

Abstract
Mechanical properties of tissue scaffolds have major effects on the morphology and differentiation of cells. In contrast to two-dimensional substrates, local biochemical and mechanical properties of three-dimensional hydrogels are difficult to control due to the geometrical confinement. We designed synthetic 3D hydrogels featuring complexes of four-arm poly(ethylene glycol) (PEG) and collagen mimetic peptides (CMPs) that form hydrogels via physical crosslinks mediated by thermally reversible triple helical assembly of CMPs. Here we present the fabrication of various PEG-CMP 3D hydrogels and their local mechanical properties determined by particle tracking microrheology. Results show that CMP mediated physical crosslinks can be disrupted by altering the temperature of the gel or by adding free CMPs that compete for triple helix formation. This allowed modulation of both bulk and local stiffness as well as the creation of stiffness gradients within the PEG-CMP hydrogel, which demonstrates its potential as a novel scaffold for encoding physico-chemical signals for tissue formation.
Keywords: tissue scaffolds, collagen mimetic peptides, hydrogel, poly(ethylene glycol), microrheology, particle tracking
Introduction
Collagen is the primary protein component of the extracellular matrix (ECM) and plays a major role in the attachment, migration, and growth of cells in most tissues. The key building block of collagen’s supramolecular structure is the triple helix assembled from three individual collagen strands.1 This unique helical conformation is made possible by the prevailing (Glycine-X-Y) repeating amino acid sequence in which X and Y positions are typically occupied by proline and hydroxyproline, respectively. Collagen’s triple helical structure and subsequent assembly into larger fibrous structures lend structural integrity to the ECM of many mammalian tissues. In addition to providing structural stability, collagen presents sites for cell binding and proteolytic degradation, securing its role as a bioactive player in tissue development.
Collagen-based biomaterials, especially bovine collagen, have been used extensively in biomedical applications.2 The biocompatibility of collagen and its bio-resorbability make it a well-suited biomaterial for tissue regeneration, and collagen is one of the most cost-effective biomaterials due to its abundance in nature. Collagen can be prepared with controlled porosity to enhance cellular migration or diffusion of bioactive factors. Unfortunately, animal-derived collagen can pose inherent problems such as immunogenicity and pathogen transmission.3 In addition, regenerated collagen gels exhibit low mechanical strength that limits their use in tissue scaffolds that are subject to high stress. These shortcomings highlight the need to develop artificial scaffolds that mimic the structural and biofunctional capacity of natural collagens in the ECM.
The design of artificial scaffolds poses many challenges including properly mimicking tissue stiffness and incorporating insoluble bioactive molecules that induce cells to differentiate, proliferate, and migrate into the scaffold. Therefore, creating functional and effective synthetic collagen tissue scaffolds requires a solid understanding of the interplay between cells and the ECM. Cell surface receptors link ECM proteins to the interior cytoskeleton, enabling cells to sense the mechanical nature of their environment and to respond by converting those mechanical cues into chemical signals.4 As a result, the viscoelastic properties of the ECM control many cellular properties including shape, viability, adhesion, and migration.5-7
The ECM is comprised of a highly hydrated network of various protein fibrils and glycosaminoglycan chains.8 This suggests that crosslinked, hydrophilic polymers exhibiting viscoelastic properties similar to the natural ECM could be effective substitutes for natural ECM for tissue engineering applications. Various hydrogels have been synthesized and successfully used as tissue substitutes after implantation via minimally invasive surgery.9 PEG-based hydrogels are the most popular among these synthetic scaffolds because their use in humans has been approved by the FDA. The extreme hydrophilicity of PEG generates a water-swollen gel that mimics the high water content of the ECM and simultaneously reduces non-specific protein adsorption and cell adhesion.10 Furthermore, PEG conjugation reactions are well understood, allowing biofunctionalities to be easily incorporated into PEG-based hydrogels to spur cell activity.
Collagen mimetic peptides (CMPs) are extensively studied as a model system for understanding the triple helix formation and stability of natural collagens in the ECM.11, 12 CMPs are typically 15-40 amino acid long peptides that mimic collagen in their ability to form the triple helix. CMPs feature the characteristic Gly-X-Y repeat sequence found in natural collagen and exhibit thermally reversible triple helix melting behavior.13 When heated above their melting temperature (Tm), the CMPs exist as single strands that, unlike natural collagen, completely reform the triple helix when cooled back below the melting temperature.
More recently, the biomimetic properties of CMPs have been employed in hydrogels for tissue engineering applications. CMPs have been shown to bind to natural collagen, presumably through a triple helical strand invasion process.14 This behavior has been exploited in the studies of photo-polymerized poly(ethylene oxide)-diacrylate (PEODA) hydrogels displaying CMPs; the CMPs allowed the gel to better retain the cell-secreted collagens within the hydrogel and enhanced ECM buildup.15 Other research efforts employed temperature-sensitive CMP folding as a way to create hydrogels with temperature dependent macroscopic stability. Koide and coworkers created hydrogels consisting solely of staggered CMPs connected by cysteine disulfide bonds. At high concentrations and below the peptide’s Tm, the triple helix formation between different staggered CMP molecules led to the assembly of supramolecular peptide structures that created a self-supporting gel.16 De Wolf and coworkers employed recombinant protein methods to create triblock copolymers featuring collagen mimetic sequences.17 These triblock copolymers also exhibited temperature-sensitive gelling behavior based on the melting of the triple helical crosslinks that hold the gel together.
In this study, we present multi-armed PEG-CMP hydrogels containing triple helical crosslinks that provide a convenient means to change both the mechanical and biochemical properties of 3D scaffolds. The temperature-sensitive folding of the CMPs enables the creation and subsequent modulation of CMP-mediated triple helical physical crosslinks within the gel. Furthermore, the CMP triple helices provide an avenue to attach insoluble factors by the addition of unbound CMP complexes to a preformed PEG-CMP hydrogel.
