

Abstract
The profoundly elevated concentrations of low-density lipoproteins (LDL) present in homozygous familial hypercholesterolemia lead to symptomatic cardiovascular disease and death by early adulthood. Studies conducted in nonhepatic tissues demonstrated defective cellular recognition and metabolism of LDL in these patients. Since mammalian liver removes at least half of the LDL in the circulation, the metabolism of LDL by cultured hepatocytes isolated from familial hypercholesterolemic homozygotes was compared to hepatocytes from normal individuals. Fibroblast studies demonstrated that the familial hypercholesterolemic subjects studied were LDL receptor-negative (less than 1% normal receptor activity) and LDL receptor-defective (18% normal receptor activity). Cholesterol-depleted hepatocytes from normal subjects bound and internalized 125I-labeled LDL (Bmax = 2.2 μg LDL/mg cell protein). Preincubation of normal hepatocytes with 200 μg/ml LDL reduced binding and internalization by approx. 40%. In contrast, 125I-labeled LDL binding and internalization by receptor-negative familial hypercholesterolemic hepatocytes was unaffected by cholesterol loading and considerably lower than normal. This residual LDL uptake could not be ascribed to fluid phase endocytosis as determined by [14C]sucrose uptake. The residual LDL binding by familial hypercholesterolemia hepatocytes led to a small increase in hepatocyte cholesterol content which was relatively ineffective in reducing hepatocyte 3-hydroxy-3-methylglutaryl-CoA reductase activity. Receptor-defective familial hypercholesterolemia hepatocytes retained some degree of regulatable 125I-labeled LDL uptake, but LDL uptake did not lead to normal hepatocyte cholesterol content or 3-hydroxy-3-methylglutaryl-CoA reductase activity. These combined results indicate that the LDL receptor abnormality present in familial hypercholesterolemia fibroblasts reflects deranged hepatocyte LDL recognition and metabolism. In addition, a low-affinity, nonsaturable uptake process for LDL is present in human liver which does not efficiently modulate hepatocyte cholesterol content or synthesis.
Keywords: LDL, Hypercholesterolemia, Lipoprotein receptor, (Hepatocyte)
Introduction
The delivery of cholesterol to cells by the low-density lipoproteins (LDL) through a receptor-mediated pathway was first described a decade ago [1]. Fibroblasts grown from patients homozygous for familial hypercholesterolemia, an autosomal codominant disorder manifesting LDL cholesterol concentration greater than 500 mg/dl and accelerated atherosclerosis [2], demonstrate functional defects in this LDL receptor pathway [3–5]. Therefore, a disruption in the LDL receptor function as demonstrated in nonhepatic tissues correlated with markedly deranged lipoprotein metabolism.
Since the liver is the principal organ for cholesterol synthesis and excretion [6], as well as lipoprotein production and catabolism [7], the field of hepatic lipoprotein receptors has been an active area of investigation. Receptors recognizing LDL (the apolipoprotein B,E receptor) have been identified in the liver of a variety of animals including rats [8–10], puppies [11–13], rabbits [14,15] and swine [16,17]. In contrast to early reports which indicated that human liver demonstrated few, if any, LDL receptors, recent studies performed with fresh human hepatic membranes indicate that a receptor for LDL is present in the human liver [18,19].
In studies conducted in patients homozygous for familial hypercholesterolemia, the ability of hepatic membranes to bind radiolabeled LDL is markedly reduced but not totally absent [19]. Although the observed reduction in LDL-hepatic membrane interaction paralleled the defect observed in nonhepatic tissues, the physiologic and pathophysiologic impact of this residual binding has not been previously determined. Utilizing newly developed techniques, the in vitro LDL metabolism of hepatocytes from normal and homozygous familial hypercholesterolemia subjects was directly evaluated.
Methods
Patients
Neither patients 1 or 2 have been previously reported in the literature. Patient 1 was a 5-year-old Hispanic female who presented at age 2 years with inderdigital and Achilles tendon xanthomas. Her total plasma cholesterol concentration had always been in excess of 700 mg/dl and both of her parents had Type II hyperlipoproteinemia. Because a variety of therapeutic regimens – including cholestyramine, niacin, probucol, d-thyroxine, and fenafibrate – had not successfully reduced her total and LDL cholesterol concentrations, a portacaval anastomosis was performed at the University of Pittsburgh.
Patient 2 was a 12-year-old white female diagnosed as an familial hypercholesterolemia homozygote based on family history, tendon and tuberous xanthomas and total plasma cholesterol concentration in excess of 600 mg/dl. Although rigid adherence to bile acid sequestrant therapy reduced her plasma cholesterol levels, the side effects of drug therapy led to noncompliance and the patient underwent portacaval shunt.
Lipoprotein preparation
Plasma was obtained from normolipidemic individuals and an apolipoprotein E-deficient patient [20] by plasmapheresis, and LDL (d = 1.030–1.050 g/ml) was isolated by preparative ultracentrifugation [21]. LDL was radiolabeled by the iodine monochloride method [22] as modified for lipoproteins [23]. Labeling efficiency ranged from 22 to 38% and less than 3% of the label remained in the organic phase of a chloroform/methanol (2:1, v/v) extraction. All LDL mass determinations were based on LDL protein concentrations assessed by the method of Lowry et al. [24]. Specific activities for 125I-labeled LDL ranged from 3.2 to 4.6 Bq/mg LDL protein.
Cell culture techniques
Human skin fibroblasts were derived from skin biopsies of normolipidemic individuals and patients 1 and 2 and maintained as previously described [19].
