

Abstract
Neuroaxonal dystrophy in brainstem, spinal cord tracts, and spinal nerves accompanied by cerebellar hypoplasia was observed in a colony of laboratory dogs. Fetal akinesia was documented by ultrasonographic examination. At birth, affected puppies exhibited stereotypical positioning of limbs, scoliosis, arthrogryposis, pulmonary hypoplasia, and respiratory failure. Regional hypoplasia in the central nervous system was apparent grossly, most strikingly as underdeveloped cerebellum and spinal cord. Histopathologic abnormalities included swollen axons and spheroids in brainstem and spinal cord tracts; reduced cerebellar foliation, patchy loss of Purkinje cells, multifocal thinning of the external granular cell layer, and loss of neurons in the deep cerebellar nuclei; spheroids and loss of myelinated axons in spinal roots and peripheral nerves; increased myocyte apoptosis in skeletal muscle; and fibrofatty connective tissue proliferation around joints. Breeding studies demonstrated that the canine disorder is a fully penetrant, simple autosomal recessive trait. The disorder demonstrated a type and distribution of lesions homologous to that of human infantile neuroaxonal dystrophy (INAD), most commonly caused by mutations of PLA2G6, but alleles of informative markers flanking the canine PLA2G6 locus did not associate with the canine disorder. Thus, fetal-onset neuroaxonal dystrophy in dogs, a species with well-developed genome mapping resources, provides a unique opportunity for additional disease gene discovery and understanding of this pathology.
Keywords: brainstem, spinal cord, peripheral neuropathy, spheroid, fetal akinesia
Introduction
Neuroaxonal dystrophy (NAD) is a nonspecific, but histologically distinct, neurodegenerative pathology of the central and/or peripheral nervous system. NAD is characterized by localized swellings (spheroids) and atrophy of axons (Summers et al., 1995). NAD can occur in chronic vitamin E deficiency, insulin deficient diabetes, aging, and exposure to certain toxins (Schmidt et al., 1991, 1997). Autosomal recessive forms of NAD have been described in humans and other species, but even within a species, there is variation in age of onset, clinical manifestations, lesion distribution, and electron microscopic description of spheroids (Sisó et al., 2006). Mechanisms by which these lesions develop are unknown.
In human infantile NAD (INAD, OMIM #256600; a.k.a. Seitelberger disease) the onset of clinical signs typically occurs between 6 and 18 months of age, first showing cognitive and motor regression, hypotonia, and progressive paraplegia (Carrilho et al., 2008; Kurian et al., 2008). A few INAD patients, however, have signs of disease at birth including fetal immobility (Janota 1979;Jennekens et al., 1984; Tachibana et al., 1986; Hunter et al., 1987; Chow and Padfield 2008). Severe cerebellar atrophy and/or Purkinje cell loss is an additional pathological hallmark of human INAD and is observed in some animal NADs as well (Woodard et al., 1974; Cork et al., 1983; Carmichael et al., 1993; Bouley et al., 2006; Nibe et al., 2007). Mutations in the gene encoding phospholipase A2, group VI (PLA2G6) are found in ∼ 80 % of human INAD patients (Khateeb et al., 2006; Gregory et al., 2008; Wu et al., 2009). Engineered abrogations of Pla2g6 (a.k.a. iPLA2β) expression in mice have produced orthologous models of human INAD, but they produce late-onset disease (Shinzawa et al., 2008; Malik et al., 2008). Identification of the INAD disease gene has not yet revealed a mechanism by which axons become dystrophic because the physiologic role of PLA2G6 in the nervous system is poorly understood. At one time, deficiency of a lysosomal hydrolase, α-N-acetylgalactosaminidase (α-NAGA; Schindler disease type I), was implicated as a cause of INAD, but this conclusion has since been contradicted (Bakker et al., 2001).
Here we report a neurodevelopmental disorder in dogs characterized by fetal-onset NAD throughout the brainstem, spinal cord and peripheral nerves. The canine disorder manifests as fetal akinesia late in gestation and respiratory failure at birth, both due to lower motor neuron dysfunction, and is accompanied by cerebellar hypoplasia. The phenotype is a fully penetrant, simple autosomal recessive trait. Alleles of informative markers flanking the canine PLA2G6 locus are not associated with alleles of the disease locus in this family. Characterization of the canine disorder sets the stage for linkage mapping to determine the underlying genetic lesion and to gain further insight into the pathogenesis of neuroaxonal dystrophy.
Materials and Methods
Animals
Dogs used in this study were members of a breeding colony maintained initially at University of Pennsylvania and later at Michigan State University. All protocols for routine housing and care, breeding and whelping, cesarean sections, perfusion, and euthanasia were approved by the respective Institutional Animal Care and Use Committees of the two institutions and were designed according to the principles described in the NIH Guide for the Care and Use of Laboratory Animals. Euthanasia was performed by parenteral administration of an overdose of sodium pentobarbital.
Ultrasonography
Abdominal ultrasonographic examination was performed on trained dogs in dorsal recumbency without sedation using an Aloka 500 ultrasound system (ALOKA, Inc., Wallingford, CT). Pregnant dogs were examined between 49 and 60 days of gestation. Day of gestation was calculated in each case by monitoring changes in vaginal epithelial cytology and serum progesterone concentration to estimate the day of ovulation, a procedure that allowed prediction of the time of full-term whelping to within ± 12 hours.
Antibody characterization
Antibodies and dilutions used in this study are listed in Table 1. Anti-glial fibrillary acid protein (GFAP), anti-neuron specific enolase (NSE), and anti-calbindin antibodies were used as cell-type markers for astrocytes, neurons, and Purkinje cells, respectively. Each demonstrated cells of characteristic morphology and distribution as described previously in dog CNS tissues (Aoki et al., 1992; Sisó et al., 2003; Hwang et al., 2008; Sago et al., 2008). On western blots of newborn dog brainstem homogenate, these antibodies recognized single bands of 52, 48, and 28 kDa, respectively, as previously reported in other species (Marangos et al., 1975; Toma et al., 2001; Zhao et al., 2008).
