

Abstract
Glycogen storage disease (GSD) types I, III, and IV can be associated with severe liver disease. The possible development of hepatocellular carcinoma and/or hepatic failure make these GSDs potential candidates for liver transplantation. Early diagnosis and initiation of effective dietary therapy have dramatically improved the outcome of GSD type I by reducing the incidence of liver adenoma and renal insufficiency. Nine type I and 3 type III patients have received liver transplants because of poor metabolic control, multiple liver adenomas, or progressive liver failure. Metabolic abnormalities were corrected in all GSD type I and type III patients, while catch-up growth was reported only in two patients. Whether liver transplantation results in reversal and/or prevention of renal disease remains unclear. Neutropenia persisted in both GSDIb patients post liver transplantation necessitating continuous granulocyte colony stimulating factor treatment. Thirteen GSD type IV patients were liver transplanted because of progressive liver cirrhosis and failure. Al1 but one patient have not had neuromuscular or cardiac complications during follow-up periods for as long as 13 years. Four have died within a week and 5 years after transplantation. Caution should be taken in selecting GSD type IV candidates for liver transplantation because of the variable phenotype, which may include life-limiting extrahepatic manifestations. It remains to be evaluated, whether a genotype-phenotype correlation exists for GSD type IV, which may aid in the decision making.
Conclusion
Liver transplantation should be considered for patients with glycogen storage disease who have developed liver malignancy or hepatic failure, and for type IV patients with the classical and progressive hepatic form.
Keywords: Glycogen storage disease, Liver transplantation, Kidney transplantation, Tube feeding, Uncooked cornstarch
Introduction
Glycogen storage diseases (GSDs) are inherited disorders in which the concentration and/or structure of glycogen in body tissues is abnormal. Essentially, all known enzymes involved in the synthesis or degradation of glycogen and glucose have been discovered to cause some type of GSD [4, 12, 17]. Liver and muscle have abundant quantities of glycogen and are the most common and seriously affected tissues. The GSDs (and associated enzymatic deficiencies) affecting the liver as the major organ are types I (glucose 6-phosphatase), III (debrancher), IV (brancher), VI (liver phosphorylase), and IX (phosphorylase b kinase), as well as glycogen synthase deficiency. The hepatic GSDs vary in the age of onset, progression, other organ involvement, and clinical severity. Types I, III and IV can be associated with severe liver disease. The possible development of hepatocellular carcinoma and/or hepatic failure makes these GSDs potential candidates for liver transplantation. GSD type Ia or von Gierke disease, is caused by a deficiency of glucose-6-phosphatase activity. The clinical manifestations are growth retardation, hepatomegaly, hypoglycemia, lactic acidemia, hyperuricemia, and hyperlipidemia. A variant caused by a defect in the transport of glucose-6-phosphate (type Ib) has the additional findings of neutropenia and impaired neutrophil function, resulting in recurrent bacterial infections and oral and intestinal mucosa ulceration. Long-term complications include gout, short stature, osteoporosis, renal disease, pulmonary hypertension and hepatic adenomas, which may undergo malignant transformation [4].
During the past 20 years, major progress has been made in managing this disorder. The current treatment of type I GSD is nocturnal nasogastric infusion of glucose or orally administered uncooked cornstarch [3, 11]. With early diagnosis and initiation of treatment, the prognosis for type I GSD has improved dramatically. Normal growth and pubertal development may be expected, and patients who are now adults have fewer late complications.
GSD type III is caused by a deficiency of glycogen debranching enzyme activity. The glycogen accumulated has a structure that resembles limit dextrin (glycogen with short outer chains). Most patients with GSD type III have both liver and muscle involvement (type IIIa). However, some patients (ca. 15% of all type III cases) have only liver involvement, without apparent muscle disease (type IIIb). Hepatomegaly, hypoglycemia,hyperlipidemia, and growth retardation usually improve with age and disappear after puberty. Overt liver cirrhosis and/or hepatocellular carcinoma may occur. In patients with disease involving muscle (type IIIa), muscle weakness, though minimal during childhood, can become predominant in adults; these patients show signs of neuromuscular involvement, with slowly progressive weakness and distal muscle wasting [4]. Treatment of type III disease consists of a high-protein diet; if hypoglycemia is present, frequent meals high in carbohydrates, with cornstarch supplements or nocturnal gastric drip feeding, constitute effective therapy. Currently there is no effective treatment for the progressive myopathy or cardiomyopathy.
