

Summary
Ubiquitination plays an important role in many cellular processes and is implicated in many diseases. Experimental identification of ubiquitination sites is challenging due to rapid turnover of ubiquitinated proteins and the large size of the ubiquitin modifier. We identified 141 new ubiquitination sites using a combination of liquid chromatography, mass spectrometry and mutant yeast strains. Investigation of the sequence biases and structural preferences around known ubiquitination sites indicated that their properties were similar to those of intrinsically disordered protein regions. Using a combined set of new and previously known ubiquitination sites, we developed a random forest predictor of ubiquitination sites, UbPred. The class-balanced accuracy of UbPred reached 72%, with the area under the ROC curve at 80%. The application of UbPred showed that high confidence Rsp5 ubiquitin ligase substrates and proteins with very short half-lives were significantly enriched in the number of predicted ubiquitination sites. Proteome-wide prediction of ubiquitination sites in Saccharomyces cerevisiae indicated that highly ubiquitinated substrates were prevalent among transcription/enzyme regulators and proteins involved in cell cycle control. In the human proteome, cytoskeletal, cell cycle, regulatory and cancer-associated proteins display higher extent of ubiquitination than proteins from other functional categories. We show that gain and loss of predicted ubiquitination sites may likely represent a molecular mechanism behind a number of disease-associated mutations. UbPred is available at http://www.ubpred.org
Keywords: UbPred, protein ubiquitination sites, prediction, post-translational modification, intrinsically disordered protein, unstructured, disordered
Introduction
The reversible modification of proteins by the covalent attachment of ubiquitin is implicated in the regulation of a variety of cellular processes. During the past decade, the functions of ubiquitin have been extended far beyond its role in just directing protein degradation.1,2. It is now established that ubiquitination is a more important and widespread protein post-translational modification than previously anticipated. Regulation of transcription factor activity,3, budding of retroviral virions,4 receptor endocytosis and lysosomal trafficking,5, control of insulin6 and TGF-β signaling pathways7 are examples of just a few processes that rely on ubiquitination.
Ubiquitination of target proteins is a highly collaborative process between the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin ligases (E3).8 Ubiquitin-protein ligases catalyze the process of transfer and covalent attachment (via an isopeptide bond) of the C-terminus of activated ubiquitin to lysine side chains of the acceptor substrate. The substrate could be mono- or polyubiquitinated, and it was previously believed that canonical K48-linked polyubiquitin chains were the main signal for targeting the substrates for degradation by the 26S proteasome. However, it has recently been shown that unconventional polyubiquitin linkages may also target proteins for degradation.9
There are at least two functionally different families of E3 ubiquitin ligases, HECT-type E3s and RING-type E3s. HECT-type E3s initially form an E3-ubiquitin thioester conjugate, and then transfer ubiquitin to the substrate. RING-type E3s do not form such conjugates, but rather form E2/E3 complexes that directly ubiquitinate the target substrate. Another recently recognized class of ligases, E4, mediate ubiquitin chain elongation on pre-existing ubiquitinated substrates.10,11 Interestingly, monoubiquitination of some substrates can even occur in an E3-independent manner.12
Despite the availability of the structures for several ubiquitin-protein ligase complexes,13–18 the mechanism of the ubiquitin conjugation reaction to the target substrate is still incompletely understood. The big cavities in the structures of ligases, their highly elongated and relatively rigid shape, as well as the large distance between the E3 catalytic domain and the E2 active site complicate our understanding of the mechanism of ubiquitin transfer. One possibility is that structural disorder of the substrate could facilitate this process.
Intrinsically disordered proteins (IDPs) exist and function as ensembles of interconverting conformations under physiological conditions.19–24 They are prevalent among regulatory and signaling proteins25 and are involved in various human diseases.25,26 IDPs perform numerous important functions in the cell, and their intrinsically disordered regions (IDRs) frequently serve as sites of post-translational modifications.20,27–29
Several lines of evidence have previously implicated the disordered structure in the protein degradation process. For example, an unstructured initiation site within the ubiquitinated substrate was shown to be required for efficient proteasome-mediated degradation of the ubiquitinated proteins.30 Structural disorder has been observed within PEST motifs,31 and was also correlated with protein half-life32,33 even more strongly than with other bona fide degradation signals such as the destruction-box, KEN-box, PEST regions and N-end residues.32 Finally, it was shown that IDPs are more susceptible to 20S proteasomal degradation in vitro than are folded proteins.34
Although the involvement of disorder in protein degradation has been examined on many levels, the question about the relationships between ubiquitination and disorder is far less explored. This might be due to the inherently difficult experimental identification of protein ubiquitination (Ub) sites. Only a limited number of Ub sites from high-throughput experiments are available in the literature, and these sites are known to be biased against short-lived proteins.35,36
Here, we first identify novel Ub sites using mutant yeast strains to better target short-lived proteins. We then examine sequence and structural preferences of all available ubiquitination sites and show that they have high propensity for intrinsic disorder and flexibility. Based on this and several other distinct properties, we constructed a predictor of ubiquitination sites, UbPred. We show that UbPred predicts ubiquitination sites in many important cell cycle regulators and other short-lived proteins. We also apply UbPred to various protein functional categories, proteins with known half lives, Rsp5 ligase substrates, and proteins involved in various human diseases, including cancer. This allowed us to gain better insight into processes and functions that depend on ubiquitination.
Materials and Methods
Sample preparation
To analyze the CDC34 mutant, termed CDC34tm, Saccharomyces cerevisiae strains KS418 (MAT a, CDC34tm ura3 leu2 trp1 lys2 ade2 ade3) and KS422 (MAT a, ura3 leu2 trp1 lys2 ade2 ade3) were grown to mid-log-phase in 1 L of SD Complete media. The SD media used to grow KS422 lacked L-lysine but was supplemented with deuterium labeled d4 L-lysine to allow for relative quantitation. Cells were then pelleted and resuspended in 8 M urea + 25 mM Ammonium bicarbonate. Glass beads were added to the resuspension and cells were broken by repeated rounds of vortex mixing. Protein quantitation was accomplished by the Bradford method. The supernatant was collected and the urea concentration was reduced to 2 M by the addition of 10 mM ammonium bicarbonate. Samples were then reduced with DTT and alkylated with iodoacetamide. After reduction and alkylation the urea concentration was reduced to 1 M by addition of 10 mM ammonium bicarbonate and 1.6 mg of each extract were combined and digested with 100μg of lyophilized Glu-C. Digestion proceeded for 72 hours at room temperature. The analysis of cells deleted for GRR1 was as described above except that the strains used were DBY2059 (MAT α, leu2-3,112) and JH001 (Mat α, grr1Δ::NAT) and the amino acid label was 13C6- leucine.
The digested sample was desalted using a SepPak (The Waters Corporation, England) and resuspended in 120 microliters of HPLC buffer A (5% Acetonitrile, 0.1% Formic Acid). 10 microliters which is approximately 260 μg of total protein was bomb loaded onto a biphasic MudPIT37 column.
LC/LC-MS/MS
The MudPIT column is a 100 μM inner diameter fused silica column packed with 10 cM of C18 resin followed by 4 cm of strong cation exchange (SCX) resin. After loading the samples onto the SCX portion of the column using a pressure bomb, the peptides were subjected to a step gradient of increasing salt concentration (ammonium acetate), moving peptides into the reverse phase resin. Prior to the next increase in salt concentration, the peptides moved to the reverse resin were subjected to a continuous gradient of increasing acetonitrile. The released peptides were continuously ionized and sprayed into the LTQ (ThermoFinnigan, USA) mass spectrometer at a flow rate of 200 nanoliters/minute.
Data processing
Peptide-to-spectrum matches were generated using SEQUEST38 and were post-processed by PeptideProphet.39 The yeast protein database was downloaded from the Saccharomyces Genome Database (SGD; www.yeastgenome.org). Differential modification of +16 daltons for methionine oxidation, +57 daltons for cysteine carboxyamidomethylation, and +114.1 for lysine ubiquitination were allowed in the search. Interestingly, performing searches with the very large adduct remaining attached to lysine after Glu-C cleavage uncovered no statistically significant ubiquitin-linked peptides. This is likely due to the inability of the search algorithm to handle the complexity of these large branched peptides. On the other hand, searching with a lysine ubiquitination remnant typical of tryptic peptides uncovered numerous ubiquitin-linked peptides. We hypothesize that grr1 mutants have a hyperactive Rsp5, which leads to elevated levels of endocytosed proteins that become partially proteolyzed during vacuolar trafficking.