Table 1. Primary Antibodies.
| Antigen | Immunogen | Manufacturer, species, type, cat. # | Dilution |
|---|---|---|---|
| glial fibrillary acidic protein | Purified bovine spinal cord GFAP | Dako, Carpinteria, CA, rabbit polyclonal #Z0334, lot 00015316 | 1:500 |
| neuron-specific enolase | Purified bovine brain NSE, γγ isoenzyme | Dako, Carpinteria, CA, rabbit polyclonal, #A587, lot 0045 | 1:300 |
| calbindin D-28k | Recombinant mouse 28 kDa calbindin | Chemicon International, Inc., Temecula, CA rabbit polyclonal #AB1778 | 1:2000 |
| Phosphorylated heavy and medium neurofilament proteins [SMI- 312] | Homogenized Fisher 344 rat hypothalamus | Abcam, Inc., Cambridge, MA, mouse monoclonal cocktail, #ab24574, lot 616423 | 1:1000 |
| activated caspase-3 | KLH-linked synthetic peptide CRGTELDCGIETD adjacent to Asp175 in human caspase 3 | Cell Signaling Technology, Beverly, MA, rabbit polyclonal #9661, affinity purified on immunogen column | 1:100 |
Activated caspase-3 antibody was raised against a synthetic KLH-coupled peptide (CRGTELDCGIETD) adjacent to Asp175 in human caspase-3, and is a well-defined marker of apoptosis in mammalian tissues (Ribera et al., 2002). The immunizing peptide is identical in dog caspase 3, as well as in most other mammals. The antibody recognizes 17-19 kDA fragments, but not the full-length caspase 3 on western blots of human and mouse cell line homogenates (manufacture's fact sheet). On western blots of D-17 canine osteosarcoma cells (ATCC ® cat no. CCL-183™) treated with doxorubicin, but not of untreated cells, we detected a 17 kDa band. For antigen retrieval de-paraffinized slides were incubated for 30 min at 99 C in 10 mM sodium citrate buffer, pH 6.0. Staining was abolished by preincubating the primary antibody with cleaved caspase-3 (Asp175) blocking peptide (Cell Signaling Technology, Beverly, MA; cat. no.1050), or by substituting the primary antibody with normal goat serum.
The SMI-312 antibody used here is a pan neurofilament (NF) marker that detects phosphorylated NF (Sternberger and Sternberger, 1983) in axons of fetal and newborn humans (Ulfig et al., 1998; Haynes et al., 2005). SMI-312 is a cocktail of monoclonal antibodies directed against extensively phosphorylated epitopes on axonal NF-M and NF-H. SMI-312 stained axons throughout the CNS in the pups of this study in a pattern similar to NSE staining. In particular, however, SMI-312 did not stain neuronal perikarya, as has been described, in normal pup tissue, congruent with its specificity for phosphorylated NF. On western blots of detergent extracts of newborn dog brainstem, SMI-312 detected 200 and 160 kDA bands.
None of the antibodies used exhibited residual staining when the primary antibody was omitted or replaced with irrelevant serum in the procedure. Because expression of various proteins and tissue structures develop rapidly in fetal and newborn pup CNS and skeletal muscle, sections from affected pups were assessed in comparison to sections of tissue from normal littermates, rather than by reliance on usual patterns of staining observed in older dogs.
Histopathology
For light microscopy, affected newborn and littermate control tissues were fixed by immersion in 100 mM sodium phosphate-buffered 1.25 M (4%) formaldehyde, pH 7.0 (NBF), paraffin or plastic (glycol methacrylate) embedded, and sectioned. In some animals, rapid fixation of the CNS was accomplished by NBF perfusion via the proximal aorta. Sections were obtained from all organs and all levels of the neuraxis. The entire brainstem and cerebellum of some dogs were examined in serial frontal and sagittal sections at 100 μm intervals. Routine stains included hematoxylin-eosin (H&E), luxol-fast blue, Klüver-Barrera, periodic acid-Schiff (PAS), Gomori's iron stain (Prussian blue), Holme's silver stain, Masson's trichrome, and cresyl echt violet in paraffin sections, and toluidine blue in plastic sections. For electron microscopy, tissues were fixed by overnight immersion in100 mM sodium phosphate-buffered (pH 7.2) 400 mM (4 %) gluteraldehyde, postfixed in 100 mM sodium phosphate-buffered (pH 7.2) 40 mM (1 %) osmium tetroxide, dehydrated in graded alcohols, and embedded in Poly/Bed 812-Araldite resin. Sections were cut, stained sequentially in uranyl acetate and citrate-buffered lead nitrate, and examined on a Philips 301 transmission electron microscope. Sections comparing normal and affected pup tissues were stained identically, and tissue comparisons between dogs were reproduced at identical total magnifications. Photomicrographs were taken with a Kodak DCS Pro 14n digital camera. Uneven illumination was corrected by background division in Image Arithmetic (http://www.t3i.nl/myblog/?page_id=7) and contrast and brightness were adjusted globally in Adobe® Photoshop® CS2. Multiple-image figures were assembled in the Microsoft® PowerPoint® 2000 SP-3 program and cropped, sized, and saved as TIFF files in Adobe® Photoshop® 6.0
Statistics
Significance of quantitative data comparing organs from affected pups and normal littermates was determined using Student's t-test. Significance of data from prospective breeding experiments was determined using the χ2 (Chi squared) test for goodness-of-fit. Where numbers or diameters of axons in a nerve are reported, they were determined on only one side of an affected or normal pup, rather than on both sides of the same individual.
Analysis of molecular markers
The canine PLA2G6 locus was located on dog chromosome 10 (chr10:29,581,962-29,631,860) by BLAT search (Kent, 2002) of the May 2005 assembly (http://genome.ucsc.edu/) using the human cDNA (NM_003560.2) as query sequence. Polymorphic markers flanking the locus and separated by 15.34 Mb were analyzed in a subset of dogs in the pedigree. Primers for FH2293 were 5′-GAATGCCCTTCACCTTGAAA-3′ and 5′-AGGAAAAGGAGAGATGATGCC-3′. Primers for C10.781 were 5′-ACCTCCAAGATGGCTCTTGA-3′ and 5′-ACGTCGAGCTCCTGGCAT-3′. PCR conditions were those recommended by Richman et al (2001) and Mellersh et al (1997). Fluorescent labels on the forward primers allowed electrophoresis and allele calling from standard capillary electrophoresis, performed by a core facility.
Results
Clinical findings
In a dog-breeding colony maintained for investigation of inherited disorders (He et al., 2003), certain matings produced a minority of offspring exhibiting characteristic malposition of limbs, scoliosis (fig. 1, panels A and B, respectively), and death at birth due to inspiratory failure. Axial and appendicular joints were fixed (arthrogryposis) at birth preventing voluntary movement, though some affected pups retained slight lateral movement of the tail and gaping motions of the mandible. Jaw motion was interpreted to be part of an inspiratory reflex, but there was no coordinated excursion of the thoracic wall or diaphragm, and the lungs did not inflate spontaneously. When positive pressure ventilation (PPV) was applied, however, the thorax expanded, the diaphragm was displaced abdominally, and the lungs inflated. Tissues of the mouth and paws that were cyanotic before PPV turned pink and stayed so as long as PPV was maintained, indicating that pulmonary gas exchange and peripheral blood circulation were intact. The heart contracted, and blood circulated for many minutes after birth in affected pups, even when PPV was not applied, but no affected pup ever developed spontaneous respiration.
Figure 1.