We used particle tracking microrheology to investigate the viscoelastic nature of PEG-CMP hydrogels and to assess the variations in local stiffness of the PEG-CMP gels. Mason et al.18 and Gittes et al.19 developed single-particle tracking techniques to study local viscoelastic properties of solutions and gels, and such microrheology approaches have become valuable tools to investigate the viscoelastic properties of small volume samples such as cells. Video-based particle tracking techniques can monitor the Brownian motion of multiple nanoparticles suspended in the investigated medium.20 The stiffness or compliance of the suspending medium is determined by tracking particle positions and calculating the mean squared displacement (MSD) as a function of time lag.18 Particle tracking has been extensively used to study intracellular viscoelastic properties using ballistic intracellular nanorheology.21-24 It has also been used to study the local and global heterogeneity of reconstituted lamin in biological gels and to measure their micromechanical properties.25 Unlike bulk rheology (e.g. cone and plate rheology), particle tracking techniques can investigate localized variations in stiffness within a small sample, which can prove valuable when analyzing stiffness gradients in novel tissue engineering scaffolds.
Experimental Section
Materials
Peptide synthesis reagents including Fmoc-amino acids, 2-(1H-benzo-triazole-1-yl)-1,1,3,3-tetra-methyluronium hexafluorophosphate (HBTU), and N, N-diisopropylethylamine (DIPEA) were purchased from Advanced Chemtech (Louisville, KY). Rink type Tentagel R RAM resin was purchased from Peptides International (Louisville, KY). Four-arm PEG-NHS [pentaerythritol tetra(succinimidyl carboxypentyl) polyoxyethylene] of average molecular weight 11,000 Da was purchased from NOF Corporation (Tokyo, Japan). Dialysis tubes (MWCO = 10 kDa) were purchased from SpectraPor Inc (Rancho Dominguez, CA).
Methods
Synthesis and Purification of Collagen Mimetic Peptides
CMPs were synthesized on Tentagel resin via conventional Fmoc mediated coupling chemistry using 4.5-fold molar excess Fmoc-protected amino acids, HBTU as activating agent, and piperidine as deprotection agent. The coupling and deprotection reactions were monitored by ninhydrin or chloranil test. The CMPs were cleaved from resin via incubation in a mixture of trifluoroacetic acid (95%), triisopropylsilane (2.5%), and deionized (DI) water (2.5%) for 2 to 3 hr. The resulting mixture was filtered and added dropwise to cold ethyl ether to precipitate crude CMP products. After overnight incubation at -20°C, the peptides were pelleted by centrifugation and dried under vacuum.
Peptides were purified using reverse-phase high performance liquid chromatography (HPLC) on a Varian Polaris 210 series equipped with a semi-prep Vydac reverse-phase C18 column. Deionized water and acetonitrile were used in a gradient mixture for the mobile phase at a flow rate of 4 mL/min, and peptides were detected by UV absorption at 220 nm. The elutions containing target peptides were collected and lyophilized. The purity of the peptides was determined by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Voyager DE-STR, Applied Biosystems, Foster City, CA).
Synthesis of PEG-CMP9 Hydrogels
PEG-CMP complexes were synthesized by reacting N-hydroxysuccinimide-activated four-arm PEG (PEG-NHS) with G3-(POG)9 (CMP9) peptides in a 1:4.5 molar ratio. PEG-NHS and CMP9 peptides were weighed out in separate vials and dissolved in 1 mL of 50 mM NaHCO3 buffer solution at 4°C and 75°C, respectively. The hot CMP solution was then pipetted into the cold PEG-NHS solution, and the reaction mixture was placed in a 75°C incubator for 24 hr during which the PEG-CMP reaction occurred. After the reaction, the PEG-CMP solution was placed in a dialysis tube. The dialysis took place in deionized water at 75°C to ensure complete melting of the CMPs such that unconjugated CMPs could diffuse out leaving only the desired PEG-CMP product. The dialyzed PEG-CMP was recovered from the dialysis tube and lyophilized.
1H NMR (in D2O) was used to determine the average number of CMPs conjugated to each four-arm PEG as reported previously.14 Comparison of δ resonances at 3.70 ppm (1000 H of PEG backbone–CH2CH2O–, s) and 1.73-2.70 ppm (192 H of CβHl,h and CγH2 of proline and CβHl,h of hydroxyproline, m),26, 27 indicated conjugation of 3.04 CMPs per single four-arm PEG-NHS. This corresponded to 81.1% yield and an average molecular weight of 18,540 Da for the PEG-CMP product (Supporting Information, Figure S1).
Gel permeation chromatography (GPC) was used to verify the number average molecular weight of the PEG-CMP complexes (Supporting Information, Figure S2). An Agilent 1200 series GPC with three tandem columns (Agilent PL aquagel-OH 30, 40, and mix) was run at 40°C using RI and light scattering detectors. Using a calculated dn/dc value for PEG-CMP,28, 29 the PEG-CMP complex average molecular weight was determined to be 18,450 Da, which is almost identical to that determined by NMR.
PEG-CMP9 (10 wt%, 100 μL) hydrogels for particle tracking experiments were produced by dissolving the lyophilized PEG-CMP in deionized water containing 100 nm diameter red fluorescent carboxylate-modified polystyrene particles (Invitrogen Molecular Probes, Carlsbad, CA) at approximately 3.6 × 108 particles per mL. This solution was placed into the wells of a 96-well plate and heated to 80°C for 30 min. Once the 96-well plate cooled down to room temperature, a self-supporting gel was formed. For circular dichroism (CD) measurements, an 80°C solution of PEG-CMP in DI water (1 wt%, 400 μL) was added to a CD cuvette and allowed to sit at room temperature overnight.