Datasets
Positive examples of ubiquitination sites were extracted from two large-scale proteomics studies,35,36 our own experiments and an ad-hoc literature search. These lysine ubiquitination sites were present in 201 proteins from S. cerevisiae. From these proteins we extracted 272 ubiquitinated (positive) fragments, each containing up to 12 upstream and downstream residues around the central lysine residue. The set of 4,651 non-ubiquitinated (negative) fragments were extracted from 124 mitochondrial matrix proteins. We reasoned that mitochondrial matrix proteins would serve as a good negative control dataset because inner membrane of mitochondria is the only cellular membrane that is not exposed to the cytosolic compartment and therefore not accessible for the ubiquitin/proteasome system.40 Therefore, we expect that this dataset would be a clean negative dataset, e.g. it would be less likely contaminated with non-annotated Ub sites. Proteins annotated with Gene Ontology (GO) term41 “mitochondrial matrix” and its children terms were extracted from the SGD database. Non-Ub sites dataset was formed by extracting fragments around each lysine within this dataset. In total, each fragment contained 25 residues (or less for the near-terminal lysines). Both sets were then filtered for similarity to prevent over-representation of any particular fragment and overestimated performance accuracy during predictor construction and evaluation.
To obtain a non-redundant dataset, no two fragments within the positive or negative datasets, as well as across the two datasets, were allowed to share >40% sequence identity. When a similar pair between a positive and negative example occurred, the negative site was always removed as less reliably labeled. The sequence identity cutoff of 40% lies well below those that provide accurate functional inference by homology transfer,42 thus allowing us to consider our dataset to be non-redundant. The resulting datasets contained 265 positive and 4,431 negative fragments.
Several other datasets for UbPred application were collected from the literature. The confident and relaxed Rsp5 ligase substrates datasets were extracted from Gupta et al.43 The datasets with protein half lives were extracted from Belle et al.44 Protein functional categories were extracted from the Swiss-Prot database (release 56.6) using the organism “human” and a list of keywords: “biosynthesis” (436 proteins), “cell cycle” (479), “cytoskeleton” (388), “G-protein coupled receptor” (828), “inhibitor” (190), “kinase” (639), “metabolism” (270), “regulation” (2055), “ribosomal” (205), “transport” (1638). The “cancer” (388) dataset was extracted by using a combination of keywords “anti-oncogene OR oncogene OR proto-oncogene OR tumor” and organism “human”. The redundancy within (but not between) functional datasets was removed based on 40% sequence identity. Disease mutations were downloaded from the Swiss-Prot database (as of September 2006) and combined with the missense disease mutations from the Human Gene Mutation Database (HGMD; www.hgmd.cf.ac.uk) as of September 2006.
Data representation and predictor construction
We calculated 586 sequence attributes for each lysine of the positive and negative datasets. The first group contained a set of 20 amino acid compositions constructed over symmetric windows of length win∈ {3, 7, 11, 21} centered at each lysine. In addition to compositional attributes, we also calculated various physicochemical and other properties within win: net charge, total charge, aromatic content, charge/hydrophobicity ratio22 and sequence complexity using Shannon and generalized β-entropy.45 Another set of attributes was derived from several sequence-based predictors of protein properties. We used predictors of flexibility,46 high B-factor,47 amphipathic moment,48 phosphorylation,27 and four predictors of intrinsic disorder.49–52 These prediction values were averaged within win∈ {1, 7, 11, 21}. Evolutionary information was exploited through position-specific scoring matrices (PSSMs), obtained via a PSI-BLAST search53 against the non-redundant GenBank database (parameters: −h 0.0001 −j 3). The 42 outputs in each PSSM row were averaged over win∈ {1, 7, 11, 21} for each lysine, thus generating 168 evolutionary attributes. Finally, position specific amino acid content was encoded for positions −3 to +3 as binary attributes.
Before model optimization, we applied a t-test attribute selection filter and retained only statistically significant attributes. A predictor was then built using a random forest approach.54 In each member of the ensemble, the set of negative examples was equal in size to the set of positive examples to achieve the highest accuracy on a class-balanced test set. Even though such training is indicative of class separability, it can cause significant over-prediction on the majority (here negative) class. This problem, however, can be addressed by changing decision thresholds or adjusting the outputs of the predictor.55
Model evaluation and performance measures
To evaluate UbPred, 100-fold cross-validation strategy was chosen. This process was further repeated 10 times to obtain stable estimates. We measured accuracy on a per-residue level by estimating sensitivity (sn) and specificity (sp). Sensitivity represents the percentage of true positives predicted to be positive (ubiquitinated), while specificity represents the percentage of true negatives predicted to be negative (non-ubiquitinated). In addition to sn and sp, we also report accuracy on a balanced sample (acc), defined as an average of sn and sp, and area under the ROC curve (AUC). The ROC curve represents a mapping of (1– sp) to sn and in our case was estimated by varying the decision thresholds.
GO annotations
To functionally annotate proteins regulated by ubiquitination, we downloaded a set of 5,884 verified ORFs (5817 sequences of length ≥50) from the SGD website and applied UbPred. A major challenge in finding proteins that are most likely to be ubiquitinated is a possibility that a direct application of UbPred to any proteome would favor longer proteins, as a consequence of <100% prediction accuracy. Thus, to extract a set of proteins with strongest predictions, we proceeded as follows.
First, a threshold t was determined such that only 100·p% of all prediction scores over all proteins were greater than t. For a sufficiently high t, or similarly, sufficiently low p, such scores can be considered as strong predictions of ubiquitination, which is supported by the low false positive rate in the bottom left-hand corner of the estimated ROC curve. Then, with a reasonable assumption, we introduced a null model in which a randomly selected lysine from any protein had 100·p% chance of being predicted as strong. Under this model, the number of strong predictions (with scores above threshold t) in each protein would be proportional to the number of lysines it contains. Therefore, using the null model assumption, the probability that, in a protein containing K lysines, the number of strong predictions that occurred by chance is k or greater, can be expressed as
where p is the probability that a randomly selected lysine has a strong prediction of being ubiquitinated. Thus, proteins with the lowest P-value P are the most likely to contain a disproportionately larger number of strong predictions than expected by chance. We considered these proteins to be the most strongly ubiquitinated proteins (i.e. over-ubiquitinated). The potential length dependence was thus eliminated since the P-values implicitly equalize the length factor. We selected the threshold of p = 0.1 and extracted all proteins with P < 0.05, Bonferroni corrected. In addition, since consecutive lysines may not be considered to be motionally independent (possibly invalidating null model assumptions), we note that a selection of the smaller samples of lysines from each protein did not significantly influence the results reported herein.
Results
Identification of novel Ub sites using combination of MudPit and mass spectrometry
Two high-throughput datasets of Ub sites are currently available in the literature.35,36 These datasets have two major shortcomings: (1) only a small number of Ub sites was identified (127 sites from the two studies combined); (2) these sites are known to be biased against proteins with short half-lives.36 To address both of these limitations, we identified additional Ub sites using combination of MudPIT, mass spectrometry and mutant yeast strains. We: (1) used grr1Δ mutant strains that are deficient in Grr1 F-box protein, a crucial component of the SCF ubiquitin ligase (SCFGrr1); (2) used yeast strains expressing a mutant of the ubiquitin conjugating enzyme Cdc34, which conjugates polyubiquitin chains more slowly and of shorter length than the wild type enzyme. In these two independent experiments, we identified 141 high-confidence Ub sites from 108 proteins (PeptideProphet score of >0.95). The unique identified peptides containing novel Ub sites are shown in Table S1 (Supplementary Materials).
As mentioned above, the problem of the short half-life of ubiquitinated proteins was addressed using two mutant yeast strains, grr1Δ and CDC34tm. It has recently been shown that some targets of SCFGrr1 could be markedly stabilized in the grr1Δ cells.56 We have used grr1Δ mutant strains to potentially improve the detection of ubiquitinated substrates with extended half-lives.
Cdc34 is the ubiquitin conjugating enzyme of the SCF complex. A universally conserved motif in close physical proximity to the catalytic cysteine defines the Cdc34-like class of ubiquitin conjugating enzymes.57 It has recently been shown that this motif is critical for extension of the polyubiquitin chain but not necessary for addition of the first ubiquitin to substrate.58 Mutation of this motif, namely serine residues 73 and 97 along with the acidic stretch of amino acid residues 103–114, to mimic the Rad6 class of ubiquitin conjugating enzymes, decreases the rate of substrate ubiquitination and ultimately extends the half life of some SCF/Cdc34 substrates (M. Goebl, submitted). Since the substrate still bears the ubiquitin tag but the degradation kinetics are notably slower, an increased steady state of the ubiquitinated substrate is available for analysis.