GSD type IV is caused by a deficiency of branching enzyme activity which results in the accumulation of glycogen with unbranched, long outer chains in the tissues. This form of GSD typically presents in the 1st year of life, with hepatosplenomegaly and failure to thrive. Hypoglycemia is rarely seen. Progressive liver cirrhosis with portal hypertension, ascites, esophageal varices, and death usually occur before 5 years of age [4]. There are however, patients who have survived without apparent progressive liver disease [10, 16]. The neuromuscular system may also be involved. The presenting symptoms are hypotonia and muscle wasting. There is no effective treatment for the disease. Severe cardiomyopathy, as the predominant symptom has been reported [20].
In this article, we review the indications for and report the long-term outcome of liver transplantation (LT) for GSD types I, III, and IV. We also define the criteria for LT in view of the recent advance in the treatment and understanding of the natural course of these diseases.
Patients and methods
GSD patients treated by LT
A total of 25 patients with GSD types I, III, and IV having received a LT are known to us. Information on 6 of these patients was only available from the literature [1, 7, 14, 18, 23], while the remaining 18 are followed by the authors [8, 9, 13, 19]. The occurrence of liver adenoma bearing the risk for malignant transformation was the reason to opt for transplantation in seven patients with GSDIa (two females: LT at ages 16.5 and 23 years respectively; five males: LT at 11.8, 18.9, 26.8, 27.1 and 27.5 years respectively) (Table 1). In four of these patients this decision was additionally based on poor metabolic control despite conventional treatment efforts. The 26.7-year-old male patient had received a combined liver and kidney transplant (case 3, Table 1). A second LT combined with a renal transplant was necessary in the 16.5-year-old female patient when she was 30.5 years of age (case 1, Table 1). Two female patients with GSDIb (7.6 and 13.8 years old) received a liver transplant solely on the basis of ineffective dietary treatment (Table 1).
Table 1. LT for GSD type I. F female; M male; KT kidney transplantation; ? not reported; N/A not applicable.
| Diagnosis | Case | Sex | Age at LT (years) | Date of LT | Indication | Outcome | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adenoma | Poor metabolic control | Liver function | Catch-up growth | Neutropenia | Complications | Survival/follow-up | |||||
| GSDIa | 1 [19] | F | 16.5 (1st LT); 30 (2nd LT, KT) | 5/1982 20/06/1996 | + | + | Normal | No | N/A | Renal failure due to cyclosporin A toxicity | 1.3 yearsa |
| 2[7] | F | 23 | 1986 | + | − | Normal | ? | N/A | None | 0.5 years | |
| 3 [18] | M | 26.7 (LT + KT) | ? | + | + | Normal | ? | N/A | Chronic rejection | 11.3 years | |
| 4 [14] | M | 27 | 02.07.1998 | + | + | Normal | Yes | N/A | None | 2 years | |
| 5 | F | 18.9 | 22/02/1991 | + | + | Normal | b | N/A | None | 6.8 years | |
| 6 | M | 11.8 | 21/12/1993 | + | + | Normal | Yes | N/A | Rejection | 3.5 years | |
| 7 [18] | M | 27 | ? | + | + | Normal | ? | N/A | Reversible IDDM | 1 year | |
| GSDIb | 8 [9] | F | 7.4 | 21/07/1991 | − | + | Normal | b | Persistent | Hemoperitoneum/thorax, CMV infection | 6.2 years |
| 9 | f | 13.8 | 18/05/1993 | − | + | Normal | No | Persistent | Rejection | 4.4 years | |
Since re-LT combined with KT
Normal growth pre- and post-LT
Three patients with GSD type III, a 32.5-year-old female (GSD IIIb) with liver cirrhosis and hepatocellular carcinoma [13], a 44-year-old male with liver failure [J. Moser, personal communication], and another female [25] received liver transplants. The indication was not reported for the latter patient.
All 13 patients with GSD type IV who had received a liver transplant and for whom information was available, were male [1, 8, 19, 23, 24] (Table 2). The two youngest were transplanted at 11 months of age and the oldest at 12 years (mean age: 3.2 years). The indication for LT was based on the occurrence of liver failure in all cases, and for one patient also on the presence of liver adenoma.