Abnormal morphology at birth. Panel A shows the ventral view of a puppy affected with fetal-onset neuroaxonal dystrophy (FNAD) demonstrating the invariant and locked position of limbs. Panel B shows a dorsal view of the partially dissected cervical to lower lumbar segments of spinal cord lying in the scoliotic vertebral column. The pup's head is to the top of both panels.
Affected puppies were detectable by abdominal ultrasonographic and/or radiographic examination of the pregnant dam several days before birth, (canine gestation is 63 days from the day of ovulation). During ultrasonographic examinations performed variously 10-2 days prior to birth, normal fetuses responded to proximity of the ultrasound probe with increased movements of limbs and trunk, but some littermates did not respond with movement. Cardiac morphology and contraction rate appeared normal in the immobile fetuses, as did all other abdominal and thoracic organs. Scoliosis in several affected fetuses was observed by abdominal radiograph of the dams at 56 days of gestation, suggesting that fixation of axial joints had already occurred in this individual. Two fetuses were immobile when examined by ultrasound at 57 days of gestation, and their joints were fixed when obtained by cesarean section at 60 days of gestation. In contrast, at 53 days of gestation, the joints of 5 affected pups obtained by cesarean section retained passive mobility (clinical status determined by histopathologic examination retrospectively). Pterygia, indicative of joint immobility from an early stage of development (Davis and Kalousek 1988; Cox et al., 2003), were never observed in affected pups. In this disorder, therefore, immobility and subsequent fixation of joints likely occurs late in gestation.
Postmortem findings
At birth, all affected pups exhibited multiple contractures of axial and appendicular joints. Examination did not reveal airway blockage. Severing the phrenic nerve caused immediate spasmodic contraction of the ipsilateral diaphragm in normal littermates sacrificed at birth, but not in affected pups, suggesting that failure to inspire was due to lack of innervation or intrinsic dysfunction of respiratory muscles. Despite a wide range in each group, the mean birth weight of affected puppies (193 ± 36 g, mean ± SD, range 124-288 g; n = 26) was less (p<2×10-5) than that of normal littermates (237 ± 38 g, range 149-316 g; n = 38). Total lung wet weight was significantly less in the affected pups (3.8 ± 0.9 g vs. 8.6 ± 1.7 g; p<8×10-11) even when normalized to body weight (0.021 ± 0.005 vs. 0.036 ± 0.007; p<7×10-9), indicative of pulmonary hypoplasia. The difference in lung weight was far greater than could be attributed to increased blood content likely present in the normal pups' lungs.
Gross morphologic abnormalities of other internal organs were confined to the CNS. Cerebral hemispheres of affected pups showed mild generalized volume reduction compared to normal littermates euthanized at birth, but otherwise demonstrated no malformation or encephaloclastic lesions. Serial coronal sections of the cerebral hemispheres showed well-developed gray and white matter structures in both affected and control pups, and the extent of myelination was similar. The weight of the formalin-fixed cerebrum with diencephalon of affected pups was ∼ 75% of that of normal littermates sacrificed at birth (5.2 ± 0.74 g, n = 19 vs. 6.8 ± 1.1 g, n = 10; p<0.002). The brainstem of affected animals was similarly reduced in weight (∼75%) but also without focal lesions or gross evidence of malformation. Strikingly, however, the cerebellum in affected dogs was markedly reduced in size compared to those in control animals (fig. 2, panels A and C), with a rudimentary folial pattern in both the cerebellar vermis and lateral cerebellar hemispheres. Weight of the formalin-fixed cerebellum of affected pups was less than 50% of littermate controls (0.15 ± 0.02 g, n = 19 vs. 0.32 ± 0.06 g, n = 10; p<10-5). Throughout its length, the cross-sectional area of spinal cord in affected pups was ∼ 35% that of normal controls, and the reduction in area affected white and grey matter nearly equally (fig. 2, panels B and D). Dorsal and ventral spinal roots and peripheral nerves of affected pups were reduced in diameter and more translucent than those of control pups.
Figure 2.

Cerebellar and spinal cord hypoplasia. Panels A and C show the left lateral view of cerebellum and brainstem of a newborn normal pup and an affected littermate, respectively. In each panel the fat arrow indicates the caudal colliculus, and the arrowhead indicates the obex (bar in panel C = 5 mm, and panels A and C are presented at the same total magnification). Panels B and D show cross-sections of spinal cord cervical segment 6 of a newborn normal pup and an affected littermate, respectively (bar in panel D = 500 μm, and panels B and D are presented at the same total magnification). There was a similar degree of hypoplasia throughout the length of affected pup spinal cord.
Histological abnormalities of the CNS were apparent by light microscopy of routinely stained sections, but lesions were confined to specific nerve tracts and nuclei. Specifically, no microscopic differences between affected pups and controls were detected in the cerebral hemispheres. The neuronal layers were formed similarly in both groups, including the placement of large pyramidal neurons. In frontal lobe coronal sections, the frontal neocortex, cingulated gyrus, corona radiata, caudate, and putamen showed intact neuronal populations. More posteriorly, the cerebral neocortex, hypothalamus, optic nerves, mamillothalamic tract, crus of fornix, hippocampus, parahippocampal gyrus, habenular nucleus, pituitary, subthalamic nucleus, and zona incerta were anatomically intact and devoid of pathological lesions in affected and control pups. Active myelination (“myelination gliosis”) was present within the white matter, and neither qualitative nor quantitative differences in glial acidic fibrillary protein (GFAP) immunoreactivity were detected in the cerebral hemispheres of affected pups and controls.
In contrast to the above, there was extensive pathology in the cerebellum, brainstem, spinal cord and peripheral nerves. Brainstem sections in affected animals were remarkable for widespread neurodegeneration and the presence of swollen axons/spheroids (neuroaxonal dystrophy) in nuclei and tracts of the extrapyramidal components of the motor system (fig. 3). In longitudinal section, axonal swellings observed in the CNS were 5-12 μm in diameter, tapered at both ends, and 20-120 μm long. They were eosinophilic on H&E, blue/grey on cresyl violet, blue on Klüver-Barrera, grey/black on Holmes' silver, red on PAS stains before and after diastase digestion, and were almost unstained on the hematoxylin counterstain used during immunohistochemical staining. In cross-section, dystrophic axons were round, and they were variously homogeneous, palely vacuolated, or more intensely stained centrally on H&E, PAS, and Holmes' silver stained sections. The identity of spheroids as swollen axons was confirmed by intense immunostaining for neuron-specific enolase (NSE, fig. 3, panel E) and absence of staining for GFAP (fig. 3, panel F). Some, but not all, dystrophic axons in the brainstem and spinal cord stained intensely for phosphorylated neurofilaments (fig. 3, panel D). Electron microscopy of dystrophic brainstem axons revealed axon-filling accumulations of small, pleomorphic, membrane-bound vesicles containing variously electron-dense fragments of organelles and amorphous material (fig. 3, panels G and H). Some mitochondrial fragments were contained in double membrane vesicles and were in various stages of degeneration, suggestive of ongoing mitophagy. There were whorls of disorganized intermediate filaments among the vesicles in some spheroids, consonant with the variable neurofilament immunostaining indicated above.