Functional characterization of known Ub sites
We first created a joint positive dataset of experimentally verified Ub sites from S. cerevisiae (Materials and Methods). This dataset included: (1) 127 non-redundant Ub sites from 92 proteins extracted from two previous high-throughput studies,35,36 in addition to 4 sites found in the literature, referred to as DA; (2) 141 newly identified non-redundant Ub sites from 108 proteins extracted from two independent MudPIT experiments, referred to as DB. The analysis of these datasets showed that they were non-overlapping. This is likely a result of the small sample sizes derived from a large pool of existing ubiquitination sites in yeast, as well as differences in the methodological approaches used to identify Ub sites in the current and previous studies. Using GO annotations, we next examined whether we succeeded in capturing greater number of proteins with short half lives.
It was previously shown that yeast proteins with short half-lives were abundant among GO annotations including “transcription regulation”, “transcription factor activity”, “cell cycle”, “DNA metabolism”, and “DNA binding”.44 We GO-annotated both datasets and observed that proteins with the above GO annotations comprised approximately 20% of DB as opposed to only 6% of DA (Table S2, Supplementary Materials). We detected Ub sites within several important short-lived cell cycle regulators, including Tel1, as well as within numerous short-lived transcription factors and DNA-binding proteins, including Hms1, Spt16, Tfa1, Gal11, and Rad26. This suggests that we in fact were able to capture short-lived proteins using mutant yeast strains. Including Ub sites from short-lived proteins into the training set is essential for better generalizability of the predictor. Thus, we considered our joint positive dataset to be reasonably diverse and suitable for predictor construction.
Structural characterization of known Ub sites
To gain better insight into structural preferences of Ub sites, we searched the available structural information for proteins from our positive dataset (combined DA and DB, Table S3) using BLAST against the Protein Data Bank (PDB)59 with ≥70% sequence identity as a cutoff value. Our search resulted in a total of 32 homologous protein chains (with 15 of them being 100% identical with query proteins) containing 28 Ub sites (Table 1).
Table 1.
Structural analysis of known Ub sites.
| SGD ID | Protein Name | PDB chain (total chains) | Residues from protein sequence | Residues from PDB chain | BLAST identity (%) | Ub site(s) | Site on aligned PDB chain (e- extended strand, c-coil, h-helix) | Additional Information (if available) |
|---|---|---|---|---|---|---|---|---|
| YDR155C | CPR1 | 1vdn:A(2) | 1–160 | 3–162 | 100.0 | K157 | K158(e) | Residue 158 is in crystal contact with residues 5 –7, 17, 47, 156, 157 and 159. |
| YHR042W | NCP1 | 2bn4:B(2) | 32–690 | 24–682 | 99.9 | K218 | K210(c) | Residue 210 participates in crystal contact formation |
| YLR080W | EMP46 | 2a6w:B(2) | 52–275 | 3–226 | 100.0 | K183 | K134(c) | |
| YPL084W | BRO1 | 1zb1:B(2) | 1–387 | 6–392 | 99.7 | K289 | K294(c) | |
| YAL038W | CDC19 | 1a3×:B(2) | 1–499 | 2–500 | 100.0 | K85 | K86(c) | |
| YDL229W | SSB1 | 2qwm:B(2) | 8–389 | 6–387 | 70.2 | K313 | R311 | Residue is not K in the PDB chain |
| YER012W | PRE1 | 3bdm:X(28) | 1–198 | 1–198 | 100.0 | K19, K29 | K19 (c), K29 (e) | |
| YGL203C | KEX1 | 1ac5:A(1) | 23–503 | 1–481 | 100.0 | K152 | K130 (c) | |
| YHR183W | GND1 | 2p4q:A(1) | 1–487 | 11–497 | 100.0 | K74 | K83 (e) | Intra-chain crystal contacts by residue 75 |
| YLR044C | PDC1 | 1qpb:B(2) | 1–561 | 3–563 | 99.3 | K248 | K249 (c) | |
| YNL209W | SSB2 | 2qwm:B(2) | 8–389 | 6–387 | 70.2 | K313 | R311 | Residue is not K in the PDB chain |
| YDL126C | CDC48 | 3cf1:C(3) | 11–796 | 2–773 | 70.2 | K673, K594 | K663 (c), K584 (h) | Intra-chain crystal contacts by residues 568 and 586 |
| YER164W | CHD1 | 2dy7:A(1) | 172–252 | 1–81 | 100.0 | K1144 | N/A | site is not covered by structure |
| YOL145C | CTR9 | 1kt1:A(1) | 735–752 | 321–338 | 72.2 | K196 | N/A | site is not covered by structure |
| YER143W | DDI1 | 2i1a:D(4) | 178–325 | 1–148 | 98.7 | K171 | N/A | site is not covered by structure |
| YBR102C | EXO84 | 2d2s:A(2) | 523–753 | 5–235 | 100.0 | K219 | N/A | site is not covered by structure |
| YBL002W | HTB2 | 1id3:H(10) | 28–131 | 27–130 | 100.0 | K124 | K123(c) | Residues 122, 126 and 128 participate in crystal contacts |
| YGR136W | LSB1 | 1zx6:A(1) | 56–111 | 3–58 | 73.2 | K41, K79 | K26(e) | Residues 22 – 26, 29 – 31 participate in crystal contacts |
| YHR042W | NCP1 | 2bn4:B(2) | 33–691 | 24–682 | 99.9 | K666 | K657(c) | Residues 1– 23 are disordered; no contribution to crystal contact formation; K29 is on the surface; large B-factor |
| YBR035C | PDX3 | 1ci0:B(2) | 1–228 | 1–228 | 100.0 | K29 | K29(c) | |
| YPR154W | PIN3 | 1zx6:A(1) | 56–112 | 2–58 | 98.3 | K80 | K26(e) | Residues 22 – 26, 29 – 31 participate in crystal contacts |
| YGL008C | PMA1 | 1mhs:B(2) | 51–916 | 53–918 | 78.1 | K555, K566, K644 | K555(e), R566, K644(h) | K555 is in contact with residues 376 and 377; 566K>R in the structure; residue 644 participates in crystal contacts |
| YGR135W | PRE9 | 3bdm:P(28) | 1–258 | 1–258 | 100.0 | K199 | K199(h) | |
| YDL140C | RPO21 | 2yu9:A(13) | 1–1537 | 1–1537 | 100.0 | K695 | K695(h) | |
| YHL015W | RPS20 | 1s1h:J(17) | 23–120 | 3–100 | 96.9 | K8 | N/A | site is not covered by structure |
| YLR167W | RPS3 | 1s1h:C(17) | 2–193 | 1–192 | 100.0 | K212 | N/A | site is not covered by structure |
| YAL030W | SNC1 | 3b5n:I(12) | 27–86 | 2–61 | 100.0 | K63 | K38(h) | |
| YOR327C | SNC2 | 3b5n:I(12) | 26–85 | 2–61 | 88.3 | K62 | K38(h) | |
| YAL005C | SSA1 | 7hsc:A(1) | 382–540 | 1–159 | 79.3 | K536, K521 | K155(c), K140(h) | |
| YLL024C | SSA2 | 3hsc:A(1) | 1–382 | 1–385 | 81.7 | K556 | N/A | site is not covered by structure |
| YEL021W | URA3 | 1dqx:D(4) | 1–267 | 1–267 | 98.9 | K209, K253, K93 | K209(c), K253(h), K93(c) | Residue 257 contributes to crystal contact formation; K253 participates in crystal contact formation |
| YBR088C | POL30 | 1plr:A(1) | 1–258 | 1–258 | 100.0 | K164 | K164(c) | Residues 161, 163 and 164 contribute to crystal contact formation |
A more detailed analysis of the available structures showed that only 8 structures (1ac5, 2p4q, 2dy7, 1kt1,1zx6, 7hsc, 3hsc and 1plr) consisted of a single chain representing protein monomers, whereas the remaining proteins were homooligomers, heterooligomers, or complexes with other proteins or ligands, including DNA. Conclusions about the structural preferences of Ub sites when they are found in complexes should be made carefully because of structural rearrangements upon binding. Moreover, crystal contacts60,61 could further obscure the true structural preferences of Ub sites. Our analysis showed that 10 out of 28 Ub sites (or their neighbors 5 residues upstream or downstream) were in crystal or interchain/intrachain contacts, and therefore the assignment of these sites to a specific structural element should be made with caution. Of the 18 sites that could be confidently assigned to ordered regions, 11 were located within coils (2 of which were close to the observed disordered regions), 4 within helices, and 3 within strands. The majority of the sites within coils and helices were surface exposed and had high B-factor values indicating high flexibility.