The Human Experimentation Committee of the University of Pittsburgh School of medicine approved the use of hepatic biopsies obtained at the time of surgery for research purposes. Hepatic biopsies and hepatocyte isolation were undertaken after informed consent had been obtained. Hepatocytes were removed from the biopsies by sterile mincing of the tissue followed by collagenase-hyaluronidase-dispase treatment as previously described [25]. Briefly, biopsies (1–4 g) were immediately perfused by subcapsular injection of 15 ml Ca2+-free Hanks' balanced salt solution supplemented with insulin (10 μg/ml) and hydrocortisone (10−6 M) and gassed with 5% CO2/95% O2 (pH 7.4) (digestion medium) at 4°C. After transport to the laboratory, the specimen was placed in 5 ml cold Ca2+-free digestion medium, dissected free of connective tissue and minced with crossed scalpels to 1 mm3 fragments. The mince was washed twice with cold medium and then placed in a 250-ml conical flask in 10 ml Ca2+-free medium at 37°C. The flask was agitated at 60 rpm at 37°C with continuous oxygenation by 5% CO2/95% O2 bubbling throughout the isolation procedure. The pH was monitored by the phenol red indicator and maintained at approximately pH 7.4 by titration with 1 N NaOH. After 15 min, the tissue fragments were allowed to settle and the medium was gently drawn off and replaced. After a second 15-min incubation, the medium was replaced with digestion medium plus 5 mM CaCl2, 0.05% collagenase (Sigma type IV or Worthington type II) and 0.1% hyaluronidase (Sigma type I-S). After 30 min, fresh enzyme solution was added. After 30 more minutes the medium was replaced with digestion medium with 5 mM CaCl2, 0.05% collagenase and 0.05% dispase. Three 30-min incubations were performed with this solution. Exhausted enzyme solution from each digestion was examined for viable hepatocytes which began to appear in small numbers in the last two collagenase-dispase digestions. After enzyme digestion, the tissue fragments were placed in a petri dish lined with sterile Nylon mesh (100 μm). Cold culture medium supplemented with 10% fetal calf serum, insulin (10 μg/ml) and hydrocortisone (10−6 M) was poured over the tissue which was then gently dispersed with the plunger of a 12 cm3 syringe. The remaining debris was centrifuged at 50 × g for 3 min. Sedimented cells were suspended in fresh medium and sedimented three times at 20 × g for 2 min. The resulting cell preparation was examined for viability by Trypan blue exclusion (final concentration 0.2%). Cell were suspended in culture medium and plated in 24-well Primaria tissue culture dishes at a density of 105 cells/cm2 (2 · 105 cells per well).
The cells were cultured in a humidified 5% CO2/95% air incubator at 37°C. After 24 h, the medium was changed to 5% fetal calf serum medium plus the components of serum-free medium (Table I). At 72 h, the medium was replaced with serum-free medium. Thereafter, the medium was replaced at least every 48 h with freshly prepared serum-free medium.
TABLE I. Serum-Free Medium for Cell Culture.
Components were added to RPMI-1640 to achieve the indicated final concentration components stored in sterile aliquots and the serum-free medium was prepared and used daily.
| Components | Final concentration |
|---|---|
| Insulin | 10 μg/ml |
| Glucagon | 10 μg/ml |
| Somatotropin | 1 μg/ml |
| Prolactin | 100 ng/ml |
| Hydrocortisone | 106 M |
| Epidermal growth factor | 50 ng/ml |
| Linoleic acid/bovine serum albumin | 5 μg/ml per |
| 0.5 mg per ml | |
| Thyrotropin releasing factor | 106 M |
| Glutamine | 1 ml/100 cm3 |
| CuSO4 | 10−7 M |
| ZnCl2 | 10−10 M |
| Selinous acid (Na) | 10−7 M |
| Penicillin | 100 U/ml |
| Fungizone | 0.25 μg/ml |
| Streptomycin | 100 μg/ml |
Hepatocyte albumin synthesis
Albumin synthesis of hepatocytes was studied by incorporation of [3H]leucine into albumin. Hepatocytes in serum-free medium were washed with leucine-free RPMI-1640 twice. Complete leucine-free serum-free medium with 1 μCi of [3H]leucine was then added to the well and the cells were incubated for 16 h. The medium was harvested and dialyzed extensively against phosphate-buffered saline/0.01% EDTA at 4°C. The amount of label incorporated into protein was determined by 10% trichloroacetic acid precipitation. Protein content was determined by the method of Lowry et al. [24]. An aliquot containing 50 μg of protein was lyophilized, reconstituted in 50 μl of 0.5% SDS/0.05 M Tris-HCl (pH 8.2), and heated in an oil bath at 100°C for 3 min. The 0.2 M Tris and distilled water were added to adjust the final concentrations to 0.05 M Tris and 0.1% SDS. Bromophenol blue tracking dye and one drop of glycerol were added and the samples were then applied to 1.5 mm polyacrylamide gels (3% stacking gel) in 0.385 M Tris-HCl/0.1% SDS (pH 8.8) and electrophoresed at 5 mA per gel overnight with 0.05 M Tris-HCl/0.384 M glycine/0.1% SDS (pH 8.3). Bio-Rad protein standards were included in each analysis. The gels were fixed in 12.5% trichloroacetic acid, stained in 0.05% Coomassie blue G-250 in 12.5% trichloroacetic acid, and destained in 5% acetic acid. Each 10-cm lane was cut into 2-mm slices which were placed in scintillation vials. Aliquots (0.5 ml) of 30% H2O2 were added, the vials heated for 4 h at 80°C and 10 ml Aquasol II scintillation fluid added for radioactivity quantitation. Regions of radioactivity corresponding to newly synthesized protein were compared to the molecular weight of known protein standards.
The rate of albumin synthesis was determined by immunoprecipitation of albumin in cell lysates after a [3H]leucine pulse-chase. Hepatocytes in serum-free medium were washed twice with leucine-free RPMI-1640 and then pulsed for 30 min with leucine-free RPMI-1640 with 1 μCi [3H]leucine (New England Nuclear, Boston, MA). The medium was removed, the cells were washed twice with complete RPMI-1640, and finally the cells were incubated for an additional 60 min with RPMI-1640. The medium was harvested and cooled on ice. The cells were washed three times with cold phosphate-buffered saline and lysed with 0.25 ml phosphate-buffered saline-1% Triton X-100. The lysate was centrifuged at 5000 × g for 1 min. The medium and lysate supernatant were used for immunoprecipitation by mixing 50 μl with 100 μl unlabeled human albumin carrier (0.1 mg/ml in 50 mM Tris, 150 mM NaCl 5 mM EDTA) followed by 50 μl anti-human albumin (Sigma A-1151). After mixing and incubation (18 h) at 4°C, the precipitates were harvested by centrifugation at 5000 × g for 1 min with two washes in the Tris buffer. The radioactivity in the precipitate was quantitated and expressed as dpm per mg cell protein.