Synthesis of PEG-CMP9-K Hydrogels with Chemical Crosslinks
Chemically crosslinked PEG-CMP9-K hydrogels were assembled by weighing out, in separate vials, a 1:3 molar ratio of PEG-NHS and bifunctional CMPs featuring two free amines, one at amino terminus and the other at lysine side chain: G3-(POG)9-K (CMP9-K). For particle tracking experiments, hydrogels (10 wt%, 100 μL) were made by first adding 50 μL of 4°C NaHCO3 buffer solution containing red fluorescent particles (same as above) to each vial. The PEG solution was added to the well of a 96-well plate, and the CMP9-K solution was then added to the same well. Thorough mixing was ensured by quickly stirring the mixture solution with a micropipette. The plate was then heated to 70°C for 30 minutes during which time a rigid gel was formed through the PEG-CMP-PEG crosslinking process. For CD measurements, PEG-NHS and CMP9-K each dissolved in 4°C NaHCO3 buffer were mixed (1 wt%, 400 μL) as described above just prior to transfer to the CD cuvette. The cuvette containing the sample was heated to 70°C for 30 min and allowed to cool to room temperature overnight.
Particle Tracking Microrheology Experiments of PEG-CMP Hydrogels
Rheological properties of hydrogels were measured using microrheology particle tracking techniques at 25°C unless otherwise noted. The fluorescent particles dispersed in the gels were filmed using a Roper Scientific Cascasde 1K CCD camera mounted on a Nikon TE2000E epifluoresence microscope using a 60× Plan Fluor oil-immersion lens (NA 1.4, Nikon, Melville, NY). Videos of the fluorescent particles embedded in the gels were filmed at 30 Hz for 20 s. At least 100 particle paths were filmed for each particle tracking data shown. The filmed images were first analyzed using MetaMorph software (Universal Imaging, West Chester, PA), and a customized software was used to obtain rheological parameters that describe the viscoelastic properties of the gels.30 Briefly, for each nanoparticle, the time-averaged mean squared displacement (MSD), defined as <Δr2(τ)> = <[x(t + τ) − x(t)]2 + [y(t + τ) − y(t)]2>, using τ as the time lag and t as the elapsed time, was determined by tracking the light intensity-weighted centroid of the nanoparticle. Statistical analysis was performed to obtain the mean values and standard error of the mean (SEM) of MSD data and the results were plotted using Graphpad Prism (Graphpad Software, San Diego, CA). Two-tailed unpaired Student’s t-tests were conducted to determine the statistical significance of MSD differences among particle populations in various hydrogels. The MSD data for these gels can be related to the complex viscoelastic modulus G*(ω),18, 31
and this complex shear modulus related to storage and loss moduli,
Here, kB is the Boltzmann’s constant, T is the absolute temperature, a is the nanoparticle radius, α(ω) is the logarithmic slope at the frequency ω = 1/τ, and Γ is the gamma function. The storage (G′) and loss (G″) moduli represent a gel’s propensity to store energy by resisting stress or to dissipate energy by flowing under stress, respectively. Hydrogel networks often resist stress elastically at high frequency shear rates and flow viscously at low frequency shear rates. The crossover frequency is defined as the point at which G′(ω) = G″(ω).
CD Thermal Melting Temperature Measurements
Circular dichroism (CD) spectra were acquired using a JASCO 710 spectrometer (JASCO Inc, Easton, MD) equipped with JASCO temperature controller. Samples (100 μM of peptides in deionized water or 50 mM sodium carbonate buffer) were studied in a Starna Cells cuvette (400 μL and 0.1 cm path length). The thermal melting curves were obtained by monitoring the mean residue molar ellipticity peak that occurred in the samples between 225-228 nm with heating rate of 1°C/min. Tm was assigned to the minimum of the first derivative of the ellipticity versus temperature curve.32 Enthalpy of melting was calculated using two state model.33
Results and Discussion
Design of PEG-CMP Hydrogels
We synthesized and characterized two types of PEG-CMP hydrogel systems (Figure 1). PEG-CMP9 consisted of four-arm PEG conjugated to G3-(POG)9 (CMP9) via the peptide’s terminal amine. NMR measurements indicated that the PEG-CMP9 complexes have on average approximately three CMPs conjugated to each PEG molecule (see methods section). At low concentrations, the PEG-CMP9 can form intramolecular triple helices among three CMPs conjugated to the same PEG. At high concentrations, intermolecular triple helices can form across different PEG-CMP9 complexes.14 These are physical crosslinks that allow formation of a self-supporting hydrogel at temperatures below CMP’s melting temperature.
Figure 1.
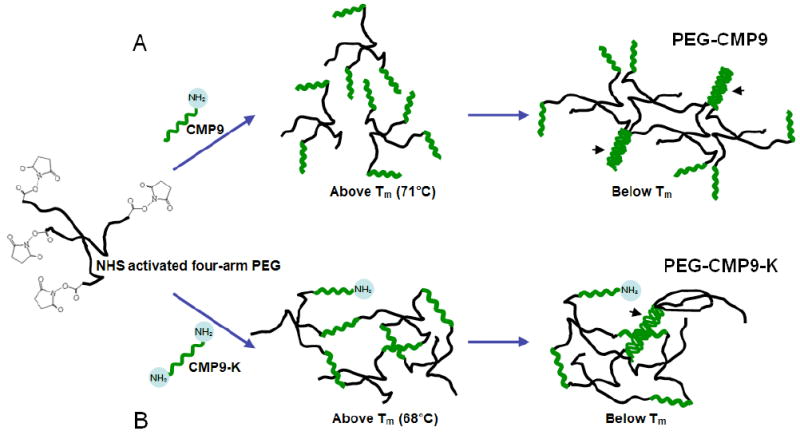
Schematics of PEG-based CMP hydrogel synthesis. (A) Reacting PEG-NHS with CMP9 yielded the target product with average conjugations of 3.04 CMP9 per PEG. PEG-CMP9 forms physical crosslinks mediated by CMP triple helices (arrowheads). (B) PEG-CMP9-K hydrogel contains covalent crosslinks due to the bifunctionality of CMP9-K. In addition to chemical crosslinks, PEG-CMP9-K can form triple helix mediated physical crosslinks (arrowhead).