In summary, despite the presence of more than 50,000 structures in PDB, reliable structural assignments can be made for only ~7% of the available Ub sites (18 out of 265 non-redundant sites). This indicates that very limited structural information is currently available for proteins that comprise known ubiquitination substrates.
Examples of ubiquitination sites located in disordered protein regions
Along with the lack of structural information for the majority of experimentally detected Ub sites, there are several examples of Ub sites located in the experimentally confirmed disordered regions (Table 2). Site-directed mutagenesis of six lysine residues to arginine near the C-terminus of p53 generates a molecule with potent transcriptional activity that is extremely resistant to Mdm2- and E6-AP-mediated ubiquitination and degradation.62 This suggests that ubiquitination sites of p53 are located in its C-terminal regulatory domain. At the same time, p53 contains large unstructured regions in its N- and C-terminal parts.63 The ubiquitin-mediated proteolysis of the c-Myc protein is governed by its transcriptional activation domain,64 which is shown to be unstructured in the absence of its binding partner TBP.65 Ubiquitination sites of histones H2A66 and H2B67 are contained within C-terminal regions that are susceptible to proteolysis68 and are unstructured.69 Multiple ubiquitination sites of α-synuclein, a completely unfolded protein,70,71 were found to be located within its N-terminus.72 Similarly, the regions of disorder and ubiquitination coincide for three cyclin-dependent kinase inhibitors, p21Cip1, p27Kip1, and p57Kip2, as well as for IκBα and the component of the yeast SNARE complex, Snc1. Finally, the Ub sites of cyclin B and securin are also located in their disordered N-termini.73 These examples demonstrate that Ub sites could be mapped to the experimentally confirmed unstructured regions in several proteins.
Table 2.
Examples of Ub sites located in the experimentally confirmed disordered regions.
| Protein name | DR location | Ub site(s) location | References |
|---|---|---|---|
| p53 | 1–70, 363–393 | C-terminus | 62,63 |
| c-myc | 1–143 | unknown, between residues 1 and 128 | 64,65 |
| Histones | |||
| H2A | 1–22, 92–128 | 119 | 66,67,69 |
| H2B | 1–32, 100–122 | 120 | |
| α-synuclein | 1–140 | 21, 23, 32, 34, 6, 10, 12 | 70,72 |
| Cell cycle inhibitors | |||
| p21 | 1–164 | Unknown | 94–97 |
| p27 | 23–106 | ||
| p57 | 1–316 | ||
| Cyclin B | 1–100 | N-terminus | 73,98 |
| Securin Pds1 | 1–100 | N-terminus | 73 |
| IκBα | 1–70 | 21, 22 | 99,100 |
| Synaptobrevin homolog SNC1 | 1–93 | 63 | 36,101 |
Sequence analysis of the position-specific and non-position-specific attributes
To determine whether Ub and non-Ub sites have distinct sequence properties, we calculated statistically significant differences in the distribution of amino acid residues surrounding ubiquitinated (265 examples) and non-ubiquitinated (4,431 examples) lysines (see Materials and Methods for datasets description).
This analysis showed 38 compositional differences between Ub and non-Ub sites (Figure 1). The most pronounced feature of Ub sites is the abundance of charged and polar amino acids, especially negatively charged D and E, and the depletion of hydrophobic residues, such as L, I, F and P around Ub sites. Interestingly, disordered proteins/regions are also characterized by similar properties, e.g. a high absolute value of the net charge and low hydrophobicity.74
Figure 1.

A Two Sample Logo93 of the compositional biases around Ub sites compared to the non-Ub sites. Only amino acid residues significantly enriched and depleted (P < 0.05; t-test) from the positive dataset are shown.
Another interesting feature is the absence of additional lysines at positions that are immediately adjacent to the Ub site. For example, lysines are depleted at positions (−4), (−3), (+1), (+2), (+3), (+4) and (+7) (Figure 1). This suggests that Ub sites do not have a tendency to cluster, perhaps due to the structural constrains that would prevent simultaneous attachment of two or more bulky ubiquitin molecules in close proximity to each other on the same substrate. This is in contrast to phosphorylation sites that often cluster, as indicated by both, experiments75,76 and predictions.27 However, we note that the depletion of lysines may also exist as an artifact of mass spectrometry-based identification.
Besides position-specific frequencies, we have also compared the overall amino acid compositions of intrinsically disordered regions, Ub and non-Ub sites (Figure 2). This graph shows the composition of these three datasets relative to the composition of completely ordered proteins from PDB-Select-25.77 Ub sites and IDRs are enriched overall in flexible residues (positive bars) and depleted in rigid residues (negative bars). For example, unlike non-Ub sites, both Ub sites and IDRs are depleted in aromatic residues, I and L, and they are enriched in D and E. At the same time, Ub sites have some common features with non-Ub sites, such as depletion of R, G, A and M and enrichment of N. In addition, Ub sites are considerably more depleted in C than both non-Ub sites and IDRs.
Figure 2.

Relative amino acid compositions of three studied datasets. Amino acid compositions are shown relative to the composition of ordered proteins from O_PDB_S25 database. Amino acids are arranged from left to right in order of increasing flexibility as defined by Vihinen et al.46 The error bars represent 95% confidence intervals.
Predictor of ubiquitination sites UbPred
Using 586 sequence-based attributes, we constructed a predictor of ubiquitination sites from protein sequence, UbPred. The analysis of the properties of Ub and non-Ub sites has shown that several attributes were positively and negatively correlated with Ub sites (Table 3). The predictions of disorder,49–52 high B-factors,47 conservation of D, E, N, S, and flexibility by Vihinen et al.46 were positively correlated with Ub sites, whereas net charge, frequency of K, hydrophobic moment, conservation of I, V and F and several other attributes were negatively correlated with Ub sites (Table 3). These data clearly suggest that Ub sites have higher propensity for disorder and flexibility than non-Ub sites.
Table 3.
Ten top features positively and negatively correlated with Ub sites.
| Positively Correlated | Negatively Correlated | ||||
|---|---|---|---|---|---|
| Feature Name | Correlation Coefficient | P-value | Feature Name | Correlation Coefficient | P-value |
| Disorder VSL2B mean | 0.153368 | 0 | Net charge | 0.152072 | 0 |
| Conservation of S score | 0.13563 | 0 | Frequency of K | 0.09838 | 1.40E-11 |
| B-factor mean | 0.129643 | 0 | AA volume mean | 0.094526 | 8.50E-11 |
| Conservation of E score | 0.126196 | 0 | Hydrophobicity mean | 0.089927 | 6.60E-10 |
| Conservation of D score | 0.123398 | 0 | Hydrophobic moment 100 mean | 0.089415 | 8.30E-10 |
| Conservation of N score | 0.120295 | 1.10E-16 | Hydrophobic moment 100 max | 0.088217 | 1.40E-09 |
| Disorder VL2 max | 0.118341 | 4.40E-16 | Conservation of I percentage | 0.078733 | 6.50E-08 |
| Disorder VLXT mean | 0.115929 | 1.50E-15 | Conservation of V percentage | 0.073923 | 3.90E-07 |
| Vihinen flexibility mean | 0.115469 | 2.00E-15 | Conservation of F percentage | 0.072504 | 6.50E-07 |
| Disorder VL2 mean | 0.115005 | 2.70E-15 | Hydrophobic moment 120 max | 0.070616 | 1.30E-06 |
To construct UbPred, we used random forest-based approach that showed better performance than other tested models (logistic regression, support vector machine and neural network). The overall performance of the predictor was estimated on a per residue basis and reached a class-balanced test accuracy of 72.0%, area under the ROC curve (AUC) was estimated at 79.6% (Figure 3).
Figure 3.

Receiver operating characteristic (ROC) curve for the UbPred predictor of ubiquitination sites (solid line) vs. the performance of the random model (dotted line). The area under the curve (AUC) was estimated to be 79.6%.