Hepatic membrane studies
Hepatic membranes were isolated from 1–2 g of hepatic tissue from normal and homozygous familial hypercholesterolemia patients, and the ability of normal and homozygous familial hypercholesterolemia hepatic membranes to bind 125I-labeled LDL was assessed as outlined previously [19]. Binding is expressed as ng 125I-labeled LDL per mg hepatic membrane binding and represents the mean of triplicate assays. Control and familial hypercholesterolemia membranes were studied at the same time under identical conditions.
Cellular lipoprotein-binding studies
Fibroblast and hepatocyte 125I-labeled LDL-binding assays were conducted identically, utilizing LDL isolated from a patient with apolipoprotein E deficiency [20]. From 0.5 to 5 · 105 cells were incubated for 24 h in 2 cm2 wells containing 1 ml serum-free medium. After a further 24 h in either fresh serum-free medium or serum-free medium containing 200 μg LDL protein/ml, cells were incubated with 0.5 ml of the indicated concentrations of 125I-labeled LDL protein. The media from these cells were then harvested after 6 h, and the cells were washed with six 1-ml ice-cold phosphate-buffered saline aliquots. Cells were solubilized in 1 N NaOH. after which the cell-associated radioactivity (bound and internalized) was quantitated and the cellular protein determined by the method of Lowry et al. [24]. Specifically bound and internalized 125I-labeled LDL was defined as the difference in binding obtained in the absence and presence of 10-fold excess unlabeled LDL. The harvested medium was used to assess the degree of 125I-labeled LDL which was degraded. After trichloroacetic acid precipitation of the medium using bovine serum albumin carrier (5 mg/ml), free iodine and iodine bound to tyrosine were subjected to potassium iodide/H2O2 treatment followed by chloroform extraction as reported by Goldstein and Brown [1]. The radioactivity of aliquots of the aqueous phase was quantitated and the amount of degraded 125I-labeled LDL was normalized to the cellular protein content. In parallel dishes, fluid phase endocytosis was quantitated by [14C]sucrose uptake [25].
Morphological studies
The light microscopic appearance of the cells in culture was monitored by phase-contrast microscopy. Documentation was obtained by micrographs taken on Polaroid Type 55 P/N 4 × 5″ sheet film. Original magnifications were 100 × or 250 ×.
Cells for ultrastructural examination were harvested from 25-mm2 culture dishes after fixation in equal volumes of conditioned medium and 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for a minimum of 2 h. Cells were harvested either within a day of plating (for analysis of the primary culture cell population) or after 7 days in culture (for analysis of the cell population on which the functional studies were performed).
These cells were pelleted in 1.5-ml conical centrifuge tubes, dehydrated and embedded in 100 nm sections were stained with uranyl acetate and lead citrate and examined in an electron microsope, where a minimum of a hundred cells was examined and photographed from each patient at each time point.
Other assays
Cellular cholesterol content was determined by an enzymic, fluorimetric assay as previously described [19,28]. The 3-hydroxy-3-methylglutaryl-CoA reductase activity was determined by the production of mevalonate [29] and normalized to cellular protein content [24]. Plasma lipid concentrations were determined using enzymic, colorimetric assays [30,31]. Lipoprotein concentrations were derived utilizing the methods of the Lipid Research Clinics [32] except that HDL cholesterol concentrations were assessed after dextran-sulfate precipitation [33]. Acylcholesterol: acyltransferase activity was determined in skin fibroblast culture [34].
Results
The plasma lipid and lipoprotein concentrations in the homozygous familial hypercholesterolemia patients were characteristic for this condition (Table II). Both the total and LDL cholesterol concentrations were 4–8-fold higher than the 90th percentile level for this age group established by the Lipid Research Clinics [35]. The increased VLDL cholesterol concentration in combination with the elevated LDL cholesterol levels indicated a Type IIb phenotype. In addition, the HDL cholesterol concentrations were depressed. Therefore, both study subjects manifested profoundly deranged lipoprotein metabolism as reflected in the fasting lipoprotein concentrations.
TABLE II. Plasma Lipid and Lipoprotein Concentrations in Homozygous Familial Hypercholesterolemia.
Values are the means ± S.E. of triplicate samples obtained in patients 1 and 2 off all medication. LRC normals represent the 10th and 90th percentile for girls ages 0–14 in the Lipid Research Clinical North American Prevalence Study [33].
| Cholesterol (mg/dl) | Triacylglycerol (mg/dl) |
||||
|---|---|---|---|---|---|
| Total | VLDL | LDL | HDL | ||
| Patient 1 | 950 ± 59 | 95 ± 17 | 844 ± 49 | 21 ± 1 | 141 ± 21 |
| Patient 2 | 729 ± 45 | 72 ± 14 | 600 ± 50 | 26 ± 8 | 133 ± 17 |
| LRC normals | 119–190 | 5–21 | 60–120 | 35–70 | 38 ± 114 |
The markedly abnormal lipoprotein concentrations observed in these familial hypercholesterolemia homozygotes were paralleled by the inability of cultured skin fibroblasts from these patients to metabolize LDL (Fig. 1). Normal fibroblasts (panel A) bound and internalized 125I-labeled LDL after cholesterol depletion. The cell-associated 125I-labeled LDL was reduced by preincubating the cells with 200 μg LDL protein/ml. The LDL metabolism of the skin fibroblasts in patient 1 (panel B) and in patient 2 (panel C) differed from normal fibroblast LDL metabolism. The fibroblasts of patient 1 demonstrated absolutely no cell-associated LDL binding and internalization. Although the fibroblasts of patient 2 did demonstrate some regulatable 125I-labeled LDL binding and internalization, the degree of binding was less than 50% that of normal fibroblasts.
Fig. 1.
