcb.200808137. doi:10.1083/jcb.200808137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin H., Wang Z., Diener D., Rosenbaum J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr. Biol. 2007;17:193–202. doi: 10.1016/j.cub.2006.12.040. doi:10.1016/j.cub.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. doi:10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 17.Ganem N.J., Godinho S.A., Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. doi:10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steigemann P., Wurzenberger C., Schmitz M.H., Held M., Guizetti J., Maar S., Gerlich D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. doi:10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 19.Li F., Ambrosini G., Chu E.Y., Plescia J., Tognin S., Marchisio P.C., Altieri D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. doi:10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 20.Reed J.C., Bischoff J.R. BIRinging chromosomes through cell division—and survivin' the experience. Cell. 2000;102:545–548. doi: 10.1016/s0092-8674(00)00076-3. doi:10.1016/S0092-8674(00)00076-3. [DOI] [PubMed] [Google Scholar]
- 21.Uren A.G., Wong L., Pakusch M., Fowler K.J., Burrows F.J., Vaux D.L., Choo K.H. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 2000;10:1319–1328. doi: 10.1016/s0960-9822(00)00769-7. doi:10.1016/S0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- 22.Yang D., Welm A., Bishop J.M. Cell division and cell survival in the absence of survivin. Proc. Natl Acad. Sci. USA. 2004;101:15100–15105. doi: 10.1073/pnas.0406665101. doi:10.1073/pnas.0406665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvalho A., Carmena M., Sambade C., Earnshaw W.C., Wheatley S.P. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J. Cell Sci. 2003;116:2987–2998. doi: 10.1242/jcs.00612. doi:10.1242/jcs.00612. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda S., Foster R.G., Porter S.B., Pelus L.M. The antiapoptosis protein survivin is associated with cell cycle entry of normal cord blood CD34(+) cells and modulates cell cycle and proliferation of mouse hematopoietic progenitor cells. Blood. 2002;100:2463–2471. doi: 10.1182/blood.V100.7.2463. doi:10.1182/blood.V100.7.2463. [DOI] [PubMed] [Google Scholar]
- 25.Sommer K.W., Schamberger C.J., Schmidt G.E., Sasgary S., Cerni C. Inhibitor of apoptosis protein (IAP) survivin is upregulated by oncogenic c-H-Ras. Oncogene. 2003;22:4266–4280. doi: 10.1038/sj.onc.1206509. doi:10.1038/sj.onc.1206509. [DOI] [PubMed] [Google Scholar]
- 26.Tran J., Master Z., Yu J.L., Rak J., Dumont D.J., Kerbel R.S. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl Acad. Sci. USA. 2002;99:4349–4354. doi: 10.1073/pnas.072586399. doi:10.1073/pnas.072586399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borradaile N.M., Pickering J.G. Polyploidy impairs human aortic endothelial cell function and is prevented by nicotinamide phosphoribosyltransferase. Am. J. Physiol. Cell. Physiol. 2010;298:C66–C74. doi: 10.1152/ajpcell.00357.2009. doi:10.1152/ajpcell.00357.2009. [DOI] [PubMed] [Google Scholar]
- 28.Battini L., Macip S., Fedorova E., Dikman S., Somlo S., Montagna C., Gusella G.L. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum. Mol. Genet. 2008;17:2819–2833. doi: 10.1093/hmg/ddn180. doi:10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burtey S., Riera M., Ribe E., Pennenkamp P., Rance R., Luciani J., Dworniczak B., Mattei M.G., Fontes M. Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol. Int. 2008;32:1193–1198. doi: 10.1016/j.cellbi.2008.07.021. doi:10.1016/j.cellbi.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 30.Kakinuma Y., Ando M., Kuwabara M., Katare R.G., Okudela K., Kobayashi M., Sato T. Acetylcholine from vagal stimulation protects cardiomyocytes against ischemia and hypoxia involving additive non-hypoxic induction of HIF-1alpha. FEBS Lett. 2005;579:2111–2118. doi: 10.1016/j.febslet.2005.02.065. doi:10.1016/j.febslet.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 31.Chen J., Jin S., Tahir S.K., Zhang H., Liu X., Sarthy A.V., McGonigal T.P., Liu Z., Rosenberg S.H., Ng S.C. Survivin enhances Aurora-B kinase activity and localizes Aurora-B in human cells. J. Biol. Chem. 2003;278:486–490. doi: 10.1074/jbc.M211119200. doi:10.1074/jbc.M211119200. [DOI] [PubMed] [Google Scholar]
- 32.Pan J., Snell W.J. Kinesin II and regulated intraflagellar transport of Chlamydomonas aurora protein kinase. J. Cell Sci. 2003;116:2179–2186. doi: 10.1242/jcs.00438. doi:10.1242/jcs.00438. [DOI] [PubMed] [Google Scholar]
- 33.Pan J., Wang Q., Snell W.J. An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev. Cell. 2004;6:445–451. doi: 10.1016/s1534-5807(04)00064-4. doi:10.1016/S1534-5807(04)00064-4. [DOI] [PubMed] [Google Scholar]
- 34.Pugacheva E.N., Jablonski S.A., Hartman T.R., Henske E.P., Golemis E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. doi:10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van der Heiden K., Hierck B.P., Krams R., de Crom R., Cheng C., Baiker M., Pourquie M.J., Alkemade F.E., DeRuiter M.C., Gittenberger-de Groot A.C., et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis. 2008;196:542–550. doi: 10.1016/j.atherosclerosis.2007.05.030. doi:10.1016/j.atherosclerosis.2007.05.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.