

Abstract
Human anal cancers are associated with high-risk human papillomaviruses (HPVs) that cause other anogenital cancers and head and neck cancers. As with other cancers, HPV16 is the most common high-risk HPV in anal cancers. We describe the generation and characterization of a mouse model for human anal cancer. This model makes use of K14E6 and K14E7 transgenic mice in which the HPV16 E6 and E7 genes are directed in their expression to stratified squamous epithelia. HPV16 E6 and E7 possess oncogenic properties including but not limited to their capacity to inactivate the cellular tumor suppressors p53 and pRb, respectively. Both E6 and E7 were found to be functionally expressed in the anal epithelia of K14E6/K14E7 transgenic mice. To assess the susceptibility of these mice to anal cancer, mice were treated topically with dimethylbenz[a]anthracene (DMBA), a chemical carcinogen that is known to induce squamous cell carcinomas in other sites. Nearly 50% of DMBA-treated HPV16 E6/E7 transgenic mice showed overt signs of tumors; whereas, none of the like treated non-transgenic mice showed tumors. Histopathological analyses confirmed that the HPV16 transgenic mice were increased in their susceptibility to anal cancers and precancerous lesions. Biomarker analyses demonstrated that these mouse anal cancers exhibit properties that are similar to those observed in HPV-positive precursors to human anal cancer. This is the first mouse model for investigating the contributions of viral and cellular factors in anal carcinogenesis, and should provide a platform for assessing new therapeutic modalities for treating and/or preventing this type of cancer.
Introduction
The incidence of anal cancer has been rising over the past thirty years with overall incidence in the general population now at 1.5 persons per 100,000 persons (1). In some subpopulations at higher risk, for example men who have sex with men, the incidence has reached over 20 per 100,000 persons (2). While 50% of patients present with disease confined to the primary site and these patients have a favorable, 80% five year survival rate (1), the remainder of the patients present with regional or distant metastasis correlating with less favorable 61% and 21% survival rates, respectively (1). Anal cancer treatment has essentially remained static; more effective clinical treatments for patients with advanced stages of disease are needed. A laboratory animal model for human anal cancer would provide an experimental platform for better understanding anal cancer and identifying novel approaches for preventing and/or controlling this debilitating disease.
Anal cancer, like cervical cancer, is associated with human papillomavirus (HPV). Seventy-eight percent of squamous cell anal cancer cases have some type of HPV present and 66% are positive for the high risk HPV16 genotype (3). HPV 16 encodes two oncoproteins that target a number of cellular factors involved in cancer, including key cellular tumor suppressors. E6 targets p53 leading to deregulation of DNA damage and apoptotic pathways. E7 binds and targets for degradation pRb, leading to an increase in cell proliferation and genomic instability. A third viral oncogene, E5, also contributes to carcinogenesis though its mechanism of action has yet to be defined. These oncogenes are thought to be necessary but not sufficient to cause anal cancer. Evident of this, the incidence of anal cancer, even in subpopulations at highest risk of this cancer, is very low compared to the much higher incidence of HPV infections in these same subpopulations. For example, men with AIDS have one of highest known incidence of anal cancer even in the era of antiretroviral treatment at 42 per 100,000 persons (4). This compares to a 72% prevalence of high risk HPV in anal swabs and a 43% prevalence of high grade anal intraepithelial neoplasia (AIN) amongst HIV-positive men who have sex with men, who represent a population at high risk of developing AIDs (5). HIV seropositivity is among the risk factors for developing anal cancer; others include low CD4 count, persistent high risk HPV infection, infection with multiple HPV types, anoreceptive intercourse, history of cervical cancer or dysplasia, cigarette smoking and immunosuppression for organ allograft (6).