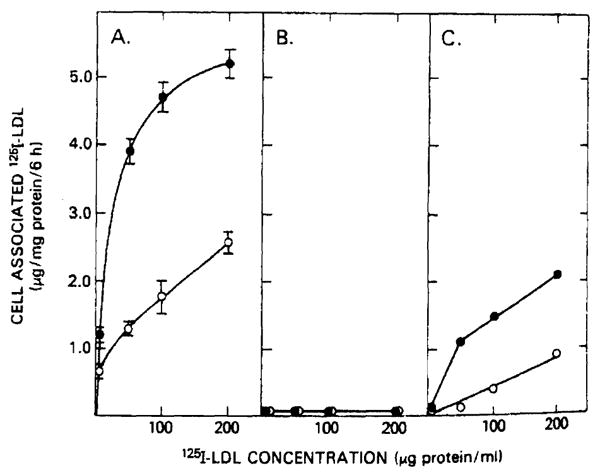
125I-labeled LDL binding and internalization by fibroblasts from normal and homozygous familial hypercholesterolemia patients. 5–7·105 fibroblasts per 2 cm2 well were incubated for 48 h in serum-free medium (●) or for 24 h in serum-free medium with 200 μg LDL protein/ml (○). After a 6-h, 37°C incubation with the indicated concentrations of 125I-labeled LDL, cells were washed six times with ice-cold phosphate-buffered saline (pH 7.0). Protein and radioactivity determinations were performed on cells solubilized with 1 N NaOH. Values represent specifically bound and internalized 125I-labeled LDL defined as the difference in binding observed in the absence and presence of 10-fold excess of unlabeled LDL. Normal fibroblasts (panel A) and fibroblasts from patient 1 (panel B) and patient 2 (panel C) were evaluated.
Utilizing the ability of LDL cholesterol to stimulate the activity of the intracellular enzyme acylcholesterol: acyltransferase, the fibroblast LDL receptor activity was quantitated. Fibroblasts from patient 1 demonstrated less than 1% normal LDL receptor activity and patient 2's fibroblasts had 18% normal activity. Therefore, these individuals reflect the variability of the degree of the loss of LDL receptor activity in homozygous familial hypercholesterolemia [4,5,34] and can be classified as receptor-negative (patient 1) and receptor-defective (patient 2).
The loss of normal LDL receptor function in cultured fibroblasts of familial hypercholesterolemia subjects has been demonstrated to parallel reduced LDL binding to hepatic membranes from familial hypercholesterolemia patients [19]. The binding of 125I-labeled LDL to hepatic membranes from patients 1 and 2 was compared to that observed in normal hepatic membranes (Table III). Total and specific 125I-labeled LDL binding was reduced from 18–40% in the familial hypercholesterolemia homozygotes. In contrast, the nonspecific binding was comparable to that observed in normal membranes. Since the coefficient of variation for the specific binding of a single membrane preparation was less than 3%, these reductions in specific binding to the membranes are significant and support previous studies [18,19]. In addition, just as in the fibroblast studies, the hepatic membranes from patient 1 demonstrated an even greater defect in LDL recognition than was observed in patient 2. Therefore, the defective fibroblast LDL metabolism in these patients was associated with reduced LDL binding to hepatic membranes.
TABLE III. The Binding of 125I-Labeled LDL to Hepatic Membranes from Normal and Homozygous Familial Hypercholesterolemia Subjects.
Hepatic membranes (56–144 μg protein) were incubated at 37°C for 30 min with 10 μg 125I-labeled LDL in the absence (total) and presence (nonspecific) of 20-fold excess unlabeled LDL. Values for the normal membranes represent the mean ± S.E.
| Binding (ng/mg membrane protein) | ||||||
|---|---|---|---|---|---|---|
| total | (% normal) | nonspecific | (% normal) | specific | (% normal) | |
| Normal (n = 3) | 1757 ± 88 | 78 ± 66 | 975 ± 100 | |||
| Patient 1 | 1440 | (82%) | 853 | (109%) | 588 | (60%) |
| Patient 2 | 1391 | (79%) | 684 | (87%) | 707 | (73%) |
In order to evaluate the role that the reduced hepatic membrane LDL binding might play in homozygous familial hypercholesterolemia hepatocyte lipoprotein metabolism, hepatocytes from both study subjects and from normolipidemic subjects were isolated and cultured in vitro.
Morphologic studies
Cells from both patient 1 (Fig. 2, left panel) and 2 (Fig. 2, right panel) taken within a day of harvest were unmistakably hepatocytic. Fewer than 3% of the cell population in either patient were nonhepatocytic determined by counting 300–400 cells; no fibroblasts were identified by either phase-contrast or electron microscopy. By electron microscopy, cells were characteristically rounded with short microvilli, identical to those normally identified within the space of Disse. Cell organelle content was typical of hepatocytes, with a preponderance of rough endoplasmic recitulum, mitochondria, and lipid droplets. In addition, lysosomes were readily identified, and occasional, highly characteristic peroxisomes were found. These findings, present in over 97% of the cells, provide irrefutable evidence for hepatocytic origin of the vast majority of the cells originally placed into culture.
Fig. 2.
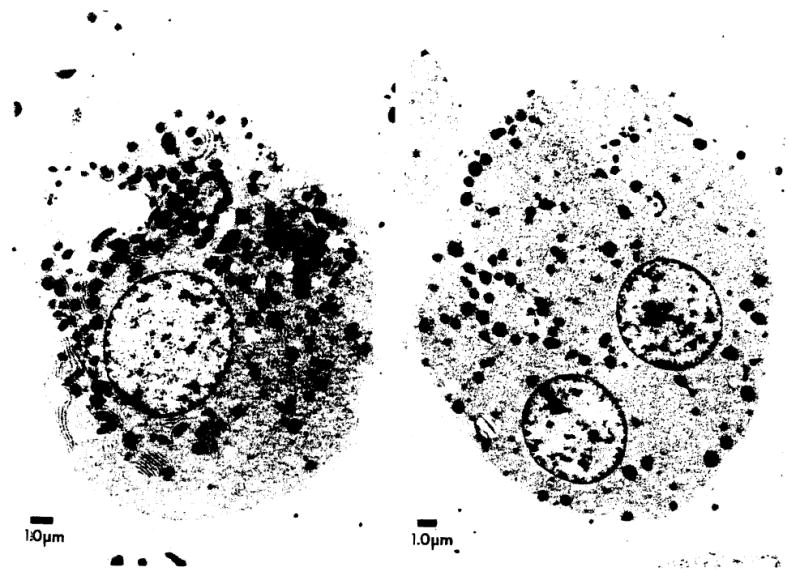
Typical cells initially isolated from patient 1 (left panel) and patient 2 (right panel) demonstrate the electron microscopic characteristics of hepatocytes.