The second type of hydrogel, PEG-CMP9-K, consisted of chemical crosslinks that are produced by the conjugation of four-arm PEG to a bifunctional CMP, G3-(POG)9-K (CMP9-K). Two free amines on each terminus of G3-(POG)9-K, one from the N-terminus and the other from the lysine side chain, allow a single CMP9-K peptide to be conjugated to two PEG arms, enabling the formation of a chemically crosslinked network. In addition to the chemical crosslinks, the CMPs within the gels can form triple helices that add physical crosslinks to the hydrogel. For comparison, we also synthesized a chemically crosslinked hydrogel using control peptides (RCMP-K) that feature the same amino acid content and the bifunctionality of CMP9-K peptides, but in a scrambled sequence that precludes triple helix formation. As a result, these hydrogels can only form the chemically crosslinked network and not the triple helix mediated physical crosslinks.
Evaluation of Triple Helical Structures of PEG-CMPs
To investigate the triple helix mediated physical crosslinks within these hydrogels, we conducted CD studies to identify the triple helical conformation of CMPs after conjugation to four-arm PEG.34 Figure 2 shows the CD melting curves for CMP9 and CMP9-K peptides as free peptides in solution as well as after PEG conjugation. The CD melting curves for CMP9 and CMP9-K in solution consist of a sigmoidal shape characteristic of triple helix structure. This sigmoidal shape is also seen for 1 wt% PEG-peptide samples, suggesting that the CMPs retain their ability to form triple helices even after PEG conjugation. The presence of triple helices after conjugation to four-arm PEG supports the notion that CMPs can create physical crosslinks through formation of triple helices in the hydrogels.
Figure 2.

Circular dichroism melting curves of CMP9, CMP9-K, RCMP-K, and their PEG conjugates. CMP9 and CMP9-K peptides exhibited sigmoidal melting transitions indicative of a triple helical structure both as free peptides in solution (left) and after PEG conjugation (right). RCMP-K peptides with a scrambled CMP sequence, G3-PGOGPGPOPOGOGPOPGOOPGGOOPPG-K, did not show triple helical melting transition upon heating.
The Tm of CMP9 (69°C) is slightly higher than that of CMP9-K (63°C). This is most likely due to the charge-charge repulsion of positively charged lysine residues on CMP9-K that destabilize the triple helix. Upon conjugation to PEG, the CMP9’s Tm slightly increases to 71°C, likely due to the templating effect that stabilizes the triple helix.35 By comparison, the Tm of CMP9-K is elevated by 5°C to 68°C after PEG conjugation. The larger increase in Tm for PEG-CMP9-K can be explained by the elimination of coulombic repulsion from charged lysine after PEG conjugation, in addition to the templating effect. The breadth of the sigmoidal curve for the two PEG-CMP hydrogels seems to reveal the nature of the triple helical interactions in the hydrogels. The sigmoidal CD curve of PEG-CMP9 shows a sharp melting transition indicative of homogenously templated triple helical peptides. In contrast, the broader sigmoidal curve for PEG-CMP9-K supports a more heterogeneous melting transition. The peptides within the PEG-CMP9-K sample may be sterically hindered from forming completely-aligned helices due to the strains associated with chemically crosslinked PEG. Here, incompletely aligned and thermally unstable triple helices could melt before the melting of fully aligned triple helices. This was also evidenced by the marked change in enthalpy of melting (ΔHs in Fig. 2) calculated from the CD melting curves. Figure 2 also shows the CD melting curve of the scrambled sequence peptide, RCMP-K, in solution and after PEG conjugation (PEG-RCMP-K). Neither graph shows a sigmoidal curve indicating that there is no melting transition originating from the triple helical conformation. These CD results validate that the PEG-RCMP-K hydrogels do not contain triple helices and therefore cannot form triple helix mediated physical crosslinks.
Particle Tracking Microrheology of PEG-CMP Hydrogels
We employed particle tracking rheology experiments to characterize the viscoelastic nature of PEG-CMP hydrogels. For a viscous liquid, nanoparticles in the medium undergo unrestricted motion, and the MSD increases linearly with time similar to the classic random walk.22 For a completely elastic material, the nanoparticles display highly restricted oscillations and the MSD, <Δr2(τ)>, is independent of time. Most gels feature both viscous and elastic behavior; the time dependence of MSD is intermediate between the MSD of a viscous liquid, <Δr2(τ)> ~ kτ, and that of a perfectly elastic material, <Δr2(τ)> ~ C, where k and C are constants.36 Here, the time dependence of MSD can be described by <Δr2(τ)> ~ kτα, and the exponent α (0<α<1) describes the viscoelastic nature of the material.23, 37
We first investigated the microrheological properties of PEG-CMP9-K and PEG-RCMP-K hydrogels. Figure 3 shows sample nanoparticle trajectories as well as MSD and α exponent data for 10 wt% PEG-CMP9-K and PEG-RCMP-K hydrogels. Figure 3D shows that nanoparticles embedded in PEG-CMP9-K hydrogels have a lower MSD (Student’s t-test p < 0.05) than those in PEG-RCMP-K hydrogels for all time lags. The ensemble-average MSD for each hydrogel type was used to calculate the best fitting α exponent by linear regression. Figure 3E shows the calculated α values from 0.1-1 and 1-10 s time lags for each hydrogel. The lower α values associated with PEG-CMP9-K hydrogels (α = 0.39 - 0.52) indicate that they behave more as a viscoelastic solid than the PEG-RCMP-K hydrogels (α = 0.58 - 0.74).
Figure 3.