HPV is thought to play a critical role in the development of most anal cancers. We previously developed E6/E7 transgenic mice in which the E6 and E7 genes are linked to the K14 promoter targeting their expression to stratified epithelium. These HPV transgenic mice do not spontaneously develop anal cancer. However in prior studies we found that, when dorsal skin of these mice was treated with DMBA and TPA, a classic tumor initiation/promotion regimen, they displayed heightened susceptibility to skin tumors compared to nontransgenic mice (7). Furthermore, these mice, when treated with just DMBA, also displayed increased susceptibility to skin carcinogenesis, suggesting that E6 and E7 can substitute for a promoting agent such as TPA (7). DMBA can also act as a complete carcinogen: multiple applications of DMBA to the skin results in earlier onset of carcinoma when compared to a one time treatment with DMBA followed by repeated application of TPA (8). To learn whether HPV oncogenes cause increased susceptibility to anal cancer in mice, we treated the anus of HPV16 transgenic mice topically with DMBA. Anal carcinogenesis occurred selectively in E6/E7 mice topically treated with 0.12μmoles DMBA once weekly for 20 weeks: 7/31 (23%) developed anal cancer and an additional 9/31 (29%) developed atypia. Atypia in the mouse model is analogous to AIN in humans. In the same group of mice, 15/31 (48%) had overt tumors which correlated to 73% of the atypia and cancer seen on microscopic evaluation. None of the DMBA-treated nontransgenic control mice developed anal cancer or atypia. These findings demonstrate that HPV16 E6 and E7 increase the susceptibility of mice to anal carcinoma. Initial characterization of this mouse anal cancer model indicates it shares histopathological and molecular properties with HPV-positive precursors to human anal cancer.
Materials and Methods
Mice
Generation of K14E6 and K14E7 mice has been previously described (9, 10). These mice were maintained on the inbred FVB/N genetic background. E6/E7 transgenic mice were generated by crossing K14E6 females with K14E7 males. All mice were kept in American Association for Accreditation of Laboratory Animal Care-approved McArdle Laboratory Cancer Center Animal Care Facility and studies with them were carried out in accordance to an approved animal protocol.
Acute Phenotypes
All mice received intraperitoneal injections of bromo deoxyuracil (BrdUrd)12.5 mg/mL in PBS at 10 μL/g body weight one hour prior to sacrifice. Irradiated mice were exposed to 12 Gy of ionizing radiation from a 137Cs source twenty-four hours prior to sacrifice. Nuclei in anal epithelium was scored as positive (brown) or negative (blue) for BrdUrd based upon BrdUrd-specific immunohistochemistry. A minimum of five high power fields were counted for each group.
Immunohistochemistry
Immunohistochemistry staining was carried out as previously described (11). Primary antibodies were applied overnight at 4°C at the following concentrations in 2.5% horse serum (1:50 BrdUrd, Calbiochem; 1:200 antiMCM7, Neomarkers; 1:50 p16 (M156), Santa Cruz).
E7 Western Blot
Anal tissue from untreated E6/E7 transgenic mice was harvested and homogenized in lysis buffer with protease and phosphatase inhibitors. Lysates were placed on ice for 30minutes with intermittent agitation. After 20 minutes of centrifugation at 14K, the supernatant was collected. Samples were run on a 12% SDS-PAGE gel, transferred to polyvinlidene difluoride membrane, and probed for E7 (1:200 Santa Cruz).
E6 Pull-down and Western Blot
Harvested anal tissue from individual untreated E6/E7 transgenic mice were homogenized in E6 specific lysis buffer with a final concentration of 50 mM HEPES pH 7.5, 150 mM NaCl, 1.1% Triton X-100, 1 mM EGTA, 10% glycerol, 1 mM PMSF and 1X protease inhibitor. After cold agitation and centrifugation as described above for E7 lysates, the supernatent was collected. E6 protein was isolated from anal tissue lysates using a PDZ interaction mediated E6 precipitation (E6 pull-down) technique. For the E6 pull-down, recombinant GST-Magi1 PDZ1 fusion protein was coupled to Glutathione Sepharose 4B beads (GE Healthcare) at a ratio of 200 μg total protein to 200 μl of beads (bed volume). The final volume was adjusted to 1 ml with PBS. Bead coupling to GST-Magi1 PDZ1 occurred for 6 hrs at 4°C with constant end over end rotation. Coupled beads were washed 3 times with 1 ml PBS prior to application in the E6 pull-down. Test lysates were then combined with 25μl of the conjugated bead-GST-Magi1 PDZ1 mixture. The final volume of each pull-down sample was adjusted to 1 ml with lysis buffer. Precipitation of E6 was carried out for 21.5 hrs at 4°C with constant end over end rotation followed by 2 washes in 1 ml PBS. Beads recovered from the E6 pull-down were boiled for 10 minutes in reducing sample buffer (50 mM Tris pH6.8, 10% Glycerol, 2% SDS, 0.145% Bromphenol Blue and 100 mM DTT) prior to separation on a Criterion 10-20% gradient Tris-HCl polyacrylamide gel. Sample blotting onto a PVDF membrane (Immobilon-P, Millipore, pore size 0.45 um) occurred at 15 V constant voltage for 42 minutes under semi-dry conditions followed by appropriate blocking of the membrane (0.05% TBS-t with 1% Amicase and 2% Albumin, O/N at 4°C).