After 7 days in culture, the light and low magnification ultrastructural appearance of the cultured cells was somewhat altered. Cells were generally larger and more adherent (Fig. 3), yet detailed ultrastructural examination revealed the same organelles noted originally. Specificially, parallel arrays of rough endoplasmic reticulum, often forming concentric whorls with glycogen particles, as well as large numbers of mitochondria and lipid droplets, provided unmistakable evidence for the persistence of hepatocytes in culture (Fig. 3, left panel). These findings are inconsistent with fibroblasts in culture. An additional feature (not illustrated) was the appearance of gland or duct lumina with junctional complexes, which appeared to be intercellular bile canaliculi. In addition, bundles of keratin filaments were seen, often terminating in membrane densities resembling ill-formed desmosomes. These features offer further evidence of the hepatocytic origin of these cells and are never observed in fibroblasts in culture. Therefore, the biochemical studies performed on liver biopsy samples reflected hepatocellular lipid and lipoprotein metabolism.
Fig. 3.
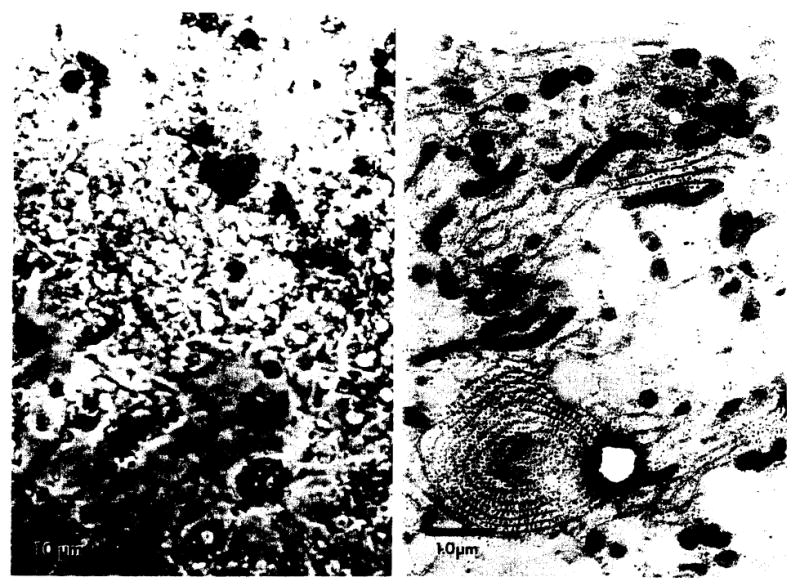
After 7 days in tissue culture, hepatocytes adhered to the tissue culture plates, flattened out and increased in size as shown by phase-contrast microscopy (left panel). The elaboration of cellular lipid, rough endoplasmic reticulum and mitochondria were observed by electron microscopy (right panel).
Both normal and familial hypercholesterolemia cells continuously synthesized albumin for up to 7 days. Utilizing the incorporation of [3H]leucine into immunoprecipitable albumin, the 42 451 ± 2 761 dpm/mg cell protein precipitated on day 3 was virtually identical to the 41 660 ± 1 660 dpm/mg cell protein precipitated on day 7 in normal cells. Albumin synthesis was also observed in hepatocytes from patients 1 and 2 which were respectively 88% and 108% normal. Therefore, the cultured cells were characterized as hepatocytes by morphologic as well as biochemical characteristics. Normal human hepatocytes have been shown to remain viable in vitro for 17 days [25].
The ability of normal and homozygous familial Hypercholesterolemia hepatocytes to bind and internalize 125I-labeled LDL was determined after 5–6 days in tissue culture (Fig. 4). After a 48-h incubation in cholesterol-free familial hypercholesterolemia, normal hepatocytes (panel A) saturably bound LDL. Preincubation of hepatocytes reduced the degree of cell-associated 125I-labeled LDL by at least 30%. Panel A represents the mean values obtained from four different human hepatocyte cell lines with an interassay variability of 6–14%. Hepatocytes from both patient 1 (panel B) and patient 2 (panel C) bound less 125I-labeled LDL than normal. Although patient 1 demonstrated no apparent up-regulation of 125I-labeled LDL binding with cholesterol depletion, patient 2 did manifest the ability to enhance 125I-labeled LDL binding.
Fig. 4.
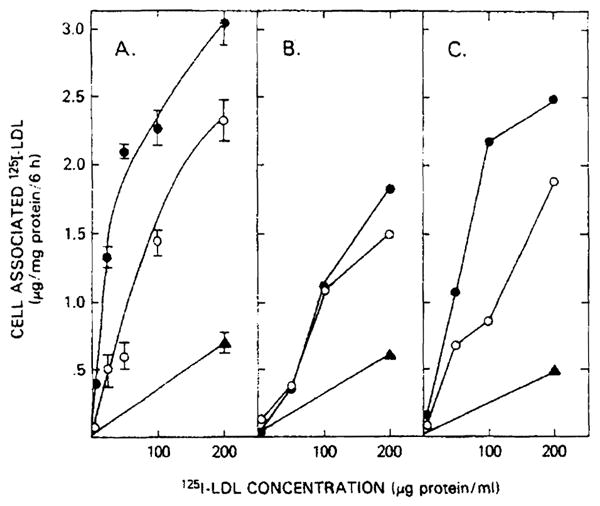
125I-labeled LDL binding and internalization by hepatocytes from normal and homozygous familial hypercholesterolemia patients. Hepatocytes (2–3·105/well) were incubated for 48 h in serum-free medium. After a further 24 h in either serum-free medium (●) of serum-free medium containing 200 μg LDL protein/ml (○), hepatocytes from normal (panel A), patient 1 (panel B) and patient 2 (panel 2) were incubated at 37°C with the indicated concentration of 125I-labeled LDL. After 6 h, the cells were washed six times with ice-cold phosphate-buffered saline (pH 7.0) and solubilized with 1 N NaOH. Total cell-associated binding and binding in the presence of 10-fold excess of unlabeled LDL (nonspecific binding, ▲) is shown. Normal values represent the mean ± S.E. for five different. hepatocyte preparations.
This LDL-cell interaction could not be ascribed to fluid phase endocytosis. In companion experiments in which hepatocytes were handled identically, the rate of fluid phase endocytosis was determined using [14C]sucrose. The observed fluid phase endocytic rate of 0.17–0.24 μl medium/mg cell protein per 6 h of 37°C incubation could account for less than 5% of the observed binding Therefore, not only are the differences in LDL metabolism present in fibroblasts of receptor-negative and receptor-defective familial hypercholesterolemia homozygotes also seen in hepatocyte LDL uptake, but also the residual association is dependent on a process(es) other than fluid phase endocytosis.