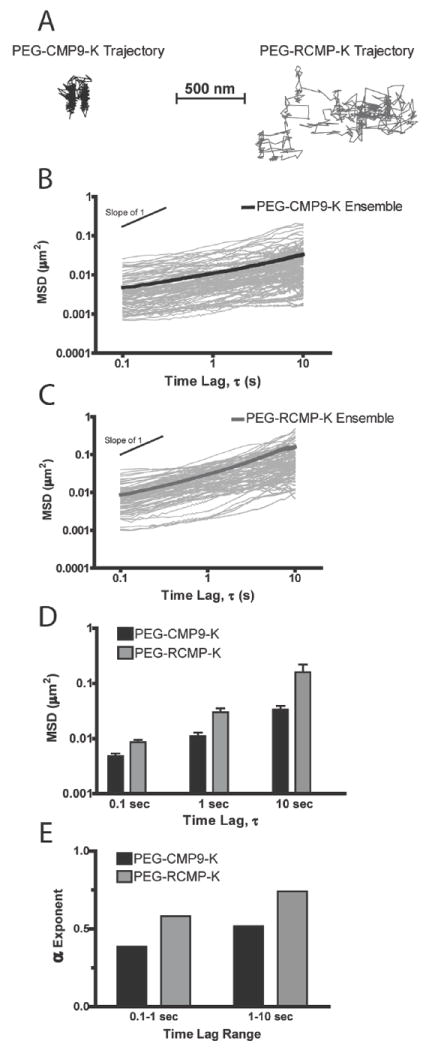
Particle tracking data for PEG-CMP9-K and PEG-RCMP-K hydrogels (10 wt%) at room temperature. (A) Sample particle trajectories. (B) and (C) Over 50 representative particle MSDs and the ensemble-average values. (D) Comparison of ensemble-average MSD between PEG-CMP9-K and PEG-RCMP-K hydrogels with data reported as mean ± SEM (n ≥ 4). (E) α exponents of PEG-CMP9-K and PEG-RCMP-K hydrogel calculated by fitting ensemble-average MSDs to <Δr2(τ)> ~ kτα.
Figure 4 shows the MSD data converted into storage and loss moduli over a range of frequencies. The phase angle for PEG-CMP9-K hydrogels (Inset, Figure 4A) falls in the viscoelastic solid regime (δ < 45°) for a wider range of frequencies than for the PEG-RCMP-K hydrogels (Figure 4B). Furthermore, as shown in Figure 4C, at ω = 7.5 Hz, which corresponds to the approximate crossover frequency for PEG-RCMP-K hydrogels where G′ and G″ are of same magnitude, the PEG-CMP9-K hydrogel’s storage modulus is significantly higher than the loss modulus (Student’s t-test p < 0.01). These results indicate that the PEG-CMP9-K hydrogel is the more elastic one between the two chemically crosslinked gels. Together with the CD data (Figure 2), the results suggest that the CMPs of the PEG-CMP9-K gel generate triple helix mediated physical crosslinks that increase the stiffness of the hydrogel.
Figure 4.

Comparison of viscoelastic properties for PEG-CMP9-K and PEG-RCMP-K hydrogels (10 wt%). (A) and (B) Storage (G′) and loss (G″) moduli of the two hydrogels as a function of frequency with phase angle shown in the inset. (C) Storage (G′) and loss (G″) moduli at ω = 7.5Hz (PEG-RCMP-K crossover frequency) with data reported as mean ± SEM error bars (n ≥ 4). (D) Ensemble-average MSDs and storage moduli G′ (at ω = 30 Hz) of PEG-CMP9-K, PEG-RCMP-K, and PEG-CMP9-K after free CF-CMP9 addition.
To further study the triple helix mediated physical crosslinks of PEG-CMP9-K hydrogels, single-stranded carboxyfluorescein-labeled CMPs (CF-CMP9) that can compete for triple helical assembly were added to the PEG-CMP9-K gel. One hundred and fifty μL of 7.25 mM CF-CMP9 solution was added on top of the PEG-CMP9-K gel surface at 80°C. The gel was then incubated at 80°C for 30 min, allowing CF-CMP9 to diffuse into the gel in a single strand form. The solution was cooled to room temperature to allow CF-CMP9s to form heterotrimeric complexes with CMP9-K domains within the gel, which can lead to a reduced number of triple helical physical crosslinks. The gel was washed with room temperature DI water to remove unbound CF-CMP9s, and the concentrations of released CF-CMP9 were determined by UV-vis absorbance at 493 nm. The release study showed that 45% of added CF-CMP peptides were retained in the gel, corresponding to a ratio of 1 CF-CMP9 peptide for every 3.2 CMP9-K peptides in the gel. Particle tracking microrheology analysis indicated that CF-CMP treated PEG-CMP9-K displayed intermediate MSDs between the PEG-CMP9-K and PEG-RCMP-K hydrogels (Figure 4D). The storage modulus at ω = 30 Hz also fell between the two gels. These results indicate that CF-CMP9 addition disrupted the CMP-mediated physical crosslinks, which led to softening of the hydrogel. The mechanical properties of this hydrogel remained unchanged over 2 days suggesting that there is no slow redistribution of the CMPs that can compromise the modification after long time period.
We next conducted particle tracking experiments on PEG-CMP9 hydrogels, which have only triple helical physical crosslinks and no chemical crosslinked network. Although this gel exhibited macroscopic viscoelastic behavior with an exponent α = 0.43, the distribution of particle MSDs showed two distinct regimes (Figure 5, A and B). While most particles displayed the typical viscoelastic MSD ~ τα, a subset of nonoparticles predominantly behaved elastically with MSDs independent of τ. The ensemble-average MSD for elastic, viscoelastic, and total ensemble cases were converted into storage and loss moduli (Figure 5C). This analysis indicated the purely elastic nature of the nanoparticles with time-independent MSD, which constituted about 25% of particles analyzed. Nevertheless, the majority of nanoparticles displayed viscoelastic MSD ~ τα dependence indicating both elastic and viscous behavior of the gel. This heterogeneity in MSD suggests the existence of regions in the gel where particles are tightly encaged by the physically crosslinked PEG-CMP9 to a degree that significantly confines the nanoparticle’s movement during the tracking time period. Previously, two-state nanoparticle Brownian motion behavior similar to the one we experienced here has been observed in the particle tracking studies of heterogeneous reconstituted lamin B125. It is possible that the local entrapment of particles was caused by PEG-CMP9 preferentially interacting with the particle surface. However, this result demonstrates the power of particle tracking techniques in studying the local mechanical properties of a 3D hydrogel system. Identification of the heterogeneous nature of purely physically crosslinked hydrogels would not have been possible with traditional macroscopic rheology techniques.
Figure 5.