Since the set of negative data points have been extracted from the mitochondrial matrix proteins, we decided to train another model in which we kept the same positive examples, but the set of negatives was chosen from all yeast proteins, giving in total 34,844 sites, of which 30,847 were non-redundant (<40% sequence identity). This predictor had accuracy of 70.7% and area under the ROC curve of 77.5% (not shown). The output score of this predictor had correlation of 0.81 with the predictor developed using the mitochondrial matrix proteins as a negative dataset. In addition, the two predictors output different class in 7.6% of cases for the default threshold of 0.5 and 1.9% of cases for the highly confident predictions (≥0.75) that provide a false positive rate of 5%. Thus, we concluded that the particular selection of the negative data set did not significantly influence predictor output.
Prediction of precise Ub sites in Rsp5 ubiquitin ligase substrates
Global identification of the substrates for E3 ligases, and especially their precise Ub sites, on a proteome scale remains a challenging problem. Protein microarrays were recently employed to identify numerous ubiquitinated substrates of yeast Rsp5 ligase.43 In total, 150 substrates were identified (e.g. “relaxed” set), among which 40 were defined as a “high-confidence” set based on either previous studies, or complementary confirmation of ubiquitination/binding to Rsp5. Although global identification of the Rsp5 substrates is valuable, the precise Ub sites within these substrates remain unknown.
Here, we asked a question whether Rsp5 substrates identified by Gupta et al.43 are ubiquitinated to a greater extent (i.e. are over-ubiquitinated) compared to the remaining proteins from the yeast proteome (see Materials and Methods for definition of over-ubiquitinated proteins). When we applied UbPred to this dataset, we found that the high-confidence Rsp5 substrates (but not the relaxed set) were significantly over-ubiquitinated as compared to other yeast proteins (P = 5.9·10−3, Wilcoxon test) (Table 4). This does not necessarily indicate that the substrates from the relaxed set lack Ub sites, but rather that the number of such sites is not unusually high compared to an average yeast protein. Such proteins can still be ubiquitinated at a smaller number of sites.
Table 4.
Ubiquitination analysis of Rsp5 substrates. The datasets were extracted from 43
| Datasets | Number of proteins | Over-ubiquitinated (%) | P-value |
|---|---|---|---|
| High confidence Rsp5 substrates | 40 | 14 (35.0%) | 5.88E-03 |
| Relaxed Rsp5 substrates | 149 | 32 (21.5%) | 3.00E-01 |
| All yeast proteins | 5817 | 1059 (18.2%) | n/a |
It has been shown that Rsp5 substrates are significantly enriched in PPxY and/or LPxY motifs,43 even though 27.5% of the high-confidence substrates and 67.3% of the relaxed substrates do not carry either of these two motifs. The analysis of UbPred predictions for the substrates with the above motifs showed that the presence of PY motifs is associated with slightly higher UbPred scores (not shown). However, examination of sequence distances between Ub sites and the PY motifs has not detected any trends, suggesting that spatial rather than sequence proximity of PY motifs and Ub sites may be important for Rsp5 binding.
Correlation of predicted Ub sites with protein half life
A recent study has determined the half lives of more than 3,750 yeast proteins.44 The availability of these data created an opportunity to ask whether proteins with shorter half-lives were over-ubiquitinated. The correlation between protein disorder and half life was previously investigated in two separate studies that arrived at the conclusions that disorder and protein half life are weakly32 and strongly33 correlated. However, the correlation between ubiquitination and protein half life has not been previously addressed.
Here, we found that short-lived proteins (with half lives of ≤4 minutes) were significantly over-ubiquitinated as compared to other yeast proteins (P = 1.9·10−10, Wilcoxon test) and to other proteins for which half life was determined (P = 9.5·10−5, Wilcoxon test, not shown) (Table 5). This suggests that the majority of yeast short-lived proteins are likely to be degraded via the ubiquitin-proteasomal pathway.
Table 5.
Ubiquitination analysis of proteins with various half lives. The datasets were extracted from 44
| Datasets | Number of proteins | Over-ubiquitinated (%) | P-value |
|---|---|---|---|
| Proteins with short half life ( ≤ 4 min) | 159 | 49 (30.8%) | 1.92E-10 |
| Proteins with longer half life (> 4 min) | 3185 | 585 (18.4%) | 7.81E-01 |
| All yeast proteins | 5817 | 1059 (18.2%) | n/a |
It has previously been observed that proteins with very short half lives have an increased incidence of PEST motifs.32 When we correlated the presence of PEST sequences with predicted Ub sites in the short-lived proteins, we observed that 38 out of 88 (43.2%) PEST sequences within these proteins carried predicted Ub sites with the UbPred score of ≥0.75. Given high disorder and flexibility content of PEST regions,31,78 it is not surprising that we find PEST sequences to be highly ubiquitinated, in addition to other previously detected modifications.78
Predictor application to the entire yeast proteome
To estimate the extent of ubiquitination and to functionally annotate predicted ubiquitinated proteins, we applied UbPred to the entire yeast proteome. Only the sites with high prediction scores have been considered in this analysis, and only the proteins for which the number of sites with high prediction scores was unlikely to have occurred by chance (Materials and Methods) have been selected for GO annotation.
The analysis of the “molecular function” annotation shows that proteins with numerous putative Ub sites span several functional categories (Figure 4A). These categories may be combined into three broader classes: (1) signaling and regulatory proteins (signal transducers, transcription, and enzyme regulators); (2) proteins involved in binding (protein, DNA, RNA and lipid binding); and (3) proteins involved in catalysis (hydrolases, transferases, protein kinases etc.). Among these classes we observed significant enrichment of proteins annotated as transcription and enzyme regulator activities, protein and DNA binding, as well as protein kinase activity. Many well-known yeast global transcriptional regulators including Swi5, Swi6, Ace2, Fkh2, Sla1, and Clb2 are present within these GO categories.
Figure 4.

GO annotations for the highly ubiquitinated proteins from S. cerevisiae proteome (colored bars) with occurrence of >5% (Bonferroni corrected) as compared to the entire yeast proteome (black bars). Top 20 (whenever available) GO Slim terms are shown. A. Molecular function; B. Biological process; C. Cellular component. The proteins are arranged in order of the decreasing fraction of proteins with a specific GO annotation present in the predicted highly ubiquitinated dataset. P-values were calculated using the hypergeometric distribution and corrected for multiple hypothesis testing. *** -P<0.0001; ** - P<0.001; * - P<0.05.
The “biological process” annotation shows that over-ubiquitinated proteins are enriched within such GO processes as transcription, cell cycle, cell budding, signal transduction, cytokinesis, and pseudohyphal growth (Figure 4B). Indeed, many transcription factors and cell cycle proteins are unstable, and their degradation is known to occur via Ub-mediated proteolysis.79
Within the “cellular component” category, over-ubiquitinated proteins are prevalent within GO annotations such as cellular bud, site of polarized growth, cytoskeleton and cell cortex (Figure 4C).
In summary, the GO annotations of over-ubiquitinated proteins generally agree with known functions and processes that depend on ubiquitination, thereby strengthening the biological significance of our predictions. The application of the UbPred to the yeast proteome allowed us to predict new targets of ubiquitination as well as to predict precise Ub sites in proteins that were previously known to be degraded by the ubiquitin-proteasomal pathway. The examples of confidently predicted Ub sites in various yeast proteins are shown in Table 6.
Table 6.
Examples of yeast proteins with highly confidently (UbPred score ≥ 0.95) predicted Ub sites.