The blocked membrane was then probed for 2.8 hrs at RT with primary anti HPV16-E6-specific monoclonal antibody 4C6 at a concentration of 2.0 ug/ml in the presence of 1.5% BSA and 0.1% TBS-t (4C6 mAb generated by Dr. Etienne Weiss et al. and made available to Arbor Vita Corp). The membrane was washed 4 times with 0.1% TBS-t before incubation with the secondary anti-mouse IgG mAb (Jackson Immunoresearch, 115-035-062, Goat anti-mouse IgG/HPR), diluted to 1:10,000 in a 0.1% TBS-t and 3.6% blocking solution. Incubation with secondary mAb occurred for 1 hr at RT followed by 6 washes with 0.1% TBS-t. For western development, ECL Advance chemiluminescent substrate (GE Healthcare) was used according to the manufacturer's recommendations. Visualization and quantification occurred via a Fujifilm Luminescent Image Analyzer LAS-3000 and Multigauge Software.
DMBA Induced Anal Carcinogenesis
Male and female adult E6/E7 transgenic mice and age matched nontransgenic control mice received topical treatments of DMBA (dimethylbenz[a]anthracene) dissolved in 100% DMSO (dimethylsulfoxide) to the anal canal once per week for twenty weeks. Treatments were initiated between 5 and 7 weeks of age. Prior to topical treatment, the mouse anal canal was evacuated of feces using manual, external pressure to the pelvic brim. Mice were treated with 0.12, 0.04 or 0.012 μmoles DMBA or DMSO without DMBA. All treatments had 4 μL of liquid inserted 5-10mm into the anal canal using a standard pipette tip. After twenty weeks of treatment, mice had an eight-week hiatus before being euthanized and anal tissue harvested for histological analysis. Mice were monitored weekly for appearance of overt tumors though only overt tumors present at sacrifice were included in the tumor phenotype data (Table 2). Overt tumors were defined as any neoplastic lesion in the anal region capable of being seen by the examiner's naked eye that proved to be a tumor upon histopathological assessment, regardless of disease classification (papilloma, atypia, carcinoma). Overt tumor size (diameter in mm) was measured at the time of sacrifice.
Table 2.
Overt anal tumors in E6/E7 mice treated with 0.12 μmole DMBA are more likely to be carcinoma
| Phenotype | n | Histological Classification of Disease | ||||
|---|---|---|---|---|---|---|
| Normal | Hyperplasia | Papilloma | Atypia (%) | Cancer (%)* | ||
| tumors | 14 | 0 | 0 | 3 | 5 (36) | 6 (43) |
| no tumors | 17 | 6 | 5 | 1 | 4 (24) | 1 (6) |
p<0.05 comparing presence of cancer in E6/E7 mice with vs without tumors
Histological Analysis
The anal canal tissue was fixed in 10% formalin, paraffin embedded, and serially sectioned at 5μm. Every 30th section was stained with H&E and histopathologically analyzed for hyperplasia, papilloma, atypia, or carcinoma.
H-ras Mutation Analysis
DNA isolated from DMBA-induced anal carcinomas was subjected to codon 61 mutation specific PCR followed by Southern blot analysis and compared to mouse skin carcinoma known to be mutant at H-ras codon 61 (12). Specific sections of DNA were isolated from formalin fixed paraffin-embedded tissues based on H&E of adjacent sections for 7/7 anal cancers and 2/9 atypias, 3/4 papillomas, and 1/6 normal anal canals which were randomly selected from 0.12 μmole DMBA treated E6/E7 mice and one normal anal canal of a dose matched nontransgenic mouse. The reaction conditions and primers for codon 61 mutation specific PCR have been previously described (12) with the following modifications to protocol: this study used 25ng of template DNA, 30 PCR cycles and ran 20μL of product on an aragose gel for ethidium bromide staining. Gels were denatured, neutralized, and DNA transferred to charged nylon membrane overnight. After UV crosslinking, the membrane was blocked and probed with radioactively labeled wildtype H-ras probe generated with the same PCR primers.