Particle tracking data for PEG-CMP9 hydrogels (10 wt%). (A) Distribution of particle MSDs in PEG-CMP9 showing viscoelastic behavior, elastic behavior, and the total combined ensemble-average values. (B) Sample particle trajectories in viscoelastic and elastic gel regions. (C) Storage (G′) and loss (G″) moduli (ω = 30 Hz) corresponding to viscoelastic regions, elastic regions, and combined average values.
We then studied the effect of temperature on the mechanical properties of PEG-CMP9 gel. The temperature at which a PEG-CMP9 gel loses its self-supporting nature was dependent on the polymer concentration. Ten wt% PEG-CMP9 hydrogels were self-supporting up to 55°C, while 7 wt% samples collapsed around 35°C. Since our LiveCell incubator (Pathology Devices, Westminster, MD) has an upper working temperature limit of 50°C, we conducted particle tracking of 7 wt% PEG-CMP hydrogel at 25°C and 45°C. The results showed a large MSD increase (Student’s t-test p < 0.001) after raising the temperature (Figure 6A). Figure 6B shows that this MSD increase led to a change in the α exponent. The PEG-CMP9 hydrogel went from a viscoelastic solid at 25°C to a primarily viscous liquid at 45°C (Δα = 0.41). By comparison, PEG-RCMP-K hydrogels that lack triple helix mediated crosslinks showed a smaller decrease in elasticity upon heating (Δα = 0.16). These results indicate that increasing the temperature of PEG-CMP hydrogels destabilizes the intermolecular triple helices that contribute to the gel elasticity.
Figure 6.

Effect of temperature on the elasticity of physically crosslinked PEG-CMP9 hydrogels (7 wt%). (A) Emsembles-average MSDs of PEG-CMP9 at 25°C and 45°C with SEM error bars (n > 100). (B) α exponents determined by fitting MSDs to <Δr2(τ)> ~ kτα for PEG-CMP9 and PEG-RMCP-K.
Next, we tested the effect of adding free CMPs that can compete for triple helix formation. After conducting the particle tracking study on a PEG-CMP9 (10 wt%, 100 μL) hydrogel, we added 40 μL of 27.2 mM CMP9 into the 80°C PEG-CMP9 sample. We chose to use 80°C solution since it is the temperature that can melt PEG-CMP9 triple helices (Tm = 71 °C) within the hydrogel and initiate the strand exchange process. After cooling to room temperature, the particle tracking measurements were conducted. The result (Figure 7) showed an order of magnitude increase in MSD for the PEG-CMP hydrogel after adding the free CMP solution (Student’s t-test p < 0.001). A control experiment was conducted using only DI water. The MSD data and corresponding α exponents and viscoelastic moduli for these experiments showed that while there is a small decrease in elasticity (Δα = 0.05) for the PEG-CMP9 after adding H2O due to dilution effect, the loss in elasticity (Δα = 0.62) for the PEG-CMP9 hydrogel upon addition of unbound CMPs was much more significant (Figure 7B). As we had expected, the free CMPs were able to compete for triple helix formation against the CMPs of the PEG-CMP9 hydrogel and disrupted the triple helix mediated physical crosslinks. These results not only demonstrate facile modulation of 3D hydrogel stiffness by addition of free CMPs but also represent a novel way to immobilize bioactive molecules to PEG-CMP hydrogel via CMP-CMP hybridization.
Figure 7.
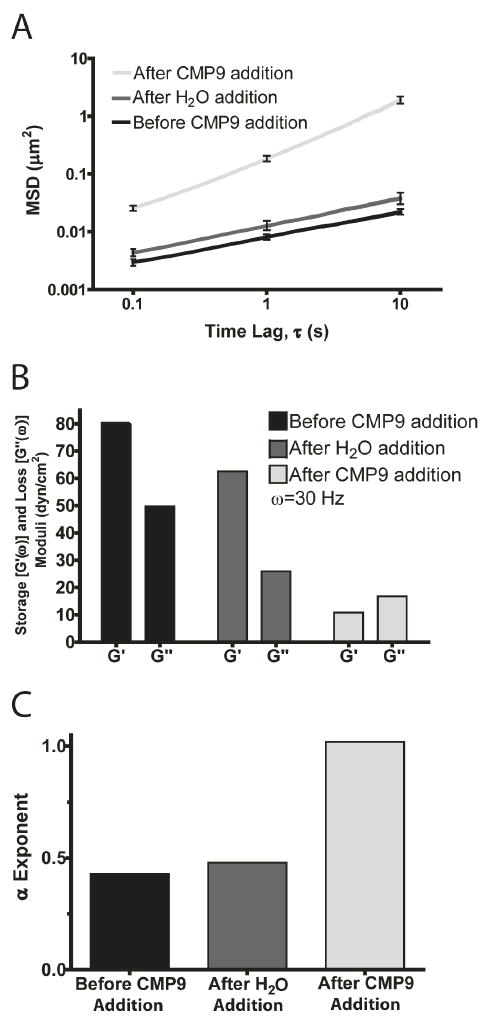
Modulation of mechanical properties of PEG-CMP9 hydrogels by addition of free CMP9 that can compete for triple helix formation. (A) Ensemble-average MSDs with SEM error bars (n > 100) shown for PEG-CMP9 hydrogels (10 wt%) before and after addition of 40 μL of 27.2 mM free CMP9 solution or H2O (control experiment). (B) Corresponding storage (G′) and loss (G″) moduli (ω = 30 Hz) for ensemble-average MSDs. (C) Corresponding α exponents determined by fitting the ensemble-average MSDs to <Δr2(τ)> ~ kτα.