The altered LDL binding in familial hypercholesterolemia hepatocytes led to abnormal cellular cholesterol homeostasis. In normal hepatocytes incubated in cholesterol-free medium, addition of LDL to the culture medium increased total and free cholesterol by 70% and 35%, respectively, and the initial esterified cholesterol content went from 2% to 12% of the total cellular cholesterol (Table IV). Familial hypercholesterolemia hepatocytes incubated in cholesterol-free medium contained significant quantities of cholesterol in the esterified form and, in contrast to normal hepatocytes, exposure of familial hypercholesterolemia to LDL increased total and free cholesterol only 21% and 29%, respectively. Furthermore, the total and free cholesterol content in familial hypercholesterolemia hepatocytes was only 69% and 45% that of normal cells after LDL incubation.
TABLE IV. Cholesterol Content of Hepatocytes from Normal and Homozygous Familial Hypercholesterolemia Patients.
Hepatocytes (3–5·105 cells/well) were incubated for 48 h in serum-free medium. After another 24 h in either serum-free medium or serum-free medium + 200 μg LDL protein/ml, cells were harvested and exhaustively extracted with chloroform/methanol (2:1, v/v). After evaporation of the organic solvent under nitrogen, cellular lipid was resuspended in isopropanol and the cholesterol was determined by an enzymic, fluorimetric assay before (free cholesterol) and after (total cholesterol) cholesteryl ester hydrolysis. The difference between total and free cholesterol is defined as esterified cholesterol, and values represent the mean ± S.E. of three normal cell lines.
| Cholesterol content (nmol/mg cell protein) | |||
|---|---|---|---|
| total | free | esterfied | |
| Normal hepatocytes (n = 3) | |||
| serum-free medium | 127 ± 11 | 123 ± 10 | 3 ± 3 |
| serum-free medium + LDL | 216 ± 15 | 190 ± 10 | 26 ± 8 |
| Patient 1 | |||
| serum-free medium | 116 | 62 | 54 |
| serum-free medium + LDL | 140 | 80 | 60 |
The activity of the rate-limiting enzyme of cholesterol biosynthesis, 3-hydroxy-3-methylglutaryl-CoA reductase, was determined in normal and familial hypercholesterolemia hepatocytes (Table V). The suppressed 3-hydroxy-3-methylglutaryl-CoA reductase activity in normal hepatocytes was reduced by 81% by preincubation in the presence of LDL. The familial hypercholesterolemia hepatocytes, however, did not reduce their 3-hydroxy-3-methylglutaryl-CoA reductase activity with exposure to LDL. Therefore, the altered familial hypercholesterolemia hepatocyte binding and internalization of LDL led to a decreased cellular cholesterol content and increased 3-hydroxy-3-methylglutaryl-CoA reductase activity.
TABLE V. 3-Hydroxy-3-Methylglutaryl-CoA (Hmg-CoA) Reductase Activity in Hepatocytes from Normal and Homozygous Familial Hypercholesterolemia Patients.
Hepatocytes (27–60 μg cellular protein/well) were incubated in serum-free medium for 48 h followed by another 24 h in serum-free medium or serum-free medium with 200 μg LDL protein/ml. Cells were harvested and the ability to generate mevalonate was determined [27].
| HMG-CoA reductase activity (pmol/mg protein/min) |
||
|---|---|---|
| serum-free medium | serum-free medium + LDL | |
| Normal hepatocytes (n = 3) | 3.1 ± 0.6 | 0.6 ± 0.2 |
| Patient 1 hepatocytes | 4.8 | 5.6 |
| Patient 2 hepatocytes | 6.4 | 6.2 |
Discussion
The present studies were undertaken to determine the effect of the loss of the LDL receptor pathway on cultured human hepatocyte cholesterol metabolism. Normal cultured human hepatocytes, like fibroblasts, expressed enhanced LDL binding and uptake with cholesterol depletion (Figs. 1 and 4). Also, like fibroblasts, this LDL uptake led to cellular cholesterol accumulation (Table III) and an inhibition of cholesterol biosynthesis (Table IV). Therefore, normal human hepatocytes share many of the characteristics of the LDL receptor pathway that have previously been defined for a variety of nonhepatic tissues.
Although normal human hepatocyte LDL metabolism was similar to fibroblast LDL metabolism, homozygous familial hypercholesterolemia hepatocytes demonstrated differences in addition to similarities. Although patient 1 was LDL receptor-negative based on fibroblast 125I-labeled LDL uptake (Fig. 1) and acylcholesterol: acyltransferase activity, her hepatocytes bound and internalized considerable quantities of 125I-labeled LDL (Fig. 4). The hepatocytes from the receptor-defective familial hypercholesterolemia homozygote (patient 2) also demonstrated a remarkable degree of regulatable 125I-labeIed LDL uptake. Therefore, despite striking reductions in fibroblast LDL metabolism in these familial hypercholesterolemia subjects, a relatively high degree of LDL binding by familial hypercholesterolemia hepatocytes was observed. Since this interaction was independent of fluid phase endocytosis, an LDL-cell interaction is present in human hepatocytes which is independent of the classical LDL-receptor pathway.
This LDL-receptor-independent binding differs from LDL-receptor binding in several ways. First, it is nonsaturable. As demonstrated by both familial hypercholesterolemia hepatocytes as well as normal hepatocytes, even with a constant 10-fold excess of unlabeled LDL competing for binding, the cells which had been preincubated with LDL (open circles, Fig. 4) demonstrated nearly linear uptake with increasing 125I-labeled LDL concentration. Second, this binding was not regulated. In the LDL receptor-negative subject, cholesterol loading by LDL preincubation and depletion of cellular cholesterol did not affect LDL uptake. Third, the non-LDL receptor binding did not lead to LDL degradation (Fig. 5) or effective delivery of free cholesterol to hepatocytes (Table IV). Although hepatocytes manifesting the LDL-receptor pathway increased total and free cellular cholesterol content by 67% and 48%, respectively, the residual binding led to a free cellular cholesterol content that was less than 50% normal (Table IV). Finally, unlike the LDL-receptor pathway which reduced the activity of the rate-limiting enzyme for cholesterolgenesis, 3-hydroxy-3-methylglutaryl-CoA reductase, the residual binding did not lead to reduction in the activity of this enzyme. Therefore, the current studies establish that LDL-hepatocyte binding is present in man which is distinct from the classic fibroblast LDL-receptor pathway.