Statistical Analysis
Two-sided Wilcoxon rank-sum test was used to determine significant differences in BrdUrd counts. Fisher's two-sided exact test was used to determine differences in rates of disease in each DMBA treated mouse group.
Results
Anal epithelia phenotype is consistent with viral oncogene expression and function
The K14 promoter targets E6 and E7 gene expression to the stratified squamous epithelium of our E6/E7 transgenic mice, analogous to HPV infection in humans. To assess transgene expression in anal tissue, western analysis for E7 was performed. E7 protein was found to be expressed in anal tissue at levels similar to that found in the human cervical cancer cell line SiHa (Fig. 1A). E6 protein was also detected by Western using a PDZ pulldown technique (Fig. 1B) or using an E6 strip test (supplemental Fig 1).
Figure 1. HPV oncoproteins are expressed in the anus of K14E6/K14E7 mice.
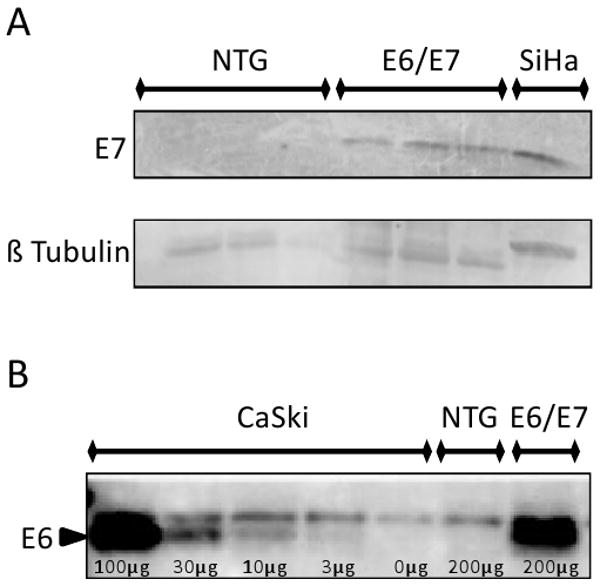
Panel A. E7 western blot analysis. Note that E7 protein is expressed in E6/E7 anal tissue at levels similar to that found in an HPV16-positive human cervical cancer cell line (SiHa) whereas none was found in nontransgenic (NTG) mice. Panel B. E6 protein detected by Western blot analysis of protein lysates subjected to E6 pulldown assay using GST MAGI1-PDZ domain. In this experiment the HPV16-positive cervical cancer cell line CaSki was used as a positive control.
HPV16 E6 and E7 oncoproteins confer acute phenotypes in the skin (7), cervix (13), and oral mucosa (14) of these HPV16 trasngenic mice. To monitor the influence of E6 and E7 on acute phenotypes of the perianal epithelium, immunohistochemistry for minichromosome maintenance 7 (MCM7) and bromo deoxyuracil (BrdUrd) was performed on anal tissue harvested from transgenic and nontransgenic mice that were or were not subjected to radiation one day prior to sacrifice. E7 is known to activate expression of MCM7 through its inactivation of pRb and consequent activation of the E2F family of transcription factors. E6 and E7 are also known to induce cell proliferation in other squamous epithelia in mice. Anal tissue of E6/E7 transgenic mice displayed heightened MCM7 staining indicative of E7's inactivation of Rb (Fig. 2A). E6/E7 transgenic mice also displayed a statistically significant increase (p<0.01) in cells supporting DNA synthesis (assessed by measuring the incorporation of BrdUrd into newly synthesized DNA) within the suprabasal compartment (26%) in comparison to nontransgenic mice (0.5%) (Fig. 2C).
Figure 2. Acute phenotypes of HPV16 E6/E7 transgenic mouse anal epithelium.