Table 2. LT for GSD Type IV. M male; ? not reported.
| Case [reference] | Sex | Age at LT (years) | Date of LT (day/month/year) | Reason for LT | Outcome | |||
|---|---|---|---|---|---|---|---|---|
| Liver failure | Adenoma | Complications/survival | Follow-up | Cardio-/myo-/neuropathy | ||||
| 1 [1] | M | 0.9 | ? | + | + | Rejection | 0.7 years | None |
| 2 [19, 24] | M | 0.9 | 28.08.1985 | + | − | Deatha | ||
| 3 [23] | M | 1.2 | ? | + | − | Deathb | Cardiomyopathy | |
| 4 | M | 1.3 | ? | + | − | None | 13.5 years | None |
| 5 [19, 24] | M | 1.7 | 21.03.1989 | + | − | − | 8.8 years | None |
| 6 [19,24] | M | 1.8 | 24.10.1987 | + | − | Deathc | ||
| 7 [19, 24] | M | 2.6 | 06.09.1984 | + | − | − | 13.2 years | None |
| 8 [19, 24] | M | 2.8 | 26.05.1989 | + | − | 8.7 years | None | |
| 9 [19, 24] | M | 3.0 | 05.09.1985 | + | − | Deathd | ||
| 10 | M | 3.7 | 22.04.1992 | + | − | None | 5.5 years | None |
| 11 [19, 24] | M | 3.8 | 25.11.1985 | + | − | 11.9 years | None | |
| 12 | M | 5.5 | 26.09.1986 | + | − | Portal vein thrombosis, rejection | 9.9 years | None |
| 13 [8] | M | 12.0 | 25.09.1993 | + | − | Re-transplant (27.09.1993)e | 3.9 years | None |
Death 7 days post-LT due to sepsis following bowel perforation
Death 9 months post-LT due to heart failure
Death 5.4 years post LT due to meningococcal sepsis
Death at 1 month post-LT following hepatic artery thrombosis
Due to non-functioning 1st donor liver
GSD type I patients treated conventionally
This population consists of 29 GSDIa patients and one GSDIb patient older than 17 years of age. All patients are being followed at Duke University Medical Center. They were divided into three groups according to age at initiation of night-time nasogastric tube feeding or cornstarch treatment. 1.) “Early”: 13 patients (including the one GSDIb patient) who were diagnosed and started on dietary treatment within the first 2 years of life (average age: 21.0 years; age range: 17.3 – 26.0 years; 8 females); 2.) “intermediate”: seven patients treated as of 2 to 15 years old (average age: 25.2 years; age range: 17.8 – 32.2 years; 4 females); and 3.) “late”: ten patients who were either diagnosed late and treatment did not begin before 15 years old and/or who were not compliant (average age: 34.5 years; age range: 25.5 – 48.4 years; 3 females). Success of treatment was assessed using the occurrence and number of adenoma and presence or level of proteinuria as parameters.
Results and discussion
GSD type I
Seven patients with GSDIa and two with GSDIb have received liver transplants (mean age 19.2 years, range 7.6–27.5 years) because of multiple liver adenomas bearing the risk of malignant transformation, and/or poor metabolic control. None, however, had carcinomas confirmed in the explanted livers. All patients are alive 0.5 to 11.3 years post-transplant (Table 1). Hypoglycemia, hyperuricemia, hyperlactic acidemia and hyperlipidemia have been corrected in all of these patients, liberating them from their strict dietary regimen. Catchup growth, however, was observed in only two patients. Normal growth before and after transplantation was reported for one GSDIa and one GSDIb patient while for two, no benefit of transplantation on growth was documented. No information on growth development was available for the remaining three patients.