Fig. 5.
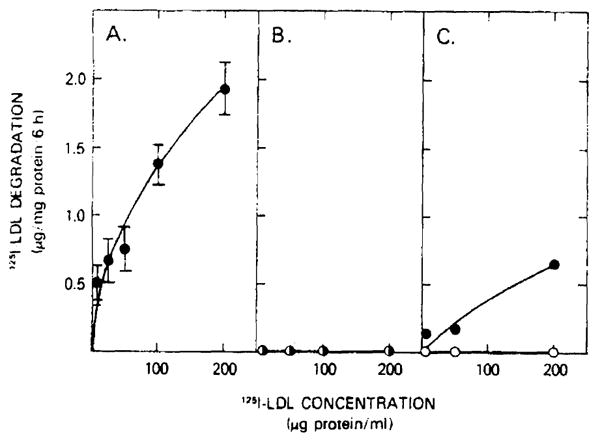
125I-labeled LDL degradation by hepatocytes from normal and homozygous familial hypercholesterolemia patients. The media from the normal hepatocytes (panel A), patient 1 hepatocytes (panel B), and from patient 2 (panel C) were harvested after a 6-h incubation with the indicated concentrations of 125I-labeled LDL. The non-chloroform-extractable 125I-label counts in the media (trichloroacetic acid supernatant) defined the degradation as outlined in Methods. Hepatocytes were incubated for 48 h prior to the exposure to 125I-labeled LDL in either the absence (○) or presence (●) of 2 mg unlabeled LDL protein/ml. Values for normal hepatocytes represent the mean ± S.E. for five different hepatocyte preparations.
A similar binding, independent of the LDL receptor, has been defined in other species. An animal model for homozygous familial hypercholesterolemia is the Watanabe Heritable Hyperlipidemic (WHHL) rabbit. Despite the defect in the fibroblast LDL-receptor pathway [36], [14C]-sucrose LDL still accumulated in the liver to a normal extent, indicating an uptake process genetically distinct from the LDL-receptor pathway [37]. Since more than 90% of this WHHL hepatic uptake was in hepatocytes [37] rather than Kupfer cells and since WHHL hepatocytes did not regulate 125I-labeled LDL degradation, Attie and coworkers concluded that the rabbit liver manifests an LDL uptake process independent of the classic LDL-receptor pathway termed the alternate pathway [7].
Although the residual binding we observed could reflect this alternate pathway, it provides an inefficient means of LDL cholesterol delivery to the cell in vitro or it may represent other processes of significance to hepatocyte lipoprotein metabolism. It could reflect another receptor pathway which partially recognizes the apolipoprotein B of LDL. An apparently unregulated uptake of apolipoprotein E-containing lipoprotein particles by an apoprotein recognition pathway has been characterized in dogs [13] and man [38], and such a residual pathway could explain these results in human hepatocytes. Another possibility revolves around the unique role of the hepatocyte in lipoprotein synthesis and secretion. Nascent lipoproteins require transport from the Golgi to the cell surface. The residual uptake of LDL by familial hypercholesterolemia hepatocytes could reflect a recycling pathway generally used for lipoprotein secretion which has been unmasked in this mutant cell line. The residual specific LDL binding present in hepatic membranes in these patients (Table III) and in previously reported cases of homozygous familial hypercholesterolemia [19] could be explained by this latter hypothesis.
The present observations have clinical as well as theoretical ramifications. Based on studies in the WHHL, it has recently been proposed that the treatment of hypercholesterolemia may be affected through the modulation of hepatic LDL receptors [39]. The present study indicates that the concept of hepatic LDL receptor modulation may indeed have therapeutic relevance in man. In addition, the different degrees of residual LDL receptor modulation in familial hypercholesterolemia hepatocytes may account for the variable response to therapy and prognosis in this disease [40]. Finally, these observations lend support to the concept that this inborn error in lipoprotein metabolism may be effectively treated by liver transplantation.
In conclusion, the direct study of normal and familial hypercholesterolemia hepatic LDL metabolism demonstrates that, in addition to fluid phase endocytosis and a regulatable LDL-receptor pathway, a nonsaturable LDL binding process is present in human hepatocytes. Although this binding could reflect an apolipoprotein B secretory system analogous to the mannose 6-phosphate receptor pathway for lysosomal enzymes [41], the importance and physiologic role of this alternative recognition remains to be elucidated.
Acknowledgments
We thank Drs. Sangvi and Warty at the University of Pittsburgh for allowing us to use their facilities for a portion of these studies; Dr. Z. Beg and Mr. J.A. Stonik for determining 3-hydroxy-3-methylglutaryl-CoA reductase activity in the hepatocytes, and Mr. Gilbert Olsen and Mr. Stephen Merlin for their excellent technical assistance.