Figure 3.

Staining, antigenic, and morphologic characteristics of dystrophic axons. Axonal swellings stained pink with H&E (panel A, red nucleus) and PAS (panel B, rostral cerebellar peduncle shown, stained after diastase digestion), and grey to black with Holme's silver stain (panel C, arrowhead indicates a spheroid in the rostral cerebellar peduncle). Immunohistochemical staining was either strongly positive or negative (arrowheads) when the primary antibody (SMI 312) was directed at phosphorylated neurofilaments (panel D). Panels E and F show serial cross-sections of the ventral medial funiculus of cervical spinal cord segment 6 immunostained for neuron-specific enolase (NSE) and GFAP, respectively. Dystrophic axons throughout the CNS uniformly stained positive for NSE but never for GFAP (see also fig. 4). Bars in panels A-F = 20 μm. Transmission electron microscopy of dystrophic axons in the red nucleus (panel G) and medullary tegmentum (panel H) showed accumulations of membrane-bound vacuoles containing variably electron-dense amorphous material or organelles in various stages of degeneration (bars in panels G and H = 1 μm.
Neuroaxonal dystrophy was prominent throughout the mesencephalic, pontine, and medullary tegmentum. Swollen axons were observed in lateral and medial ventral thalamic, red, and caudal olivary nuclei; the cerebellorubral tract, the rubrospinal tract, the mesencephalic tract of the trigeminal nerve, and rostral and caudal cerebellar peduncles; and diffusely distributed in the reticular formation (fig. 4). They were particularly obvious at midline decussations of the rostral and caudal cerebellar peduncles and the myencephalic reticular formation. The medullary reticular formation was conspicuously devoid of intact large neuron perikarya of the gigantocellular tegmental field in affected pups, with only remnants of cells remaining in normal positions. There were signs of neuronal degeneration and dystrophic axons in cranial nerve (CN) nuclei and tracts, respectively, of CN V, VII, IX, X, XI, and XII. These lesions were not observed in the cerebellar white matter, crus cerebri, pontine corticospinal tract, colliculi, pyramidal tracts, rostral olives, pontine nuclei, or CN III, IV, and VI. Iron deposition was not observed in the brainstem or any other part of the CNS.
Figure 4.

Brainstem neuroaxonal dystrophy and astrocytic reaction. Transverse sections through the red nucleus and nucleus of CN III (panels A and B), genu of CN VII nerve tract and nucleus of CN VI (panels C and D), nucleus and ascending fibers of CN VII (panels E and F), and caudal olive at the level of the obex (panels G and H) are shown. Panels A, C, E, and G were H&E stained; panels B, D, F, and H were GFAP immunostained (bars in all panels = 100 μm). In each panel, filled arrowheads point to examples of degenerating neuronal perikarya, and open arrowheads indicate swollen axons or spheroids. The roman numerals in some panels indicate cranial nerve nuclei. The dotted lines in panel B outline the apparently normal tract of CN III passing through the red nucleus. GFAP staining was greater than normal in panels B and D but was no different from staining in control pup sections in panels F and H.
In comparison to controls, the affected pup brainstem sections exhibited variably increased GFAP staining and astrocyte hypertrophy in the areas of the red nucleus, the decussation of the cerebellorubral tract, the transverse pontine fibers, throughout the medullary tegmentum, ascending and decussating fibers and the septae of the caudal olives, all along the course of CN VII, and throughout the nucleus ambiguous (fig. 4). Areas that were consistently spared included the pyramids, superior olives, and the pontine nuclei. Despite widespread morphologic evidence of apoptotic cells (condensed, fragmented nuclei and darkly eosinophilic cytoplasm), there was very little activated caspase-3 staining of brainstem neurons and no more so in affected than in control pup sections.
Sections of cerebellar cortex, including vermis and hemispheres, from affected pups were remarkable for reduced foliation, decreased neuronal precursor populations in the external granular cell layer, and decreased Purkinje cells and internal granular neurons (fig. 5). Purkinje cell loss was patchy; many were degenerating or absent, but a subset of residual Purkinje cells were present in their usual position between the external and internal granular cell layers. Calbindin immunostain also revealed a dearth of Purkinje cell axons in the arbor vitae. None of the routine or immunohistochemical stains applied revealed swollen axons/spheroids in the cerebellum. The external granular cell layer varied in thickness from complete absence in some areas up to 35 microns in others, while the external granular layer in control pups was of uniform thickness throughout, at approximately 40 microns. Affected and normal pups showed approximately equal numbers of mitotic figures in the external and internal granular layers and of activated caspase-3 stained cells in the internal granular layer and deeper in the arbor vitae. Similarly, in both affected and normal pups there was exuberant GFAP staining of astrocytes throughout the arbor vitae with fine extensions through the internal granular, Purkinje, and external granular cell layers. In contrast, only affected pups showed morphologic or activated caspase-3 evidence of increased Purkinje cell apoptosis, and only in affected pups were there patches of astrocyte hypertrophy in the Purkinje and external granular layers (fig. 5, panels F and H). Positions of neurons of (deep) cerebellar nuclei were often observed as holes in the neuropil or mere remnants of cells (fig. 5, panel J). Remaining neurons in these nuclei exhibited pyknotic nuclear fragmentation and deeply eosinophilic cytoplasm on H&E stained sections. Some, but only a small subset of cells with such morphologic evidence of apoptosis also stained for activated caspase-3. GFAP staining was attenuated in the deep nuclei of normal pups relative to the surrounding neuropil but was somewhat increased in the deep nuclei of affected pups.
Figure 5.

Cerebellar histopathology. Tissue sections derived at birth of normal littermates (panels A, C, E, G) and FNAD affected pups (panels B, D, F, H, I, and J) are shown. Panels A-H and I show near-midline sagittal sections of cerebellum with the dorsal surface to the top and rostral to the left of each panel. Panels A-D show sections from pups taken by cesarean section at 60 days of gestation; other panels are from full-term pups. Sections in panels A and B are cresyl violet stained, (bars = 1 mm) and in panels C and D are calbindin immunostained (for Purkinje cells) with cresyl violet counterstain (bars = 200 μm). Sections in panels E and F are activated caspase-3 immunostained. Adjacent sections in panels G and H are GFAP immunostained. Panel I shows an H&E stained section of affected pup cerebellum adjacent to sections in F and H, and panel J shows an H&E stained transverse section through the dentate nucleus (bars in E-J = 100 μm).