Whether LT results in reversal and/or prevention of renal disease remains unclear. The first GSDIa patient to receive a liver transplant in 1982 (case 1, Table 1) [15], required a second liver transplant 14 years after the first, at age 30.5 years. This was necessary since she had developed end-stage liver cirrhosis secondary to hepatitis C, which was most likely acquired via an infected blood transfusion. By this time, she had also developed chronic renal failure (serum creatinine 1.6–3.3 mg/dl, BUN 29– 48 mg/dl, creatinine clearance 20–30 ml/min/1.73 m2, oliguria) with only mild proteinuria (192 mg/24 h), and was treated with a combined liver re-transplantation and kidney transplantation. Ultrasound had excluded nephrolithiasis and calcinosis. Renal failure was attributed to interstitial kidney disease due to cyclosporin A nephrotoxicity. This diagnosis was based on the kidney function tests and only minimal proteinuria. A renal biopsy was not performed. Renal impairment in GSD type I is usually manifested by focal segmental glomerulosclerosis of which hyperfiltration and proteinuria are early signs [5]. The second liver transplant recipient with GSD type I (case 3, Table 1) had received a combined liver and kidney transplant at age 26.7 years. One GSDIb patient (case 8, Table 1) had a decreased inulin (59 ml/min/1.73 m2; normal: 126 ± 30 ml/min/1.73 m2) and PAH clearance (369 ml/min/1.73 m2; normal: 648 ± 168 ml/min/1.73 m2) as well as increased albumin/creatinine ratio (7.5 mg/mmol; normal: <2 mg/mmol) prior to LT. Four years post-transplant, this patient's PAH (508 ml/min/1.73 m2) and albumin/creatinine ratio (3.1 mg/mmol) have improved, and the inulin clearance (60 ml/min/1.73 m2) has remained stable (A. Lachaux, personal communication). All other GSDI patients have been followed for a maximum of 6.8 years and have not presented any sign of renal disease to date.
Patients with GSDIb benefit from LT by gaining metabolic control. Neutropenia, however, may persist and infections may recur after transplantation necessitating continuous administration of granulocyte colony stimulating factor [9].
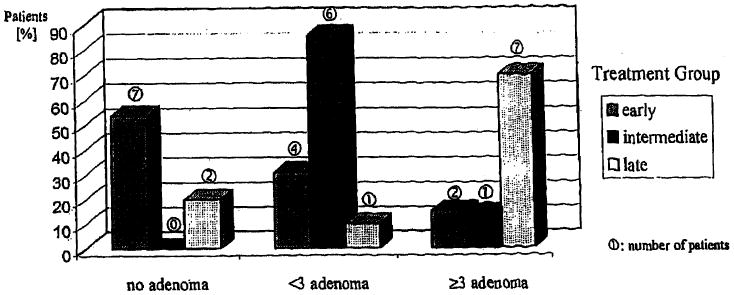
In the last 20 years, significant improvement in the diagnosis and treatment of GSD-I has been achieved. The benefit of early initiation of treatment is demonstrated in 30 GSDI patients older than 17 years. They were divided into three groups depending on the individual onset of night-time nasogastric tube feeding or cornstarch administration. These, however, small groups, were compared with respect to the presence of proteinuria and liver adenoma. It is noteworthy that only one patient in the early treatment group, who died at 20.2 years of age, had developed GSD related renal insufficiency necessitating a kidney transplant (Fig. 1). This patient had a history of recurrent urinary tract infections and pyelonephritis [5, 6]. The percentage of patients without proteinuria is highest in the early treated group. This observation is supported by the results of a recently published study on metabolic control and renal. dysfunction in GSDI patients [26]. The incidence of liver adenoma is also more rare in patients who were treated already within the first 2 years of life, which further suggests that early treatment is beneficial (Fig. 2). None of the patients had developed signs of malignant liver tumors as screened for regularly by abdominal ultrasound and computer tomography as well as determination of serum α-fetoprotein levels. However, the reliability of these tests to detect malignant transformation in an early stage has not yet been proven. LT should, therefore, be considered for those GSDI patients with findings highly suggestive of malignant liver tumors. A combined liver and kidney transplantation may be indicated when renal function is already compromised.
Fig. 1. Effect of age at treatment initiation on renal function as expressed by proteinuria.
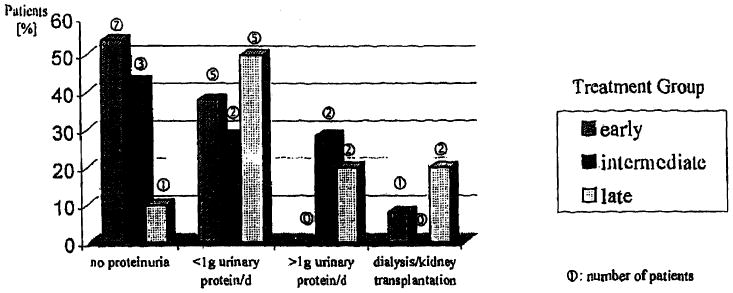
Fig. 2. Effect of age at treatment initiation on the occurrence of liver adenomas.