| ORF | Protein name | Predicted Ub site(s)* |
|---|---|---|
| Ten high confidence Rsp5 substrates | ||
| YKR021W | ALY1 | 168, 203 |
| YJL084C | ALY2 | 144, 704, 871 |
| YNR069C | BSC5 | 41 |
| YOR042W | CUE5 | 15, 39, 76, 347, 395, 396 |
| YGR136W | LSB1 | 41 |
| YPL193W | RSA1 | 10, 191, 271, 276 |
| YMR140W | SIP5 | 7, 324 |
| YJL151C | SNA3 | 125 |
| YHR131C | YHR131C | 795 |
| YGL161C | YIP5 | 46, 57 |
| Ten proteins with short half-life (≤ 4 min) | ||
| YDR421W | ARO80 | 85, 87, 826 |
| YDL149W | ATG9 | 113, 138 |
| YLR220W | CCC1 | 74 |
| YLR228C | ECM22 | 355, 362, 379, 430 |
| YOR033C | EXO1 | 462, 470, 522, 527 |
| YHR061C | GIC1 | 141, 153, 217 |
| YMR172W | HOT1 | 505, 531, 576 |
| YER104W | RTT105 | 52 |
| YJR056C | YJR056C | 19, 97, 124 |
| YPL158C | YPL158C | 205, 399 |
| Ten transcriptional regulators | ||
| YKL112W | ABF1 | 133, 156, 712 |
| YNL068C | FKH2 | 795, 828, 836 |
| YEL009C | GCN4 | 194 |
| YDL056W | MBP1 | 248, 743 |
| YOR372C | NDD1 | 345 |
| YHR206W | SKN7 | 60 |
| YMR016C | SOK2 | 714 |
| YNL309W | STB1 | 65, 119, 178 |
| YER111C | SWI4 | 752, 842 |
| YLR182W | SWI6 | 140, 186 |
Only the sites with UbPred score of ≥ 0.95 are shown
Prediction of Ub sites in human proteins from Swiss-Prot functional categories
To determine whether there are differences in the extent of ubiquitination between human proteins that carry various functions in the cell, we performed comparative analysis of the over-ubiquitinated human proteins from 11 Swiss-Prot functional categories (see Materials and Methods) and the entire human proteome (Figure 5). Significant differences in ubiquitination were observed between proteins from different functional categories. In comparison to other human proteins, cytoskeletal, cell cycle, regulatory and cancer-associated proteins were significantly over-ubiquitinated, whereas kinases, transport, metabolism, biosynthesis, GPCRs and ribosomal proteins were significantly under-ubiquitinated. These results correlate with the yeast data since yeast cell cycle and regulatory proteins were also found to be over-ubiquitinated (Figure 4). In addition, the ubiquitination predictions also correlate with the disorder content of the same functional categories,25 with proteins from highly disordered categories being over-ubiquitinated.
Figure 5.
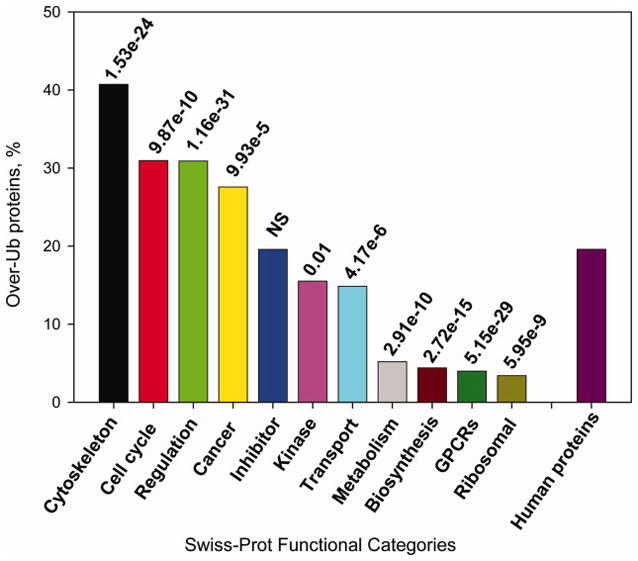
Frequencies of highly ubiquitinated proteins in eleven functional categories from Swiss-Prot as compared to the entire human proteome. P-values were calculated using the Wilcoxon test.
Gain and loss of predicted Ub sites in disease-associated proteins
Proteins involved in various human diseases carry a wide range of mutations. For example, hundreds of missense mutations, insertions and deletions have been identified in proteins involved in development of various cancers. Examples of such highly mutated proteins include p53, BRCA1, APC, RB1, ATM and others. Disease mutation can affect the Ub site directly, or it can be located in close proximity to a Ub site, but in both cases its effect would likely be aberrant degradation of a target protein. An example of altered ubiquitination of β-catenin’s oncogenic mutants clearly demonstrates such a possibility.80
Here, we investigated changes in the predicted Ub sites of disease-associated proteins extracted from Swiss-Prot and HGMD databases. Disease mutations could cause either gain or loss of predicted Ub sites. The effect of annotated disease mutation was assessed based on the difference between UbPred scores for the wild type and mutated residue, with a negative delta value signifying the loss of a Ub site and a positive delta value signifying the gain of a Ub site (Table S4). Only highly confident predictions with UbPred delta score of ≥0.75 are shown in Table S4.
Numerous mutations involved in various types of cancers could cause gain or loss of predicted Ub sites. The phenotypic effects of such mutations would be either destabilization/accelerated degradation of tumor suppressors due to gain-of-function mutations (i.e. gain of Ub sites), or stabilization/abnormal accumulation of oncoproteins and tumor growth factors due to loss-of-function mutations (i.e. loss of Ub sites). In fact, both of these mechanisms have been previously observed in human cancers.81,82 For example, the mutation K347N in human myosin-XVIIIb is known to be associated with lung small cell carcinoma,83 and this mutation also causes loss of a highly confidently predicted Ub site in this protein (Table S4). Two cancer-associated mutations in the p53 tumor suppressor, K292I and K305M, result in the loss of predicted Ub sites within p53 (Table S4). Interestingly, these two highly confidently predicted Ub sites in p53 are located at the boundary of its DNA-binding domain, whereas the majority of previously known Ub sites of p53 cluster in its C-terminal regulatory domain.62 These Ub sites could be novel, not yet experimentally identified Ub sites in p53. In addition, K305 of p53 is also known to be acetylated,84 and competition between acetylation, methylation, sumoylation and ubiquitination was previously suggested as regulatory mechanism for transcription factors activity.85,86 Therefore, lysine 305 in p53 is a good candidate for experimental verification.
We also observe gain of predicted Ub sites as a result of disease mutations (Table S4). For example, gain of Ub sites was predicted in many cancer proteins such as VHL, BRCA2, p53, as well as in proteins involved in other diseases such as CFTR, FANCA, WAS. In summary, mutations influencing ubiquitination status of a protein could serve as promising candidates for generating and testing hypotheses about altered degradation of the disease proteins.
Discussion
Although much knowledge about ubiquitination has been accumulated to date, there are still numerous unanswered questions regarding specific aspects of this highly complex system. So far, no consensus sequence that determines what specific lysine of the substrate would become ubiquitinated has been identified when non-homologous proteins are considered. In addition, the broad range of specificities of the ligases, together with the relative rigidity of their structures, raises a question about the mechanisms of substrate selection. It is difficult to assume that all substrates carry a similar preexisting structure before they bind to the components of the ubiquitination machinery.
Disorder has previously been implicated in various aspects of ubiquitination.30–33 Here, we present several lines of evidence that a significant fraction of Ub sites may be located in intrinsically disordered regions. First, we searched the literature and found a number of experimentally confirmed Ub sites located in disordered regions. Second, despite the large size of PDB, only 7% of currently known Ub sites in yeast could be confidently mapped to protein structures. Third, the use of disorder region predictors as well as the analysis of sequence, physicochemical and evolutionary properties around Ub sites showed higher propensity of Ub sites to be disordered than ordered (the average disorder prediction scores for Ub and non-Ub sites were 0.57±0.01 and 0.44±0.003, while the scores calculated on the experimentally verified disordered and ordered protein regions were 0.66±0.02 and 0.39±0.01, respectively. Fourth, the functional classes of proteins predicted to be over-ubiquitinated also show signatures of structural disorder, however, this evidence may not be independent. One previous study that also examined structural preferences of Ub sites concluded that these sites were preferentially located within loops.87 However, since the Catic et al. study was limited to only 40 Ub sites and was structure-based, it did not account for the presence of disorder, for which structural information was not available.
Locating Ub sites in unstructured regions is compelling when one takes into account the crystal structures of ubiquitin ligases. The structures of ubiquitin ligases contain large cavities and gaps13,14,17,18,88 that may serve to accommodate unstructured substrates. The Cul1 subunit of the SCF complex is rigid and elongated, and the gap between Skp2 and the E2 active site is ~50Å, supposedly to bind to a wide range of substrates of different sizes.14 Given the rigidity of the SCF complex and the diversity of proteins to which it binds, it is likely that the substrates adopt significant flexibility in order to conform to the rigid scaffold of the SCF complex. Indeed, the structure of β-TrCP1-Skp1 bound to a β-catenin peptide15 indicates that 15 out of 26 residues of the substrate peptide are disordered. Similarly, 14 out of 24 residues of the p27 Kip1 substrate in another structure are also disordered.17 In addition, a large distance between the E3 and E2 active sites suggests that the transfer of ubiquitin requires some large-scale movements. It is reasonable to speculate that movement of the substrate is required for the successful transfer and conjugation of the ubiquitin molecule. Thus, large cavities in structures of ubiquitin ligases could serve to accommodate diverse disordered substrates.