Recent research activities suggest that while overall scaffold stiffness affects cell motility, stiffness gradients within the scaffolds could produce directional cell migration and growth.38-40 Besides modifying the entire PEG-CMP9 hydrogel with unbound CMPs that compete for triple helix formation, we also attempted to spatially modify the PEG-CMP9 hydrogel with local CF-CMP9 addition. Our intention was to allow hot CF-CMP9 solution to diffuse locally into the gel creating a gradient of stiffness by breaking up the triple helical crosslinks. The hot single-stranded CF-CMP9 can provide thermal energy to locally melt the PEG-CMP9 intermolecular triple helices and induce subsequent hybridization between melted PEG-CMP9 and CF-CMP9 during cooling to permanently break the triple helical crosslinks. We injected 5 μL of CF-CMP9 solution (80°C; 1.21 mM) into one side of the self-supporting PEG-CMP9 hydrogel (10 wt%, 100 μL) sitting at 37°C. Figure 8A shows the reconstituted fluorescent micrograph of the PEG-CMP9 hydrogel in the 96-well plate after CF-CMP9 injection. The intensity variation across the gel from bottom right to upper left corner clearly shows the gradient nature of the CF-CMP9 present in the hydrogel (Supporting Information, Figure S3). Using the particle tracking microrheology, we were able to discern the spatial stiffness variations within a 5 mm diameter gel. As shown in Figure 8B, particle tracking experiments confined to two opposing locations in the PEG-CMP9 hydrogel showed significant difference in terms of MSD (Student’s t-test p < 0.001) and elasticity. The area adjacent to CF-CMP9 injection site (marked by ×) showed a storage modulus that is approximately half of that found at the opposite side of the injection, 43 versus 79 dyn/cm2. These findings demonstrate that PEG-CMP and CMP interactions can create local changes in physico-chemical properties of pre-fabricated PEG hydrogels. Beyond using CMP modification to tune the stiffness of hydrogels, our results show that CMPs could be used to introduce spatially-defined insoluble bioactive factors such as adhesion peptides and growth factors, which are known to work as spatio-temporal signals during tissue development. This modification method may not be ideal for hydrogels with encapsulated cells due to high temperature (80°C) treatment process; however it can be readily used for systems where cells are seeded on top of the hydrogel after the modification. We are currently working on developing similar hydrogels that use moderate temperature or other non-cell abusive environmental triggers for triple helical assembly.
Figure 8.

Spatial modification of PEG-CMP9 hydrogel with CF-CMP9. (A) Reconstituted fluorescence micrograph of 10% PEG-CMP hydrogel in the well of 96-well plate after injecting the CF-CMP9 solution to bottom right corner (marked ×) of the gel. (B) Ensemble-average MSD data with SEM error bars (n > 100) from the particle tracking measurements at the injection area and the opposite area of the gel. (C) Corresponding storage (G′) and loss (G″) moduli (ω = 30 Hz) for ensemble-average MSDs at injection and opposite sites of the gel.
Conclusion
We developed and characterized novel PEG-based hydrogels with CMP mediated physical crosslinks. Previous efforts have shown that hydrogels featuring CMPs can improve the bioactivity of tissue engineering scaffolds by enhancing the accumulation of cell-secreted collagens.15, 41 Based on the results shown here, we believe that the CMPs can be used not only to enhance the interaction between the scaffold and cell secreted collagen, but also to control the stiffness of hydrogels via creation and disruption of triple helical physical crosslinks. We learned that CMPs retain their triple helical nature even after conjugation to PEG both in purely physically crosslinked PEG-CMP9 and chemically crosslinked PEG-CMP9-K hydrogels. We showed that PEG-CMP hydrogels have temperature dependent elasticity due to the instability of intermolecular triple helix crosslinks at elevated temperatures. Furthermore, both PEG-CMP9 and PEG-CMP9-K hydrogels can be modified to exhibit varying degrees of mechanical stiffness or to display matrix-bound molecules such as CF by adding free CMPs that compete for triple helix formation. Our studies suggest that the PEG-CMP hydrogels can be designed with a gradient in stiffness upon local addition and diffusion of free CMPs. We envision modulating triple helix mediated crosslinks in hydrogels as a method to tune the stiffness of scaffolds to optimize cell growth, differentiation, and migration. The stiffness of the gels presented here are in line with in vitro reconstituted collagen and fibrin gels as well as other physically crosslinked matrices successfully used for endothelial cell proliferation and guided neuron growth.42-45
Supplementary Material
Acknowledgments
We thank Allen Wang for help with hydrogel synthesis, Christopher Hale and Shyam Khatau for help with particle tracking microrheology and microscopy, Yong Ren for help with gel permeation chromatography, Stephen Diegelmann and Yang Li for help with NMR, Garrett Jenkinson for help with quantitative image analysis, and Tania Chan for help with circular dichroism. This work was supported by grants from the National Science Foundation to S.M.Y. (DMR-0645411) and the National Institutes of Health to D.W. (U54CA143868) and S.M.Y. (R21GM74812) as well as fellowships to P.J.S. from HHMI Graduate Training Program (NBMed) and National Defense Science & Engineering Graduate Fellowship Program (32 CFR 168a).