GSD type III
The first report on LT in GSD type III did not provide details on the patient [25]. The second report describes a 32-year-old with GSDIIIb who underwent LT because of end-stage cirrhosis [13]. A hepatocellular carcinoma was also found in the explanted liver. At 4.2 years after transplantation, the patient has normal liver function and metabolic control without any restrictions, and remains on prednisone, cyclosporin A, and azathioprine for prophylaxis of graft rejection. She continues to have no signs of myopathy or cardiomyopathy. A third patient with GSDIII was transplanted when 44 years old following the development of liver failure. His explanted liver was negative for both, benign and malignant tumors (J. Moser, personal communication).
Liver fibrosis is a common feature of GSD type III, and in some cases micronodular cirrhosis has been found. In most cases, the fibrosis/cirrhosis appears not to progress. However, overt liver cirrhosis with portal hypertension and bleeding esophageal varices have been observed. Liver adenoma can occur in GSD type III but malignant transformation has not yet been reported. Hepatocellular carcinoma in the absence of adenoma has been observed in three cases [13, 22, Y.-T. Chen, personal communication]. LT is indicated when end-stage cirrhosis and/or hepatocellular carcinoma are detected. As GSD type III is a multi-system disorder (type IIIa), the long-term success of LT particularly with regard to myopathy/cardiomyopathy is not known. A non-invasive DNA based diagnosis can be used to identify patients having only liver involvement (IIIb) [21].
GSD type IV
Clinical data are available on 13 patients who underwent LT (Table 2). All patients were male with two sets of brothers (cases 2, 6, and 5, 7). The mean age at the time of transplantation was 3.2 years (range 0.9–12 years). All patients showed progressive liver cirrhosis with evidence of portal hypertension at the time of transplantation. Two died immediately after transplantation (days 3 and 36); one died 9 months post transplantation from heart failure [23]; the other died 5.5 years post-transplant from meningococcal sepsis while on long-term immunosuppressants (prednisone, cyclosporin A). Ten patients have not had neuromuscular or cardiac complications during follow-up periods for as long as 13.5 years. Remarkably, there was a reduction in the amount of amylopectin on myocardial biopsy in one of these patients (case 5, Table 2) [24]. This was attributed to cells migrating from the donor liver to the heart, acting as enzyme couriers. The absence of cardiac symptoms in these patients contrasts to case 3 who died 9 months post LT from heart failure with pathological infiltration of amylopectin [23]. This patient had mild left ventricular hypertrophy on ECG but a normal echocardiogram prior to LT. Possibly he had irreversible cardiac involvement pre-transplantation, although this remains speculative.
Four patients with the non-progressive hepatic form of GSD type IV previously reported and now at ages 5, 7, 15, and 22 years old, continued to have normal liver function and no skeletal muscle, cardiac or neurological involvement, despite significant deficiency of branching enzyme activity [16]. Mutation analysis showed that both hepatic and neuromuscular forms of GSD-IV are caused by mutations in the same glycogen branching enzyme gene [2]. The mutant alleles in patients with the fatal neonatal neuromuscular form and with the progressive hepatic form resulted in diminished branching enzyme activity in the transient expression system. Two patients with the non-progressive hepatic form carry a missense mutant allele (Y329S), which exhibits 45% of branching enzyme activity. This mutation was not detected in 35 unrelated normal individuals or in patients with the more severe forms of GSD type IV [2]. Further study of genotype-phenotype correlations may yield useful information in predicting the clinical outcome regarding the prognosis and necessity of an eventual LT. Liver transplantation should be considered a treatment option for those patients with hepatic GSD who developed liver malignancy or failure, and for GSD type IV patients with clearly the classical and progressive hepatic form. It needs to be remembered that transplantation may only improve the hepatic aspect of GSD while in patients with GSD type IIIa or type IV, it may progress in other organs, namely the heart, skeletal muscle and nervous system, which may be deleterious. It remains to be evaluated, whether a genotype-phenotype correlation exists for GSD type IV, which may aid in decision making.
Acknowledgments
This work was supported by grants from the National Institute of Health (DK39078 to Y.T.C.; M01-RR30 from the National Center for Research Sources, General Clinical Research Centers Program) and the Muscular Dystrophy Association.