Another important result of this work is development of the Ub sites predictor. UbPred achieved a balanced accuracy of 72%, and area under the ROC curve was estimated to be ~80%. We demonstrated the utility of UbPred by: (1) predicting precise Ub sites in a dataset of Rsp5 ubiquitin ligase substrates; (2) establishing the correlation between ubiquitination and protein half life; (3) identifying functional categories of yeast and human proteins that are likely to be regulated by ubiquitination; and (4) demonstrating potential loss and gain of Ub sites as a consequence of disease mutations in humans. Thus, the initial application of UbPred to various datasets has expanded our understanding of ubiquitination in several biological processes and human diseases.
It should be noted that UbPred algorithm does not account for E3 binding/recognition sites that in some cases have been shown to be located distantly from Ub sites. Therefore, UbPred will not predict the ultimate ubiquitination status of the site since this status would depend on whether E3 binds to a protein or not. In essence, it will output the probability that the site is ubiquitinated if other conditions (such as E3 binding) are satisfied. Currently, it is not known whether universal ubiquitination/degradation signals could successfully predict the ubiquitination status of a substrate. Recent evidence suggests that the presence of bona fide degradation signals, such as the destruction-box, KEN-box, PEST regions and specific N-end residues shows no correlation with the protein half-life, and it has hardly any influence on protein turnover.32 In agreement with this observation, the computational scan of our positive examples for the presence of two degradation signals, a KEN-box (K-E-N) and a destruction box (R-x-x-L, x = any amino acid) showed that only 8 out of 265 substrates carried KEN-box, and only 18 substrates carried destruction box motifs in their vicinity. These signals, therefore, could not serve as global predictors of substrate ubiquitination and/or degradation. The disorder status of the substrate seems to be a better global ubiquitination signal than the presence of specific motifs.
While we were working on this project, another predictor of Ub sites was developed.89 It was trained on 157 Ub sites extracted from a database of ubiquitinated proteins.90 The majority of the Ub sites in this database were extracted from the two large-scale proteomics-based publications,35,36 also used in our work. However, the developed predictor achieved poor performance on our newly identified Ub sites (Sensitivity = 50.4%; Specificity = 55.8%, Accuracy = 53.1%; AUC = 54.8%).
To summarize, the involvement of flexible and disordered protein regions into various aspects of ubiquitination process further emphasizes the functional importance of such regions. Although many functions of disordered regions have already been discovered, we provide computational evidence that ubiquitination has signatures similar to other post-translational modifications that rely on the unfolded structure.20,27,28,91 Moreover, the development of UbPred represents an attempt to identify candidate Ub sites based on the local sequence information. While the number of experimentally determined Ub sites will be growing in the future and these sites will be added to our training set to improve predictor performance, the current accuracy of UbPred is useful for predicting novel ubiquitination substrates as well as new sites in already known substrates. With an established link between the ubiquitin-proteasome system and a number of human diseases,81,82,92 such predictions, especially when confirmed by experiments, would help to target the degradation of individual proteins more precisely, and may ultimately lead to the development of better drugs.
Supplementary Material
Acknowledgments
We would like to thank Keith Dunker and Jurg Ott for helpful discussions. We also thank Jelena Radivojac and Weisha Zhu for help with the dataset assembly. This study was supported by NIH NCI 1R21CA113711 and NSF 0444818 grants to LMI and NSF grant DBI-0644017 to PR.
References
- 1.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2(3):195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 2.Pickart CM. Ubiquitin enters the new millennium. Mol Cell. 2001;8(3):499–504. doi: 10.1016/s1097-2765(01)00347-1. [DOI] [PubMed] [Google Scholar]
- 3.Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol. 2003;4(3):192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- 4.Pornillos O, Garrus JE, Sundquist WI. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 2002;12(12):569–579. doi: 10.1016/s0962-8924(02)02402-9. [DOI] [PubMed] [Google Scholar]
- 5.Terrell J, Shih S, Dunn R, Hicke L. A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol Cell. 1998;1(2):193–202. doi: 10.1016/s1097-2765(00)80020-9. [DOI] [PubMed] [Google Scholar]
- 6.Rome S, Meugnier E, Vidal H. The ubiquitin-proteasome pathway is a new partner for the control of insulin signaling. Curr Opin Clin Nutr Metab Care. 2004;7(3):249–254. doi: 10.1097/00075197-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Izzi L, Attisano L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene. 2004;23(11):2071–2078. doi: 10.1038/sj.onc.1207412. [DOI] [PubMed] [Google Scholar]
- 8.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 9.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137(1):133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96(5):635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- 11.Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120(1):73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Hoeller D, Hecker CM, Wagner S, Rogov V, Dotsch V, Dikic I. E3-independent monoubiquitination of ubiquitin-binding proteins. Mol Cell. 2007;26(6):891–898. doi: 10.1016/j.molcel.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2–E3 enzyme cascade. Science. 1999;286(5443):1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 14.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416(6882):703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 15.Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11(6):1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- 16.Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112(2):243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 17.Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20(1):9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26(1):131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 19.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 20.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573 – 6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 21.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 22.Uversky V, Gillespie J, Fink A. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41(3):415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 24.Bourhis JM, Canard B, Longhi S. Predicting protein disorder and induced folding: from theoretical principles to practical applications. Curr Protein Pept Sci. 2007;8(2):135–149. doi: 10.2174/138920307780363451. [DOI] [PubMed] [Google Scholar]
- 25.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 26.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 27.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32(3):1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6(5):1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92(5):1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11(9):830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 31.Singh GP, Ganapathi M, Sandhu KS, Dash D. Intrinsic unstructuredness and abundance of PEST motifs in eukaryotic proteomes. Proteins. 2006;62(2):309–315. doi: 10.1002/prot.20746. [DOI] [PubMed] [Google Scholar]
- 32.Tompa P, Prilusky J, Silman I, Sussman JL. Structural disorder serves as a weak signal for intracellular protein degradation. Proteins. 2008;71(2):903–909. doi: 10.1002/prot.21773. [DOI] [PubMed] [Google Scholar]
- 33.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322(5906):1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70(4):1357–1366. doi: 10.1002/prot.21614. [DOI] [PubMed] [Google Scholar]
- 35.Hitchcock AL, Auld K, Gygi SP, Silver PA. A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc Natl Acad Sci U S A. 2003;100(22):12735–12740. doi: 10.1073/pnas.2135500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotech. 2003;21(8):921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 37.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 38.Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67(8):1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 39.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 40.Arnold I, Langer T. Membrane protein degradation by AAA proteases in mitochondria. Biochim Biophys Acta. 2002;1592(1):89–96. doi: 10.1016/s0167-4889(02)00267-7. [DOI] [PubMed] [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rost B, Liu J, Nair R, Wrzeszczynski KO, Ofran Y. Automatic prediction of protein function. Cell Mol Life Sci. 2003;60(12):2637–2650. doi: 10.1007/s00018-003-3114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta R, Kus B, Fladd C, Wasmuth J, Tonikian R, Sidhu S, Krogan NJ, Parkinson J, Rotin D. Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol Syst Biol. 2007;3:116. doi: 10.1038/msb4100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A. 2006;103(35):13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daroczy Z. Generalized information functions. Information and Control. 1970;16(1):36–51. [Google Scholar]