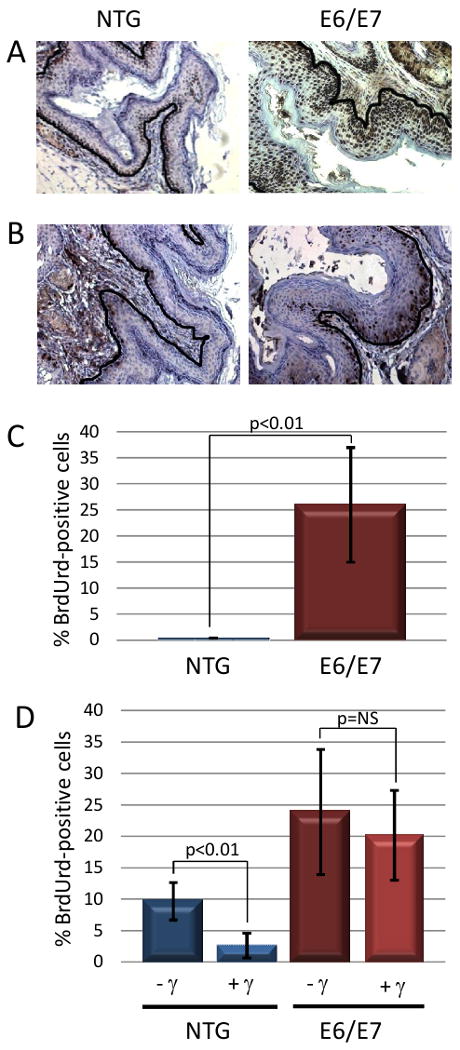
Panel A. Shown is MCM7-specific immunohistochemical staining of sections from the anus of nontransgenic (NTG) and K14E6/E7 transgenic mice. Note that in anal epithelium of E6/E7 mice, expression of MCM7, a biomarker for E7's inactivation of Rb, is strongly induced as compared to NTG mice. Brown nuclei indicate positive staining which is expected along the basement membrane (outlined in black) in NTG mice while it is seen throughout the epithelium in E6/E7 mice. Panel B. Shown is BrdUrd-specific immunohistochemical staining of sections from the anus of nontransgenic and K14E6/E7 transgenic mice. Panel C. Quantification of the percentage of suprabasal cells with BrdUrd-positive nuclei. Panel D. Response of anal epithelium to radiation-induced growth arrest. Shown is the quantification of the frequency of BrdUrd-positive basal cells in anal epithelia of nontransgenic and K14E6/E7 transgenic mice that were (+) or were not (-) treated with 12Gy (γ) external radiation.
Both E6 and E7 are known to inhibit normal DNA damage responses in other squamous epithelia of K14E6 and K14E7 mice. Therefore we also analyzed DNA damage responses to ionizing radiation. The normal cellular response to this form of DNA damage in many epithelial tissues is a reduction in the number of cells supporting DNA synthesis at 24 hours post-irradiation. This was also observed in the perianus of nontransgenic mice: whereas 10% of cells were DNA synthesis competent (BrdUrd-positive) prior to irradiation, this dropped to 2.6% at 24 hours post irradiation (Fig. 2D, p< 0.01). In contrast, DNA synthesis in irradiated E6/E7 transgenic mice did not decrease significantly following irradiation (Fig. 2D; 24% BrdUrd-positive cells prior to irradiation compared to 20% after; p = 0.64).
These data demonstrate that E6 and E7 oncogenes are expressed in the anal epithelium of K14E6/E7 mice and are functional. We therefore predicted that the anal epithelium of these mice would be vulnerable to HPV-induced carcinogenesis.
Anal carcinogenesis only occurs in E6/E7 mice treated with DMBA
E6/E7 transgenic mice do not develop anal tumors spontaneously. We therefore investigated whether the viral genes could synergize with a chemical carcinogen to cause anal cancers. We chose DMBA to induce anal carcinogenesis as it is known to induce carcinomas in the skin (8) and synergizes with E6 or E7 to induce skin carcinomas (7). The anal epithelium of E6/E7 transgenic mice and age-matched nontransgenic mice were treated with DMBA (at three different doses) for 20 weeks followed by an 8 week hiatus. Both male and female mice were included as gender is not a risk factor for human anal cancer. At the time of euthanasia, overt tumors (i.e. any tumor observable with the naked eye) were scored for size and the entire perianal region of each mouse was harvested for histopathological analysis. Every 30th 5μm section was stained with hematoxylin and eosin and scored for disease. The worst case disease for each mouse was thereby determined.
Twenty-three percent of E6/E7 mice treated with 0.12 μmole of DMBA developed carcinoma and an additional 29% had atypia on histological evaluation (Table 1). In contrast none of the DMBA-treated nontransgenic mice developed either cancer or atypia (Table 1). Representative histopathological images of neoplastic disease arising in the E6/E7 transgenic mice are shown in Figure 3A. Forty-five percent of E6/E7 mice in the 0.12 μmole DMBA group had overt tumors that could be identified with the naked eye. The mice with overt tumors were more likely than mice without overt tumors to have atypia or cancer upon comprehensive histopathological examination. Specifically, 43% of overt tumor-bearing mice had frank cancer and 36% had atypia. In comparison, of the mice not bearing an overt tumor, only 6% had frank cancer and 24% had atypia that were discovered upon histopathological analysis (Table 2). The median size of overt tumors was 2 mm and the median time of onset of overt tumors was at the 22nd week following initiation of the DMBA treatment protocol (Figure 3B and C). The incidence of overt tumors correlated with the dose of DMBA applied (Table 3).