Abbreviations
- GSD
glycogen storage disease
- LT
liver transplantation
Contributor Information
D. Matern, Department of Pediatrics, Duke University Medical Center, P.O. Box 3528, Durham, NC 27710, USA. Tel.: +1-919-684-2036, Fax: +1-919-684-8944
T. E. Starzl, Thomas E. Starzl Transplantation Institute, University of Pittsburgh, Pa., USA
W. Arnaout, Center for Liver Diseases and Transplantation, Cedar's Sinai Medical Center, Los Angeles, Calif., USA
J. Barnard, Department of Pediatrics, Vanderbilt University Medical Center, Nashville, Tenn., USA
J. S. Bynon, Department of Surgery, University of Alabama, Birmingham, Ala., USA
A. Dhawan, Pediatric Liver Service, King's College Hospital, London, UK
J. Emond, Liver Transplant Program, University California San Francisco, Calif., USA
E. B. Haagsma, Department of Gastroenterology and Hepatology, University Hospital, Groningen, The Netherlands
G. Hug, Children's Hospital Medical Center, Cincinnati, Ohio, USA
A. Lachaux, Department of Pediatrics, Hopital Edouard Herriot, Lyon, France
G.P.A. Smit, Beatrix Children's Hospital, University of Groningen, Groningen, The Netherlands
Y-T. Chen, Department of Pediatrics, Duke University Medical Center, P.O. Box 3528, Durham, NC 27710, USA. Tel.: +1-919-684-2036, Fax: +1-919-684-8944
References
- 1.Alshak NS, Cocjin J, Podesta L, van de Velde R, Makowka L, Rosenthal P, Geller SA. Hepatocellular adenoma in glycogen storage disease type IV. Arch Pathol Lab Med. 1994;118:88–91. [PubMed] [Google Scholar]
- 2.Bao Y, Kishnani P, Wu JY, Chen YT. Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest. 1996;97:941–948. doi: 10.1172/JCI118517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen YT, Bazarre CH, Lee MM, Sidbury JB, Coleman RA. Type I glycogen storage disease: nine years of management with cornstarch. Eur J Pediatr. 1993;152(Suppl 1):S56–S59. doi: 10.1007/BF02072090. [DOI] [PubMed] [Google Scholar]
- 4.Chen YT, Burchell A. The glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. McGraw-Hill; New York: 1995. pp. 935–965. [Google Scholar]
- 5.Chen YT, Coleman RA, Scheinman JI, Kolbeck PC, Sidbury JB. Renal disease in type I glycogen storage disease. N Engl J Med. 1988;318:7–11. doi: 10.1056/NEJM198801073180102. [DOI] [PubMed] [Google Scholar]
- 6.Chen YT, Scheinman JI. Hyperglycaemia associated with lactic acidaemia in a renal allograft recipient with type I glycogen storage disease. J Inherit Metab Dis. 1991;14:80–86. doi: 10.1007/BF01804394. [DOI] [PubMed] [Google Scholar]
- 7.Coire IC, Qizilbash AH, Castelli MF. Hepatic adenomata in type la glycogen storage disease. Arch Pathol Lab Med. 1987;111:166–169. [PubMed] [Google Scholar]
- 8.Dhawan A, Tan KC, Portmann B, Mowat AP. Glycogenosis type IV: liver transplant at 12 years. Arch Dis Child. 1994;71:450–451. doi: 10.1136/adc.71.5.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donadieu J, Bader-Meunier B, Bertrand Y, Lachaux A, Labrune Ph, Gougerot-Pocidalo MA, Odievre M, Gibeaud P, Yver A, Tchernia G, griscelli C, Dommergues JP. Recombinant human G-CSF (lenograstim) for infectious complications in glycogen storage disease type Ib - report of 7 cases. N Rev Fr Hematol. 1993;35:529–534. [PubMed] [Google Scholar]
- 10.Greene HL, Brown BI, McClenathan DT, Agostini RM, Taylor SR. A new varaiant of type IV glycogenosis: deficiency of branching enzyme activity without progressive liver disease. Hepatology. 1988;8:302–306. doi: 10.1002/hep.1840080219. [DOI] [PubMed] [Google Scholar]
- 11.Greene HL, Slonim AE, O'Neil JA, Burr IM. Continuous nocturnal intragastric feeding for management of type 1 glycogen storage disease. N Engl J Med. 1976;294:423–425. doi: 10.1056/NEJM197602192940805. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes J, Chen YT. The glycogen storage diseases. In: Fernandes J, van den Berghe G, Saudubray JM, editors. Inborn metabolic diseases – diagnosis and treatment. Springer Verlag; Berlin: 1995. pp. 71–87. [Google Scholar]