In spinal cord, the affected pups had fewer intact neurons in the lateral and ventral horns and dorsal root ganglia at all levels of the cord when compared to littermate controls (fig. 6, panels A and B). The neurons that remained were in various stages of degeneration as evidenced by chromatolysis with eccentric nuclei or overall light staining (ghost cells), occasional apoptotic morphology, and some neurons that were surrounded by gliotic tissue. Swollen axons and spheroids similar to those in the brainstem were seen at all levels of the spinal cord white matter and occasionally in gray matter. In the cranial cord, these were in all funiculi but most consistently observed in the fasciculi gracilis and cunteatus and the spinal tract of the trigeminal nerve. More caudally, spheroids were most often observed in the lateral and ventral funiculi. Compared to controls, there were astrocyte hypertrophy and increased GFAP-immunoreactive cell processes throughout the lateral and ventral horns of spinal cord gray matter (fig. 6, panels C and D). Neurons exhibiting activated caspase-3 staining were rare in affected pup sections of cord but were not observed at all in normal littermate sections.
Figure 6.

Spinal cord histopathology. Transverse sections of spinal cord segment C6 of a normal littermate (panels A and C) and an FNAD affected pup (panels B and D) at birth are shown with Klüver-Barrera stain (panels A and B) or GFAP immunostain (panels C and D; bars in panels A-D = 500 μm). Transverse sections of spinal cord segment L5 dorsal roots (panels E and F) and ventral roots (panels G and H) of a normal littermate (panels E and G) and an FNAD affected pup (panels F and H) at birth are shown (toluidine blue; bars = 20 μm). The greatly enlarged dorsal root axon of panel F is shown at higher magnifications in panels I (bar = 2 μm) and J (bar = 500 nm). Panel J shows the area bounded by the dashed line box in panel I.
In order to observe earlier stages of disease progression in the CNS, two pregnancies were interrupted by cesarean section at 53 and 60 days of gestation, respectively, and fetal tissues were examined. The same cerebellar and brainstem abnormalities observed in full-term affected pups were seen in affected fetuses, although there were fewer swollen axons and less neuronal degeneration. Delayed cerebellar development was already evident at 53 days of gestation, but it appeared that the folia had developed further in the full term affected pups. In these, the youngest affected fetuses examined, neurons of the deep cerebellar nuclei were present, but signs of neurodegeneration were already evident. The spinal cord of affected pups was already reduced in diameter to ∼70 % of normal controls at 53 days of gestation. The large neuronal perikarya of the ventral horn were in appropriately placed groups but already showed some mild degenerative changes, and there was increased astrocytosis in the lateral and ventral gray matter.
Dorsal and ventral spinal nerve roots in affected pups examined at birth were remarkable for the paucity of myelinated axons (fig. 6, panels E-H). For instance, at the L5 segment, an affected pup dorsal root had fewer than 50% of the myelinated axons of a normal littermate (1806 vs. 3793), and the number of ventral root axons was reduced to ∼ 60% of normal (3156 vs. 5305). Numbers and morphology of Schwann cell nuclei appeared normal, and myelin sheaths had normal appearance. Dystrophic axans were present in spinal roots of affected pups as well (fig. 6, panels F, I, and J). Electron microscopy of dorsal roots demonstrated some hugely dilated axons (5-15 μm diam. vs. 1-3 μm diam. of surrounding axons in affected pups, as well as in normal littermates). They had intra-axonal accumulation of disordered neurofilaments, degenerating mitochondria, and small vacuoles containing amorphous material, but the myelin sheaths appeared normal.
Spheroids were also observed in peripheral nerves and intramuscular nerve branches of affected pups (fig. 7). In the peroneal nerve, there were fewer myelinated axons overall (1235 ± 75 vs. 3051 ± 273), and the average diameter of remaining axons (excluding the obviously swollen axons with diameters > 6 μm) was less than that of normal pups (1.5 ± 0.5 μm vs. 2.8 ± 1.1 μm). In the phrenic nerve of affected pups, myelinated axon number was better preserved (1053 ± 65 vs. 1308 ± 27), but average axon diameters were again reduced (0.9 ± 0.3 μm vs. 1.5 ± 0.5 μm) (fig. 7, panels A and B). Myelin sheaths appeared normal in the peripheral nerves examined.
Figure 7.

Peripheral nerve and skeletal muscle histopathology. Transverse sections of phrenic (panels A and B) and sciatic (panels C and D) nerves and semimembranosus muscle (panels E-H) of a normal littermate (panels A, C, E, and G) and an FNAD affected pup (panels B, D, F, and H) at birth. Panels A and B are toluidine blue stained (bars = 20 μm), and panels C and D are toluidine blue-carmine red stained (bars = 30 μm). Panels E and F are immunostained for activated caspase-3, and panels G and H are H&E stained (bars = 50 μm).
In skeletal muscle of both affected and normal pups observed at birth, most muscle cells were small, though in some areas they varied in size with occasional hypertrophic cells, and some had central nuclei. In both, there was a mixture of myotubes and myofibers, and there were appropriate numbers of muscle spindles. Enzymatic histochemical fiber typing revealed almost entirely type II fibers, as previously described in newborn dog muscle (Braund and Lincoln 1981; Shelton et al., 1988). In affected pup muscle, however, there was increased space between fibers, sporadic or groups of small fibers, and increased numbers of fibers showing pyknotic and fragmented nuclei, suggestive of apoptosis (fig. 7, panels G and H). This was confirmed by observation of increased numbers of myocytes in affected pup muscle staining positively for activated caspase-3 (7.4 ± 3.4/high power field vs. none observed in normal littermates) (fig. 7, panels E and F).
Microscopic examination of the stifle in affected pups revealed multiple abnormalities previously described as secondary to joint immobilization (Akeson et al., 1987). The patellar tendon was 50-60 % the thickness of that in normal littermates, and femoral and tibial cortices were thinned. The origin of the patellar tendon exhibited disorganization of collagenous fibers, and its insertion on the tibia was compromised by loss of Sharpey's fibers and osteoclastic resorption of bone. There was early-stage proliferation of fibrofatty connective tissue within the joint space and among tissues caudal to the joint.