Table 1.
Anal tumors selectively arise in E6/E7 mice treated with 0.12 μmole DMBA
| Genotype | n | Histological Classification of Disease | ||||
|---|---|---|---|---|---|---|
| Normal | Hyperplasia | Papilloma | Atypia (%)* | Cancer (%)† | ||
| NTG | 32 | 32 | 0 | 0 | 0 (0) | 0 (0) |
| E6/E7 | 31 | 6 | 5 | 4 | 9 (29) | 7 (23) |
p<0.01 comparing presence of atypia in nontransgenic (NTG) mice vs E6/7 mice
p<0.01 comparing presence of cancer in nontransgenic mice vs E6/7 mice
Figure 3. Histopathology of anal tissue.

Panel A. Shown are H&E stained sections of various samples categorized as normal (N) including inflammation and hyperkeratosis, hyperplasia, papilloma (P), atypia (A), and carcinoma (C). Panel B. Frequency of overt tumors. Note that overt tumors appeared at the highest frequency in the E6/E7 mice treated with 0.12 μmole DMBA. Tumors were a median of 2mm in size at the time of sacrifice with a range from 1mm to 17mm. The largest tumor occurred when 3 smaller tumors coalesced followed by aggressive growth; as such this mouse was euthanized four days prior to the original endpoint. Panel C. Time of onset of tumors. Note that overt tumors arose at a median of 22 weeks after 0.12 μmole DMBA treatment was initiated with a range from 11 to 27 weeks. Panel D. Expression of cancer biomarkers in mouse. Shown are tissue sections from mouse anal cancer. Top images are H&E stained sections showing keratin pearls and large atypical nuclei consistent with squamous cell carcinoma. Shown at the bottom are representative images of a cancer stained with MCM7 (left bottom) and p16 (right bottom). See Table S1 for a complete reporting of the IHC for all samples evaluated.
Table 3.
DMBA dose correlates with increase in tumor incidence as well as carcinogenesis in E6/E7 anal tissue
| Genotype | Dose (μmole) | n | Phenotype | Histological Classification | |
|---|---|---|---|---|---|
| Overt Tumor | Atypia (%) | Cancer (%) | |||
| Nontransgenic | 0.12 | 32 | 0 | 0 (0) | 0 (0) |
| E6/E7 | 0.12 | 31 | 14 | 9 (29) | 7 (23) |
| E6/E7 | 0.04 | 30 | 5 | 6 (20) | 0 (0) |
| E6/E7 | 0.012 | 31 | 1 | 0 (0) | 0 (0) |
| E6/E7 | DMSO only | 10 | 0 | 0 (0) | 0 (0) |
In mouse skin, including that of HPV transgenic mice, DMBA contributes to epidermal carcinogenesis primarily by causing activating A to T transversion mutations in codon 61 of H-ras (7, 12). We therefore looked for the presence of this H-ras mutation in the anal tumors arising in the DMBA-treated E6/E7 mice. In contrast to what is commonly observed in DMBA-induced skin carcinomas, we failed to find this mutation in the anal papillomas, atypia or even frank cancers (supplemental Figure 2). At this point it remains unclear whether DMBA is causing activating mutations elsewhere in H-ras or in other members of the ras gene family, or is contributing to anal cancer through mutation of other gene(s).
In the epithelia of the lower female reproductive tract (cervix and vagina) as well as the head and neck, p16 and MCM7 are robust markers for HPV-associated neoplastic disease both in mouse models (14, 15) as well as in humans (15, 16). Importantly, in humans where only a fraction of HNSCC is associated with HPVs, high expression of MCM7 and p16 was selectively observed in the HPV-positive cancers (16). MCM7 and p16 staining was 100% sensitive and specific for high grade AIN when over 50% of cells were immunopositive (17). In addition, all of these samples were found to harbor high risk-HPV. We performed immunohistochemistry to assess whether these same markers were expressed in HPV-associated neoplastic disease of the anus in our mouse model (Fig. 3D). In the majority of the mouse cancers we observed robust MCM7 and p16 staining, though for p16, there were several cancers with low to variable signals. Prior studies in tissue culture and in mouse models for HPV-associated cervical and head/neck cancers (18-21) lead to the prediction that the induction of MCM7 and p16 seen in Fig 3 is the consequence of E7 expression.