- 46.Vihinen M, Torkkila E, Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19:141–149. doi: 10.1002/prot.340190207. [DOI] [PubMed] [Google Scholar]
- 47.Radivojac P, Obradovic Z, Smith DK, Zhu G, Vucetic S, Brown CJ, Lawson JD, Dunker AK. Protein flexibility and intrinsic disorder. Protein Sci. 2004;13(1):71–80. doi: 10.1110/ps.03128904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenberg D, Weiss RM, Terwilliger TC. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc Natl Acad Sci USA. 1984;81:140–144. doi: 10.1073/pnas.81.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 50.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins. 2003;52(4):573–584. doi: 10.1002/prot.10437. [DOI] [PubMed] [Google Scholar]
- 51.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53 (Suppl 6):566–572. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 52.Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breiman L. Random forests. Mach Learn. 2001;45(1):5–32. [Google Scholar]
- 55.Latinne P, Saerens M, Decaestecker C. Adjusting the outputs of a classifier to new a priori probabilities may significantly improve classification accuracy: evidence from a multi-class problem in remote sensing. Williamstown, MA: Morgan Kaufmann; 2001. [Google Scholar]
- 56.Benanti JA, Cheung SK, Brady MC, Toczyski DP. A proteomic screen reveals SCFGrr1 targets that regulate the glycolytic-gluconeogenic switch. Nat Cell Biol. 2007;9(10):1184–1191. doi: 10.1038/ncb1639. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Mathias N, Steussy CN, Goebl MG. Intragenic suppression among CDC34 (UBC3) mutations defines a class of ubiquitin-conjugating catalytic domains. Mol Cell Biol. 1995;15(10):5635–5644. doi: 10.1128/mcb.15.10.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123(6):1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 59.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15(4):327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 61.Dosztanyi Z, Magyar C, Tusnady G, Simon I. SCide: identification of stabilization centers in proteins. Bioinformatics. 2003;19(7):899–900. doi: 10.1093/bioinformatics/btg110. [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol. 2000;20(22):8458–8467. doi: 10.1128/mcb.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell S, Klein C, Muller L, Hansen S, Buchner J. p53 contains large unstructured regions in its native state. J Mol Biol. 2002;322(5):917–927. doi: 10.1016/s0022-2836(02)00848-3. [DOI] [PubMed] [Google Scholar]
- 64.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18(3):717–726. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McEwan IJ, Dahlman-Wright K, Ford J, Wright AP. Functional interaction of the c-Myc transactivation domain with the TATA binding protein: evidence for an induced fit model of transactivation domain folding. Biochemistry. 1996;35(29):9584–9593. doi: 10.1021/bi960793v. [DOI] [PubMed] [Google Scholar]
- 66.Bohm L, Crane-Robinson C, Sautiere P. Proteolytic digestion studies of chromatin core-histone structure. Identification of a limit peptide of histone H2A. Eur J Biochem. 1980;106(2):525–530. doi: 10.1111/j.1432-1033.1980.tb04599.x. [DOI] [PubMed] [Google Scholar]
- 67.Thorne AW, Sautiere P, Briand G, Crane-Robinson C. The structure of ubiquitinated histone H2B. Embo J. 1987;6(4):1005–1010. doi: 10.1002/j.1460-2075.1987.tb04852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bohm L, Crane-Robinson C. Proteases as structural probes for chromatin: the domain structure of histones. Biosci Rep. 1984;4(5):365–386. doi: 10.1007/BF01122502. [DOI] [PubMed] [Google Scholar]
- 69.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 70.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 71.McNulty BC, Young GB, Pielak GJ. Macromolecular crowding in the Escherichia coli periplasm maintains alpha-synuclein disorder. J Mol Biol. 2006;355(5):893–897. doi: 10.1016/j.jmb.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 72.Nonaka T, Iwatsubo T, Hasegawa M. Ubiquitination of alpha-synuclein. Biochemistry. 2005;44(1):361–368. doi: 10.1021/bi0485528. [DOI] [PubMed] [Google Scholar]
- 73.Cox CJ, Dutta K, Petri ET, Hwang WC, Lin Y, Pascal SM, Basavappa R. The regions of securin and cyclin B proteins recognized by the ubiquitination machinery are natively unfolded. FEBS Lett. 2002;527(1–3):303–308. doi: 10.1016/s0014-5793(02)03246-5. [DOI] [PubMed] [Google Scholar]
- 74.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269(1):2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 75.Patel MS, Korotchkina LG. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: complexity of multiple phosphorylation sites and kinases. Exp Mol Med. 2001;33(4):191–197. doi: 10.1038/emm.2001.32. [DOI] [PubMed] [Google Scholar]
- 76.Minella O, Mulner-Lorillon O, Bec G, Cormier P, Belle R. Multiple phosphorylation sites and quaternary organization of guanine-nucleotide exchange complex of elongation factor-1 (EF-1betagammadelta/ValRS) control the various functions of EF-1alpha. Biosci Rep. 1998;18(3):119–127. doi: 10.1023/a:1020140527930. [DOI] [PubMed] [Google Scholar]
- 77.Hobohm U, Sander C. Enlarged representative set of protein structures. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sandhu KS, Dash D. Conformational flexibility may explain multiple cellular roles of PEST motifs. Proteins. 2006;63(4):727–732. doi: 10.1002/prot.20918. [DOI] [PubMed] [Google Scholar]
- 79.Thomas D, Tyers M. Transcriptional regulation: Kamikaze activators. Curr Biol. 2000;10(9):R341–343. doi: 10.1016/s0960-9822(00)00462-0. [DOI] [PubMed] [Google Scholar]
- 80.Al-Fageeh M, Li Q, Mohaiza Dashwood W, Myzak MC, Dashwood RH. Phosphorylation and ubiquitination of oncogenic mutants of beta-catenin containing substitutions at Asp32. Oncogene. 2004 doi: 10.1038/sj.onc.1207634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ciechanover A, Schwartz AL. The ubiquitin system: pathogenesis of human diseases and drug targeting. Biochim Biophys Acta. 2004;1695(1–3):3–17. doi: 10.1016/j.bbamcr.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 82.Petroski MD. The ubiquitin system, disease, and drug discovery. BMC Biochem. 2008;9 (Suppl 1):S7. doi: 10.1186/1471-2091-9-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishioka M, Kohno T, Tani M, Yanaihara N, Tomizawa Y, Otsuka A, Sasaki S, Kobayashi K, Niki T, Maeshima A, Sekido Y, Minna JD, Sone S, Yokota J. MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc Natl Acad Sci U S A. 2002;99(19):12269–12274. doi: 10.1073/pnas.192445899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang YH, Tsay YG, Tan BC, Lo WY, Lee SC. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem. 2003;278(28):25568–25576. doi: 10.1074/jbc.M212574200. [DOI] [PubMed] [Google Scholar]
- 85.Freiman RN, Tjian R. Regulating the regulators: lysine modifications make their mark. Cell. 2003;112(1):11–17. doi: 10.1016/s0092-8674(02)01278-3. [DOI] [PubMed] [Google Scholar]
- 86.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15(2):164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 87.Catic A, Collins C, Church GM, Ploegh HL. Preferred in vivo ubiquitination sites. Bioinformatics. 2004;20(18):3302–3307. doi: 10.1093/bioinformatics/bth407. [DOI] [PubMed] [Google Scholar]
- 88.Gieffers C, Dube P, Harris JR, Stark H, Peters JM. Three-dimensional structure of the anaphase-promoting complex. Mol Cell. 2001;7(4):907–913. doi: 10.1016/s1097-2765(01)00234-9. [DOI] [PubMed] [Google Scholar]
- 89.Tung CW, Ho SY. Computational identification of ubiquitylation sites from protein sequences. BMC Bioinformatics. 2008;9:310. doi: 10.1186/1471-2105-9-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chernorudskiy AL, Garcia A, Eremin EV, Shorina AS, Kondratieva EV, Gainullin MR. UbiProt: a database of ubiquitylated proteins. BMC Bioinformatics. 2007;8:126. doi: 10.1186/1471-2105-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Daily KM, Radivojac P, Dunker AK. Intrinsic disorder and protein modifications: building an SVM predictor for methylation. San Diego, CA: 2005. pp. 475–481. [Google Scholar]
- 92.Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 93.Vacic V, Iakoucheva LM, Radivojac P. Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics. 2006;22(12):1536–1537. doi: 10.1093/bioinformatics/btl151. [DOI] [PubMed] [Google Scholar]
- 94.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Flaugh SL, Lumb KJ. Effects of macromolecular crowding on the intrinscially disordered proteins c-Fos and p27(Kip 1) Biomacromolecules. 2001;2(2):538–540. doi: 10.1021/bm015502z. [DOI] [PubMed] [Google Scholar]
- 96.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27(kip1) cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 97.Adkins JN, Lumb KJ. Intrinsic structural disorder and sequence features of the cell cycle inhibitor p57(Kip2) Proteins. 2002;46(1):1–7. doi: 10.1002/prot.10018. [DOI] [PubMed] [Google Scholar]
- 98.King RW, Glotzer M, Kirschner MW. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol Biol Cell. 1996;7(9):1343–1357. doi: 10.1091/mbc.7.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaffray E, Wood KM, Hay RT. Domain organization of I kappa B alpha and sites of interaction with NF- kappa B p65. Mol Cell Biol. 1995;15:2166–2172. doi: 10.1128/mcb.15.4.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci USA. 1995;92(24):11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rice LM, Brennwald P, Brunger AT. Formation of a yeast SNARE complex is accompanied by significant structural changes. FEBS Lett. 1997;415(1):49–55. doi: 10.1016/s0014-5793(97)01091-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.