Clinical genetics
The pedigree of dogs exhibiting the constellation of clinical and neuroanatomic abnormalities described above is shown in figure 8. An affected pup first appeared in a litter produced by a brother-sister mating between offspring of a mating between a purebred giant schnauzer and a beagle. Subsequent to outcross matings of presumptive carriers to unrelated mongrel dogs (M1-M3), affected pups were born in litters with < 3 % neonatal mortality of other causes. All offspring were examined at birth. The gross phenotype of affected pups, as described above, was consistent and unambiguous. In the 33 prospective matings of this pedigree, 59 affected puppies, 34 male and 25 female, were among 230 total offspring produced in matings between obligate carriers. These results (25.7 % affected pups with even gender distribution) were not statistically different from results expected under the hypothesis of simple autosomal recessive inheritance of a fully penetrant trait (χ2=0.82, df=1, p>0.36 for affected vs. normal clinical status; χ2=0.24, df=1, p>0.62 for gender distribution of affected offspring). These data indicate also that there was little or no embryonic or fetal loss of affected pups. Also of note is that the ratio of affected to total pups did not differ between matings in which alleles from different genetic backgrounds were segregating (26.4% vs. 25.4% on the left and right sides, respectively, of the pedigree in fig. 8).
Figure 8.
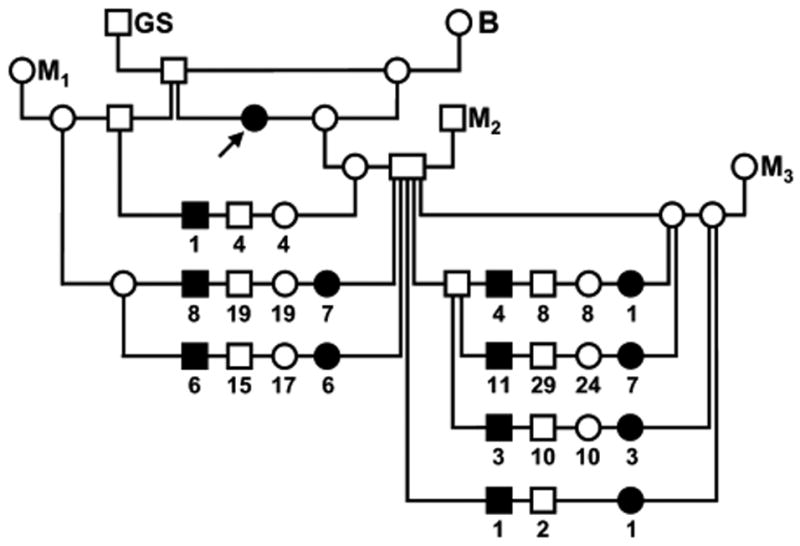
Canine FNAD pedigree. Circles indicate females, squares indicate males, filled symbols indicate pups exhibiting the FNAD phenotype and open symbols indicate phenotypically normal dogs. Offspring of matings are arranged on a horizontal line connecting vertical lines descending from symbols for the sire and dam. Numbers below symbols indicate the number of offspring of the sex and phenotype indicated produced in multiple matings of the same parents. The arrow indicates the proposita. Dogs indicated GS and B were a purebred giant schnauzer and beagle, respectively, and M1-M3 were three unrelated mongrels.
Molecular genetics
Eighty percent of human INAD patients exhibit mutations at the PLA2G6 locus on chromosome 22. Therefore, we considered the canine orthologue as a candidate gene for FNAD in this family. Polymorphic markers flanking the canine PLA2G6 locus (chr10: 29.58-29.63 Mb; CanFam2 assembly May 2005) were examined in a subset of the canine pedigree shown in figure 8. The obligate carriers, F274 and F284, were heterozygous at both markers (fig. 9). Thirty-six offspring from matings of these dogs were genotyped. As expected for markers separated by ∼15 Mb, the marker alleles recombined on 7 of 72 chromosomes (9.7 %). At least one affected pup exhibited each of the four possible unrecombined genotypes. Therefore, no hypothesized allele of PLA2G6 was uniformly homozygous in affected pups, a criterion imposed by the simple autosomal recessive inheritance of this disorder observed in the breeding experiments described above. PLA2G6 was excluded as a candidate disease gene in canine FNAD.
Figure 9.

PLAGA2 is excluded from the canine FNAD locus. Alleles of markers flanking the canine PLAGA2 locus were examined in grandparents, parents and 36 offspring. Pedigree symbol conventions are as for figure 8, and marker genotypes of each grandparent or parent are below their respective symbols. Parental haplotype phase was deduced from grandparent genotypes. The table indicates the number of normal and affected offspring, respectively, that exhibited each of the nonrecombinant haplotypes or recombination on one chromosome. At least one affected pup exhibited each of the possible haplotype pairs, thus excluding the locus bounded by these markers from the disease locus.
Discussion
In this report, fetal akinesia of affected pups, documented in utero, led to multiple joint contractures, pulmonary hypoplasia, and failure of respiration at birth. It is well demonstrated that any condition restricting fetal movement in utero may result in a constellation of morphologic abnormalities, including intrauterine growth retardation, facial anomalies, immobile joints, and limb malposition (Moessinger, 1983; Hall, 1986, 1997; Hageman et al., 1987; Folkerth et al., 1993; Bürglen et al., 1996; Riemersma et al., 1996; Brownlow et al., 2001). Such conditions can be extrinsic to the fetus (e.g. oligohydramnios) or intrinsic lesions (e.g. toxic, infectious, or genetic) that affect the kinesthetic pathways, motor neurons, the neuromuscular junction, or skeletal muscle. If fetal respiratory movements or swallowing are inhibited, the condition additionally results in pulmonary hypoplasia and polyhydramnios, respectively. Arthrogryposis multiplex congenita, multiple congenital contractures, Pena-Shokeir syndrome/phenotype, and most recently, fetal akinesia deformation sequence (FADS) have variously been used to describe these signs of etiologically heterogeneous fetal-onset disorders (Porter, 1995). At present, it is clear that when the FADS constellation of abnormalities is recognized in a patient, it is a description, rather than a diagnosis, and begs the question of cause. Therefore, fetal akinesia, joint contractures, pulmonary hypoplasia, and respiratory failure observed in the affected pups described here are interpreted as secondary features of motor unit dysfunction.
The etiology of fetal akinesia in this family is an inherited lesion that causes NAD and degeneration of spinal sensory and motor neurons, among others. It is a fully penetrant, autosomal recessive disorder. This family has genetic contributions from 5 unrelated dogs of widely different backgrounds, yet we observed no apparent alteration of the phenotype by modifying loci. The canine disorder is clinically and developmentally similar to the earliest-onset (fetal) cases of human INAD, described as connatal (Chow and Padfield 2008), but it is not caused by mutation of the canine PLA2G6 locus. We prefer the term fetal-onset neuroaxonal dystrophy (FNAD) to describe the canine disorder as well as those of human patients whose clinical signs at birth indicate onset of this pathologic process during fetal life.
We suggest that a primary or secondary effect on motor neurons causes a failure to establish or, more probably, a loss of skeletal muscle innervation and the ensuing motor dysfunction observed as fetal akinesia. Our interpretation of the histopathology is that affected pups develop normally for a period, probably through the second trimester, but subsequently experience degeneration and dysfunction of a subset of CNS cells during the late fetal period. At birth the disorder is defined by multifocal neuroaxonal dystrophy with delayed or arrested development of specific CNS structures and accompanied by lower motor neuron degeneration, skeletal muscle dysfunction, and the consequent joint remodeling caused by immobility.