Footnotes
Supporting Information Available: NMR and GPC traces of PEG-CMP9 conjugates and fluorescence intensity plot of CF-CMP9 modified PEG-CMP9 hydrogel. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Engel J, Bächinger HP. Top Curr Chem. 2005;247:7–33. [Google Scholar]
- 2.Stenzel KH, Miyata T, Rubin AL. Annu Rev Biophys Bioeng. 1974;3:231–253. doi: 10.1146/annurev.bb.03.060174.001311. [DOI] [PubMed] [Google Scholar]
- 3.Shoulders MD, Raines RT. Annu Rev Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao G, Suresh S. Nat Mater. 2003;2:715–725. doi: 10.1038/nmat1001. [DOI] [PubMed] [Google Scholar]
- 5.Galbraith CP, Sheetz MP. Curr Opin Cell Biol. 1998;10:566–571. doi: 10.1016/s0955-0674(98)80030-6. [DOI] [PubMed] [Google Scholar]
- 6.Yeung T, Georges P, Flanagan L, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey P. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 7.Discher D, Janmey P, Wang Y. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 8.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 9.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 10.Phelps E, Landazuri N, Thule P, Taylor W, Garcia A. Proc Natl Acad Sci U S A. 2009;107:3323–3328. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmgren SK, Taylor KM, Bretscher LE, Raines RT. Nature. 1998;392:666–667. doi: 10.1038/33573. [DOI] [PubMed] [Google Scholar]
- 12.Kotch F, Raines R. Proc Natl Acad Sci U S A. 2006;103:3028–3033. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mo X, An Y, Yun C, Yu SM. Angew Chem, Int Ed. 2006;45:2267–2270. doi: 10.1002/anie.200504529. [DOI] [PubMed] [Google Scholar]
- 14.Wang A, Foss C, Leong S, Mo X, Pomper M, Yu SM. Biomacromolecules. 2008;9:1755–1763. doi: 10.1021/bm701378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HJ, Lee J, Chansakul T, Yu C, Elisseeff J, Yu SM. Biomaterials. 2006;27:5268–5276. doi: 10.1016/j.biomaterials.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki C, Asada S, Kitagawa K, Koide T. Pept Sci. 2008;90:816–823. doi: 10.1002/bip.21100. [DOI] [PubMed] [Google Scholar]
- 17.Werten M, Teles H, Moers A, Wolbert E, Sprakel J, Eggink G, de Wolf F. Biomacromolecules. 2009;10:1106–1113. doi: 10.1021/bm801299u. [DOI] [PubMed] [Google Scholar]
- 18.Mason T, Ganesan K, van Zanten J, Wirtz D, Kuo S. Phys Rev Lett. 1997;79:3282–3285. [Google Scholar]
- 19.Gittes F, Schnurr B, Olmsted PD, MacKintosh FC, Schmidt CF. Phys Rev Lett. 1997;79:3286–3289. [Google Scholar]
- 20.Apgar J, Tseng Y, Fedorov E, Herwig M, Almo S, Wirtz D. Biophys J. 2000;79:1095–1106. doi: 10.1016/S0006-3495(00)76363-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panorchan P, Lee J, Kole T, Tseng Y, Wirtz D. Biophys J. 2006;91:3499–3507. doi: 10.1529/biophysj.106.084988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panorchan P, Lee JS, Daniels BR, Kole TP, Tseng Y, Wirtz D. Methods Cell Biol. 2007;83:115–140. doi: 10.1016/S0091-679X(07)83006-8. [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Hale C, Panorchan P, Khatau S, George J, Tseng Y, Stewart C, Hodzic D, Wirtz D. Biophys J. 2007;93:2542–2552. doi: 10.1529/biophysj.106.102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou X, Rowe RG, Hiraoka N, George J, Wirtz D, Mosher D, Virtanen I, Chernousov M, Weiss S. Genes Dev. 2008;22:1231–1243. doi: 10.1101/gad.1643308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panorchan P, Wirtz D, Tseng Y. Phys Rev E. 2004;70 doi: 10.1103/PhysRevE.70.041906. [DOI] [PubMed] [Google Scholar]
- 26.Feng Y, Melacini G, Taulane J, Goodman M. J Am Chem Soc. 1996;118:10351–10358. [Google Scholar]
- 27.Melacini G, Feng Y, Goodman M. J Am Chem Soc. 1996;118:10359–10364. [Google Scholar]
- 28.Wien J, Arakawa T, Philo J. Anal Biochem. 1996;240:155–166. doi: 10.1006/abio.1996.0345. [DOI] [PubMed] [Google Scholar]
- 29.Kendrick B, Kerwin B, Chang B, Philo J. Anal Biochem. 2001;299:136–146. doi: 10.1006/abio.2001.5411. [DOI] [PubMed] [Google Scholar]
- 30.Kole T, Tseng Y, Huang L, Katz J, Wirtz D. Mol Biol Cell. 2004;15:3475–3484. doi: 10.1091/mbc.E04-03-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mason T, Weitz D. Phys Rev Lett. 1995;74:1250–1253. doi: 10.1103/PhysRevLett.74.1250. [DOI] [PubMed] [Google Scholar]
- 32.Gauba V, Hartgerink J. J Am Chem Soc. 2007;129:2683–2690. doi: 10.1021/ja0683640. [DOI] [PubMed] [Google Scholar]
- 33.Persikov A, Xu Y, Brodsky B. Protein Sci. 2004;13:893–902. doi: 10.1110/ps.03501704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmgren S, Bretscher L, Taylor K, Raines R. Chem Biol. 1999;6:63–70. doi: 10.1016/S1074-5521(99)80003-9. [DOI] [PubMed] [Google Scholar]
- 35.Kwak J, De Capua A, Locardi E, Goodman M. J Am Chem Soc. 2002;124:14085–14091. doi: 10.1021/ja0209621. [DOI] [PubMed] [Google Scholar]
- 36.Xu J, Viasnoff V, Wirtz D. Rheol Acta. 1998;37:387–398. [Google Scholar]
- 37.Saxton M. Biophys J. 1994;66:394–401. doi: 10.1016/s0006-3495(94)80789-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo C, Wang H, Dembo M, Wang Y. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong J, Velasco A, Rajagopalan P, Pham Q. Langmuir. 2003;19:1908–1913. [Google Scholar]
- 40.Saez A, Ghibaudo M, Buguin A, Silberzan P, Ladoux B. Proc Natl Acad Sci U S A. 2007;104:8281–8286. doi: 10.1073/pnas.0702259104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee HJ, Yu C, Chansakul T, Hwang N, Varghese S, Yu SM, Elisseeff J. Tissue Eng. 2008;14:1843–1851. doi: 10.1089/ten.tea.2007.0204. [DOI] [PubMed] [Google Scholar]
- 42.Raub C, Suresh V, Krasieva T, Lyubovitsky J, Mih J, Putnam A, Tromberg B, George S. Biophys J. 2007;92:2212–2222. doi: 10.1529/biophysj.106.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaguchi N, Zhang L, Chae B, Palla C, Furst E, Kiick K. J Am Chem Soc. 2007;129:3040–3041. doi: 10.1021/ja0680358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raeber G, Lutolf M, Hubbell J. Biophys J. 2005;89:1374–1388. doi: 10.1529/biophysj.104.050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo Y, Shoichet M. Nat Mater. 2004;3:249–253. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.