- 13.Haagsma EB, Smit GPA, Niezen-Koning KE, Gouw ASH, Meerman L, Slooff MJH. Type IIIb glycogen storage disease associated with end-stage cirrhosis and hepatocellular carcinoma. Hepatology. 1997;25:537–540. doi: 10.1002/hep.510250307. [DOI] [PubMed] [Google Scholar]
- 14.Kirschner BS, Baker AL, Thorp FK. Growth in adulthood after liver transplantation for glycogen storage disease type I. Gastroenterology. 1991;101:238–241. doi: 10.1016/0016-5085(91)90483-2. [DOI] [PubMed] [Google Scholar]
- 15.Malatack JJ, Iwatsuki S, Gartner JC, Roe T, Finegold DN, Shaw BW, Zitelli BJ, Starzl TE. Liver transplantation for type I glycogen storage disease. Lancet. 1983;1:1073–1075. doi: 10.1016/s0140-6736(83)91910-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McConkie-Rosell A, Wilson C, Piccoli DA, Boyle J, DeClue T, Kishnani P, Shen JJ, Boney A, Brown B, Chen YT. Clinical and laboratory findings in four patients with the nonprogressive hepatic form of type IV glycogen storage disease. J Inherit Metab Dis. 1996;19:51–58. doi: 10.1007/BF01799348. [DOI] [PubMed] [Google Scholar]
- 17.Moses SW. Pathophysiology and dietary treatment of the glycogen storage diseases. J Pediatr Gastroenterol Nutr. 1990;11:155–174. doi: 10.1097/00005176-199008000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Poe R, Snover DC. Adenomas in glycogen storage disease type I. Two cases with unusual histologic features. Am J Surg Path. 1988;12:477–83. doi: 10.1097/00000478-198806000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Selby R, Starzl TE, Yunis E, Todo S, Tzakis AG, Brown BI, Kendall RS. Liver transplantation for type I and type IV glycogen storage disease. Eur J Pediatr. 1993;152(Suppl):S71–S76. doi: 10.1007/bf02072093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Servidei S, Riepe RE, Langston C, Tani LY, Bricker JT, Crisp-Lindgren N, Travers H, Armstrong D, DiMauro S. Severe cardiopathy in branching enzyme deficiency. J Pediatr. 1987;111:51–56. doi: 10.1016/s0022-3476(87)80341-4. [DOI] [PubMed] [Google Scholar]
- 21.Shen JJ, Bao Y, Liu HM, Lee P, Leonard JV, Chen YT. Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J Clin Invest. 1996;98:352–357. doi: 10.1172/JCI118799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimizu J, Shiraishi H, Sakurabayashi S, Sugiura F, Takizawa M, Miyazaki K, Matsumoto Y. A report on an adult case of type III glycogenosis with primary liver cancer and liver cirrhosis (in Japanese) Nippon Shokakibyo Gakkai Zasshi. 1982;12:2328–2332. [PubMed] [Google Scholar]
- 23.Sokal EM, Van Hoof F, Alberti D, de Ville de Goyet J, de Barsy T, Otte JB. Progressive cardiac failure following othotopic liver transplantation for type IV glycogenosis. Eur J Pediatr. 1992;151:200–203. doi: 10.1007/BF01954384. [DOI] [PubMed] [Google Scholar]
- 24.Starzl TE, Demetris AJ, Trucco M, Ricordi C, Ildstad S, Terasaki PI, Murase N, Kendall RS, Kocova M, Rudert WA, Zeevi A, Van Thiel D. Chimerism after liver transplantation for type IV glycogen storage disease and type I Gaucher's disease. N Engl J Med. 1993;328:745–749. doi: 10.1056/NEJM199303183281101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Superina RA, Pearl RH, Roberts EA, Phillips MJ, Graham N, Greig PD, Langer B. Liver Transplantation in children: the initial Toronto experience. J Pediatr Surg. 1989;24:1013–19. doi: 10.1016/s0022-3468(89)80205-2. [DOI] [PubMed] [Google Scholar]
- 26.Wolfsdorf JI, Laffel LMB, Crigler JF. Metabolic control and renal dysfunction in type I glycogen storage disease. J Inherit Metab Dis. 1997;20:559–568. doi: 10.1023/a:1005346824368. [DOI] [PubMed] [Google Scholar]