To our knowledge, there is no other animal model of FNAD. Spontaneously occurring familial disorders characterized by neuroaxonal dystrophy have been described in various dog breeds (Cork et al., 1983; Griffiths et al., 1986; Sacre et al., 1993; Diaz et al., 2007; Jäderlund et al., 2007; Nibe et al., 2007, 2009), cats (Carmichael et al., 1993), sheep (Harper et al., 1991), horses (Miller and Collatos, 1997), and mice (Bouley et al., 2006). Each of these differs in at least some aspect of distribution of CNS lesions, integrity of myelin sheaths, or spheroid ultrastructure from what we report here. It is difficult, however, to make direct comparisons of the lesions because the neuronal pathology of the affected FNAD pups begins in utero, and the resultant motor dysfunction is lethal at birth. In all other descriptions of NAD in animals the onset of disease signs is at least some months after birth. The exceptions are those few human INAD patients that show signs at birth or prenatally. In the pathologically related human disorders, including idiopathic neurodegeneration with brain iron accumulation (NBIA) type 1 (OMIM #234200) and type 2 (OMIM #610217), the onset of disease is typically not until several years of age. Nor was iron accumulation observed in brainstem of the FNAD pups.
Canine FNAD affects neurons throughout the cerebellum, brainstem, spinal cord, and peripheral nerves, but the neuronal layers and nuclei of major CNS structures are in correct positions. The bulk of pathology occurs in synaptically connected neurons of the extrapyramidal motor control system, including proprioceptive input via sensory neurons and tracts, and in cranial and spinal nerves that carry axons of lower motor neurons to other than extra-ocular muscles. One possible scenario is that sensory input is lost initially, and neurons in the motor coordinating pathways and reflex arcs subsequently lose synaptic activity or trophic molecules needed to maintain their integrity during development. Alternatively, the distribution of pathology may be due to a cell-autonomous defect that is most devastating to the particular subset of neurons involved. Most probably, however, the totality of observed pathology results from a mixture of mechanisms.
NAD is often attributed to disturbance of axonal transport (Coleman 2005; Saxena and Caroni 2007; DeVos et al., 2008), but in most cases causality has not been demonstrated. Additionally, swollen axons and spheroids are a feature of the neuropathology observed in mice expressing engineered or spontaneous mutations of genes whose products mediate basal macroautophagy and/or ubiquitin-mediated protein turnover (Saigoh et al., 1999; Komatsu et al., 2006, 2007; Hara et al., 2006). Studies in the Lurcher mouse, a model of excitotoxic neurodegeneration, indicate that an early response to axonal dystrophy in Purkinje cells is induction of autophagy to protect axons under metabolic stress (Wang et al., 2006). Whether disturbances of axonal transport overwhelm basal autophagy and/or ubiquitin-mediated protein turnover in neurons and lead to accumulation of axonal contents as spheroids, or whether these homeostatic processes interact in another way, remains to be determined.
The occurrence of dystrophic axons in the canine disorder is only partially coincident with astrocytic reaction or gliosis, suggesting that there may not be a cause and effect relationship and/or that dystrophic axons do not cause exuberant inflammation and are not rapidly cleared. The most consistent association between astrocytosis and signs of neurodegeneration (shrunken cells, eccentric nuclei, and chromatolysis) was in the caudal olives and ventral and intermediate horns of spinal cord, suggesting a somewhat different degenerative mechanism in cells of those structures. A similar lack of coincident pathologies was that most neurons exhibiting morphologic evidence of apoptosis did not stain for activated caspase-3. However, all of these observations are essentially “snapshots” of ongoing pathologic processes taken at birth, and any lack of association may simply indicate temporally discrete stages of pathology in individual cells.
Pontocerebellar hypoplasia type 1 (PCH 1; a.k.a. pontocerebellar hypoplasia with spinal muscular atrophy; OMIM # 607596) is another autosomal recessive, fetal-onset disorder of CNS development with similarities to canine FNAD. In human PCH 1, the major subdivisions of the cerebellum are usually intact, albeit small, and there is loss of Purkinje and granular cells. Degeneration of the inferior (caudal) olivary nuclei, massive loss of ventral pontine neurons, and reactive gliosis in the brainstem are prominent. Lower motor neuron dysfunction is the feature that causes early lethality and distinguishes PCH 1 from all other types of PCH (Barth, 1993, 2000). In the earliest-onset cases of PCH 1, fetal akinesia leads to prominent morphologic abnormalities of the FADS complex. Death typically ensues in the immediate postnatal period due to generalized hypotonia and respiratory insufficiency requiring ventilatory support (Gorgen-Pauly et al., 1999; Muntoni et al., 1999; Ryan et al., 2000). The genetic basis of PCH 1 is unknown and most likely heterogeneous (Barth, 1993, 2000; Rudnik-Schöneborn et al., 2003). The main similarities between canine FNAD and human PCH 1 are that both exhibit cerebellar hypoplasia and features of FADS that result from fetal-onset motor dysfunction. Two important differences between these disorders are that the pontine nuclei, which are largely intact in FNAD, degenerate in PCH 1, and NAD is not reported in PCH 1.
Mutations of PLA2G6 cause most cases of human INAD (Gregory et al., 2008), but how these cause NAD is unknown. The recessive nature of human INAD and canine FNAD indicates that, in both instances, mutations cause a loss of gene function. Because canine FNAD is not due to mutation of PLA2G6, NAD is likely a stereotyped response of neurons to a variety of inherited dysfunctions, as well as to vitamin E deficiency and other insults, perhaps defining a common mechanism or pathway (Schmidt et al., 1991, 1997). At this time we are poised to take a positional-candidate cloning approach for identification of the gene mutation responsible for canine FNAD. The availability of high-resolution canine linkage and radiation hybrid maps, the assembled canine genome sequence with 7.6 × coverage, gene and SNP arrays, and now a large kindred segregating canine FNAD complete the set of needed resources for a whole-genome scan by linkage or association of markers.
Acknowledgments
The authors thank G. Diane Shelton and Brunhilde Wirth for helpful discussions, Thomas Van Winkle for initial histopathologic assessment, Thomas Mave for caesarian sections, the MSU Investigative HistoPathology Laboratory for tissue sections and stains, and Ralph Common for electron microscopy and photomicrographs. Dr. Rosenstein's present address is VCA South Shore Animal Hospital, 595 Columbian St, Weymouth, MA 02190. We are especially grateful to Donald F. Patterson for inspiration and guidance early in these investigations.