Discussion
HPV has been implicated in human anal carcinogenesis based upon the fact that DNA from high-risk HPV genotypes are found present in a large fraction of anal cancers (3, 22). This is the first study that demonstrates that high-risk HPV oncogenes, E6 and E7, increase the susceptibility to development of anal cancer in K14E6/E7 transgenic mice genetically engineered to express these viral oncogenes in stratified squamous epithelia. As has been observed in other sites in these mice, i.e. the skin, cervix, head and neck (7, 13, 14), addition of a co-carcinogen was necessary to drive efficient development of cancers in the anus. We chose DMBA as the co-carcinogen because of its proven capacity to induce carcinoma in a broad spectrum of tissues, and the fact that there had been observed previously a strong synergy between the HPV16 E6 and E7 oncogenes and DMBA in the induction of carcinomas in the mouse skin (7). In the present study, anal cancer only arose in the DMBA treated K14E6/E7 mice, not in the like-treated nontransgenic mice. This result demonstrates that a synergy between HPV16 oncogenes and DMBA is also manifest in the anal epithelia.
The frequency of anal cancer even in subpopulations of people at high risk for developing this cancer is much smaller than frequency of high risk HPV DNA in the same cohort. Among 446 HIV positive men who have sex with men, 2.5% developed anal cancer over mean follow up of 20.5 months while 85% were positive for high risk HPV (23). This leads to the hypothesis that, as with other HPV associated cancers, genetic/epigenetic changes in the human genome must contribute together with HPV oncogenes in the development of anal cancer. Loss of heterozygosity (LOH) at 11q in anal cancers ranges from 39-87% amongst different studies (24). One group has proposed a model in which LOH at 11q together with LOH at one or more other sites including 17p (p53), 5q (APC), or 5q (DCC) contribute to anal carcinogenesis. LOH at these genes in addition to activating mutations in K-ras are commonly implicated in human colon cancer. However, unlike in colon cancer, N, K and H ras mutations at codons 12, 13 and 61 are rarely found in human anal cancers (25). In this regard, it was interesting that we failed to find mutations at codon 61 of H-ras in the cancers or precancerous lesions arising in our mouse model for anal cancer, even though such mutations are commonly observed in DMBA-induced skin cancers on the same HPV16 transgenic background (7). Activation of the AKT pathway has also been proposed as a contributor to human anal carcinogenesis (26). The absence of TGFβ Receptor II (TGFβRII) gene predisposes mice to rectal cancers (27). This observation may have relevance to HPVs role in cancer as HPV16 E7 causes a reduction in expression of TGFβRII in mouse cervical tissue (28). Our mouse model for HPV-associated anal cancer provides an experimental platform for identifying and testing roles of these or other cellular changes as well as the individual roles of E6 and E7 in anal cancer.
Supplementary Material
Acknowledgments
This study was supported by an NIH grant (R01 DE017315) funded to PFL under the AIDS-related malignancy research initiative. MKS was supported by a NIH training grant (NIH/NCI T32 CA090217) for clinical fellows.