References
- 1.Goldstein JL, Brown MS. J Biol Chem. 1974;210:5153–5162. [PubMed] [Google Scholar]
- 2.Khachadurian AK. Am J Med. 1964;37:402–407. doi: 10.1016/0002-9343(64)90196-2. [DOI] [PubMed] [Google Scholar]
- 3.Brown MS, Goldstein JL. Proc Natl Acad Sci USA. 1974;71:788–792. doi: 10.1073/pnas.71.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldstein JL, Dana SE, Brunschede GY, Brown MS. Proc Natl Acad Sci USA. 1975;72:1092–1096. doi: 10.1073/pnas.72.3.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toileshaug H, Hobgood KK, Brown MS, Goldstein JL. Cell. 1983;32:941–951. doi: 10.1016/0092-8674(83)90079-x. [DOI] [PubMed] [Google Scholar]
- 6.Dietschy JM, Wilson JD. N Engl J Med. 1970;282:1128–1138. 1241–1249. doi: 10.1056/NEJM197005142822005. [DOI] [PubMed] [Google Scholar]
- 7.Attie AD, Pittman RC, Steinberg D. Hepatology. 1982;2:269–281. doi: 10.1002/hep.1840020215. [DOI] [PubMed] [Google Scholar]
- 8.Kovanen PT, Brown MS, Goldstein JL. J Biol Chem. 1979;254:11367–11373. [PubMed] [Google Scholar]
- 9.Windler EET, Kovanen PT, Chao YS, Brown MS, Havel RJ, Goldstein JL. J Biol Chem. 1980;255:10464–10471. [PubMed] [Google Scholar]
- 10.Harkes L, Van Berkel JC. Biochim Biophys Acta. 1982;712:677–683. doi: 10.1016/0005-2760(82)90297-1. [DOI] [PubMed] [Google Scholar]
- 11.Kovanen PT, Bilheimer DW, Goldstein JL, Jaramillo JJ, Brown MS. Proc Natl Acad Sci USA. 1981;78:1194–1198. doi: 10.1073/pnas.78.2.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahley RW, Hui DY, Innerarity TL, Weisgraber KH. J Clin Invest. 1981;68:1197–1206. doi: 10.1172/JCI110365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angelin B, Raviola CA, Innerarity TL, Mahley RW. J Clin Invest. 1983;71:816–831. doi: 10.1172/JCI110835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovanen PT, Brown MS, Basu SK, Bilheimer DW, Goldstein JL. Proc Natl Acad Sci USA. 1981;78:1396–1400. doi: 10.1073/pnas.78.3.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soltys PA, Portman OW. Biochim Biophys Acta. 1979;574:505–520. doi: 10.1016/0005-2760(79)90247-9. [DOI] [PubMed] [Google Scholar]
- 16.Pangburn SH, Newton RS, Chang CM, Weinstein DB, Steinberg D. J Biol Chem. 1981;256:3340–3347. [PubMed] [Google Scholar]
- 17.Bachorik PS, Kwiterovich PO, Cook JC. Biochemistry. 1978;17:5287–5299. doi: 10.1021/bi00617a032. [DOI] [PubMed] [Google Scholar]
- 18.Harders-Spengel K, Wood CB, Thompson GR, Myant NB, Soutar AK. Proc Natl Acad Sci USA. 1982;79:6355–6359. doi: 10.1073/pnas.79.20.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoeg JM, Demosky SJ, Jr, Schaefer EJ, Starzl TE, Brewer HB., Jr J Clin Invest. 1984;73:429–436. doi: 10.1172/JCI111229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghiselli G, Schaefer EJ, Gascon P, Brewer HB. Science. 1981;241:1239–1341. doi: 10.1126/science.6795720. [DOI] [PubMed] [Google Scholar]
- 21.Havel RJ, Eder HA, Bragdon JH. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacFarlane AS. Nature (Lond) 1958;182:53. doi: 10.1038/182053a0. [DOI] [PubMed] [Google Scholar]
- 23.Bilheimer DW, Eisenberg S, Levy RI. Biochim Biophys Acta. 1972;260:212–221. doi: 10.1016/0005-2760(72)90034-3. [DOI] [PubMed] [Google Scholar]
- 24.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 25.Edge SB, Hoeg JM, Triche T, Schneider PD, Brewer HB., Jr J Biol Chem. 1986;261:3800–3806. [PubMed] [Google Scholar]
- 26.Enat R, Jefferson DM, Rinz-Opazo N, Gatmaitan Z, Leinwand LA, Reid LM. Proc Natl Acad Sci USA. 1984:1411–1415. doi: 10.1073/pnas.81.5.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverstein SC, Steinman RM, Cohn ZA. Annu Rev Biochem. 1977;46:669–722. doi: 10.1146/annurev.bi.46.070177.003321. [DOI] [PubMed] [Google Scholar]
- 28.Heider JG, Boyette RL. J Lipid Res. 1978;19:514–518. [PubMed] [Google Scholar]
- 29.Beg Z, Stonik JA, Brewer HB., Jr Proc Natl Acad Sci USA. 1978;75:3678–3682. doi: 10.1073/pnas.75.8.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zak B. Clin Chem. 1977;23:1201–1214. [PubMed] [Google Scholar]
- 31.Zeigenhorn J. Clin Chem. 1975;24:1627–1629. [PubMed] [Google Scholar]
- 32.US Department of Health, Education and Welfare. DHEW Report Number (NIH) 1974. Lipid Research Clinical Program Manual of Laboratory Operation; pp. 75–628. [Google Scholar]
- 33.Warnick GR, Benderson J, Albers JJ, Baille EE, Sexton B, Schaefer EJ, Carlson D, Hill M, Brewer HB, Jr, Wiebe DA, Hagalhurst J, Cooper GR. Clin Chem. 1982;28:1379–1388. [PubMed] [Google Scholar]
- 34.Sprecher DL, Hoeg JM, Schaefer EJ, Brewer HB., Jr Metabolism. 1985;34:294–299. doi: 10.1016/0026-0495(85)90015-0. [DOI] [PubMed] [Google Scholar]
- 35.US Department of Health and Human Services. NIH Publication No. 80-1527. 1980. The Lipid Research Clinics: Population Studies Data Book; pp. 1–136. [Google Scholar]
- 36.Tanzawa K, Shimada Y, Kuroda M, Tsujita M, Arai M, Watanabe H. FEBS Lett. 1980;118:81–84. doi: 10.1016/0014-5793(80)81223-3. [DOI] [PubMed] [Google Scholar]
- 37.Pittman RC, Carew TE, Attie AD, Witztum JL, Watanabe Y, Steinberg D. J Biol Chem. 1982;257:7994–8000. [PubMed] [Google Scholar]
- 38.Hoeg JM, Demosky SJ, Jr, Gregg RE, Schaefer EJ, Brewer HB., Jr Science. 1985;227:759–761. doi: 10.1126/science.2982214. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein JL, Kita T, Brown MS. N Engl J Med. 1983;309:288–296. doi: 10.1056/NEJM198308043090507. [DOI] [PubMed] [Google Scholar]
- 40.Sprecher DL, Schaefer EJ, Kent KM, Gregg RE, Zech LA, Hoeg JM, McManus B, Roberts WC, Brewer HG., Jr Am J Cardiol. 1984;54:20–30. doi: 10.1016/0002-9149(84)90298-4. [DOI] [PubMed] [Google Scholar]
- 41.Sly WS, Fisher HD. J Cell Biochem. 1982;18:67–85. doi: 10.1002/jcb.1982.240180107. [DOI] [PubMed] [Google Scholar]