References Cited
- 1.Ries LAG, M D, Krapcho M, Stinchcomb DG, et al., editors. SEER Cancer Statistics Review. Bethesda, MD: National Cancer Institute; 1975-2005. [Google Scholar]
- 2.Cress R, Holly E. Incidence of anal cancer in California: increased incidence among men in San Francisco, 1973-1999. Prev Med. 2003;36:555–60. doi: 10.1016/s0091-7435(03)00013-6. [DOI] [PubMed] [Google Scholar]
- 3.Hoots B, Palefsky J, Pimenta J, Smith J. Human papillomavirus type distribution in anal cancer and anal intraepithelial lesions. Int J Cancer. 2009;124:2375–83. doi: 10.1002/ijc.24215. [DOI] [PubMed] [Google Scholar]
- 4.Chaturvedi A, Madeleine M, Biggar R, Engels E. Risk of human papillomavirus-associated cancers among persons with AIDS. J Natl Cancer Inst. 2009;101:1120–30. doi: 10.1093/jnci/djp205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin-Hong PV, Berry JM, Cheng SC, et al. Comparison of patient- and clinician-collected anal cytology samples to screen for human papillomavirus-associated anal intraepithelial neoplasia in men who have sex with men. Ann Intern Med. 2008;149:300–6. doi: 10.7326/0003-4819-149-5-200809020-00004. [DOI] [PubMed] [Google Scholar]
- 6.Welton M, Sharkey F, Kahlenberg M. The etiology and epidemiology of anal cancer. Surg Oncol Clin N Am. 2004;13:263–75. doi: 10.1016/j.soc.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Song S, Liem A, Miller JA, Lambert PF. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267:141–50. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- 8.Verma A, Conrad E, Boutwell R. Differential effects of retinoic acid and 7,8-benzoflavone on the induction of mouse skin tumors by the complete carcinogenesis process and by the initiation-promotion regimen. Cancer Res. 1982;42:3519–25. [PubMed] [Google Scholar]
- 9.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73:5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balsitis SJ, Sage J, Duensing S, Munger K, Jacks T, Lambert PF. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol Cell Biol. 2003;23:9094–103. doi: 10.1128/MCB.23.24.9094-9103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maufort JP, Williams SM, Pitot HC, Lambert PF. Human papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res. 2007;67:6106–12. doi: 10.1158/0008-5472.CAN-07-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riley R, Duensing S, Brake T, Münger K, Lambert P, Arbeit J. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–71. [PubMed] [Google Scholar]
- 14.Strati K, Pitot HC, Lambert PF. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proc Natl Acad Sci U S A. 2006;103:14152–7. doi: 10.1073/pnas.0606698103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brake T, Connor JP, Petereit DG, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–80. [PubMed] [Google Scholar]
- 16.Pyeon D, Newton MA, Lambert PF, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–19. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreuter A, Jesse M, Potthoff A, et al. Expression of proliferative biomarkers in anal intraepithelial neoplasia of HIV-positive men. J Am Acad Dermatol. 2009 doi: 10.1016/j.jaad.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 18.Khleif S, DeGregori J, Yee C, et al. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc Natl Acad Sci U S A. 1996;93:4350–4. doi: 10.1073/pnas.93.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiyono T, Foster S, Koop J, McDougall J, Galloway D, Klingelhutz A. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–8. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 20.Strati K, Lambert PF. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007;67:11585–93. doi: 10.1158/0008-5472.CAN-07-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shai A, Brake T, Somoza C, Lambert PF. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007;67:1626–35. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong A, Chan R, Aggarwal N, Singh M, Nichols W, Bose S. Human papillomavirus genotypes in anal intraepithelial neoplasia and anal carcinoma as detected in tissue biopsies. Mod Pathol. 2010;23:144–50. doi: 10.1038/modpathol.2009.143. [DOI] [PubMed] [Google Scholar]
- 23.Kreuter A, Potthoff A, Brockmeyer N, et al. Anal carcinoma in HIV-positive men: results of a prospective study from Germany. Br J Dermatol. 2010 doi: 10.1111/j.1365-2133.2010.09712.x. [DOI] [PubMed] [Google Scholar]
- 24.Gervaz P, Hirschel B, Morel P. Molecular biology of squamous cell carcinoma of the anus. Br J Surg. 2006;93:531–8. doi: 10.1002/bjs.5376. [DOI] [PubMed] [Google Scholar]
- 25.Hiorns L, Scholefield J, Palmer J, Shepherd N, Kerr I. Ki-ras oncogene mutations in non-HPV-associated anal carcinoma. J Pathol. 1990;161:99–103. doi: 10.1002/path.1711610203. [DOI] [PubMed] [Google Scholar]
- 26.Patel H, Polanco-Echeverry G, Segditsas S, et al. Activation of AKT and nuclear accumulation of wild type TP53 and MDM2 in anal squamous cell carcinoma. Int J Cancer. 2007;121:2668–73. doi: 10.1002/ijc.23028. [DOI] [PubMed] [Google Scholar]
- 27.Guasch G, Schober M, Pasolli H, Conn E, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313–27. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diaz-Chavez J, Hernandez-Pando R, Lambert P, Gariglio P. Down-regulation of transforming growth factor-beta type II receptor (TGF-betaRII) protein and mRNA expression in cervical cancer. Mol Cancer. 2008;7:3. doi: 10.1186/1476-4598-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.