Grant Sponsors: National Institute of Neurologic Disease and Stroke (NS41989), the National Institute of Research Resources (RR02512), and the Purebred Dog Endowment Fund of the College of Veterinary Medicine, Michigan State University.
Abbreviations
- FADS
fetal akinesia deformation sequence
- FNAD
fetal-onset neuroaxonal dystrophy
- INAD
infantile neuroaxonal dystrophy
- NAD
neuroaxonal dystrophy
- PCH 1
pontocerebellar hypoplasia type 1
- PLA2G6
phospholipase A2 group VI gene
Literature Cited
- 1.Akeson WH, Amiel D, Abel MF, Garfin SR, Woo SLY. Effects of immobilization on joints. Clin Orthop Relat Res. 1987;219:28–37. [PubMed] [Google Scholar]
- 2.Aoki T, Tanaka T, Watabe H. Purification and characterization of gamma-enolase from various mammals. Chem Pharm Bull (Tokyo) 1992;40:1236–1239. doi: 10.1248/cpb.40.1236. [DOI] [PubMed] [Google Scholar]
- 3.Bakker HD, de Sonnaville MLCS, Vreken P, Abeling NGGM, Groener JEM, Keulemans JLM, van Diggelen OP. Human α-N-acetylgalactosaminidase (α-NAGA) deficiency: no association with neuroaxonal dystrophy? Eur J Hum Genet. 2001;9:91–96. doi: 10.1038/sj.ejhg.5200598. [DOI] [PubMed] [Google Scholar]
- 4.Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain & Devel. 1993;15:411–422. doi: 10.1016/0387-7604(93)90080-r. [DOI] [PubMed] [Google Scholar]
- 5.Barth PG. Pontocerebellar hypoplasia--how many types? Eur J Paediatr Neurol. 2000;4:161–162. doi: 10.1053/ejpn.2000.0294. [DOI] [PubMed] [Google Scholar]
- 6.Bouley DM, McIntire JJ, Harris BT, Tolwani RJ, Otto GM, DeKruyff RH, Hayflick SJ. Spontaneous murine neuroaxonal dystrophy: a model of infantile neuroaxonal dystrophy. J Comp Path. 2006;134:161–170. doi: 10.1016/j.jcpa.2005.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braund KG, Lincoln CE. Histochemical differentiation of fiber types in neonatal canine skeletal muscle. Am J Vet Res. 1981;42:407–415. [PubMed] [Google Scholar]
- 8.Brownlow S, Webster R, Croxen R, Brydson M, Neville B, Lin JP, Vincent A, Newsom-Davis J, Beeson D. Acetylcholine receptor δ subunit mutations underlie a fast-channel myasthenic syndrome and arthrogryposis multiplex congenital. J Clin Invest. 2001;108:125–130. doi: 10.1172/JCI12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bürglen L, Amiel J, Viollet L, Lefebvre S, Burlet P, Clermont O, Raclin V, Landrieu P, Verloes A, Munnich A, Melki J. Survival of motor neuron gene deletion in the arthrogryposis multiplex congentia-spinal muscular atrophy association. J Clin Invest. 1996;98:1130–1132. doi: 10.1172/JCI118895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmichael KP, Howerth EW, Oliver JE, Jr, Klappenbach K. Neuroaxonal dystrophy in a group of related cats. J vet Diagn Invest. 1993;5:585–590. doi: 10.1177/104063879300500414. [DOI] [PubMed] [Google Scholar]
- 11.Carrilho I, Santos M, Guinarães A, Teixeira J, Chorão R, Martins M, Dias C, Gregory A, Westaway S, Nguyen T, Hayflick S, Barbot C. Infantile neuroaxonal dystrophy: What's most important for the diagnosis ? Eur J Paediatr Neurol. 2008;12:491–500. doi: 10.1016/j.ejpn.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Chow G, Padfield JH. A case of infantile neuroaxonal dystrophy-connatal Seitelberger disease. J Child Neurol. 2008;23:418–420. doi: 10.1177/0883073807308710. [DOI] [PubMed] [Google Scholar]
- 13.Colman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- 14.Cork LC, Troncoso JC, Price DL, Stanley EF, Griffin JW. Canine neuroaxonal dystrophy. J Neuropathol Exper Neurol. 1983;42:286–296. doi: 10.1097/00005072-198305000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Cox PM, Brueton LA, Bjelogrlic P, Pomroy P, Sewry CA. Diversity of neuromuscular pathology in lethal multiple pterygium syndrome. Pediatr Dev Pathol. 2003;6:59–68. doi: 10.1007/s10024-002-0042-9. [DOI] [PubMed] [Google Scholar]
- 16.Davis JE, Kalousek DK. Fetal akinesia deformation sequence in previable fetuses. Am J Med Genet. 1988;29:77–87. doi: 10.1002/ajmg.1320290110. [DOI] [PubMed] [Google Scholar]
- 17.DeVos KJ, Grierson AJ, Ackerly S, Miller CCJ. Role of axonal transport in neurodegenerative diseases. Ann Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 18.Diaz JV, Duque C, Geisel R. Neuroaxonal dystrophy in dogs: Case report in 2 litters of papillion puppies. J Vet Intern Med. 2007;21:531–534. doi: 10.1892/0891-6640(2007)21[531:ndidcr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 19.Folkerth RD, Guttentag SH, Kupsky WJ, Kinney HC. Arthrogryposis multiplex congenita with posterior column degeneration and peripheral neuropathy: a case report. Clin Neuropathol. 1993;12:25–33. [PubMed] [Google Scholar]
- 20.Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, Gitschier J, Hayflick SJ. Neurodegeneration associated with genetic defects in phosphlipase A2. Neurology. 2008;71:1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths IR, Duncan ID. The central nervous system in canine giant axon neuropathy. Acta Neuropathol. 1979;46:169–172. doi: 10.1007/BF00690839. [DOI] [PubMed] [Google Scholar]
- 22.Gorgen-Pauly U, Sperner J, Reiss I, Gehl HB, Reusche E. Familial pontocerebellar hypoplasia type I with anterior horn cell disease. Eur J Paediatr Neurol. 1999;3:33–38. doi: 10.1053/ejpn.1999.0177. [DOI] [PubMed] [Google Scholar]
- 23.Hageman G, Willemse J, van Ketel BA, Verdonck AFMM. The pathogenesis of fetal hypokinesia: a neurological study of 75 cases of congenital contractures with emphasis on cerebral lesions. Neuropediatrics. 1987;18:22–33. doi: 10.1055/s-2008-1052430. [DOI] [PubMed] [Google Scholar]
- 24.Hall JG. Analysis of Pena Shokeir phenotype. Am J Med Genet. 1986;25:99–117. doi: 10.1002/ajmg.1320250112. [DOI] [PubMed] [Google Scholar]
- 25.Hall JG. Arthrogryposis multiplex congenita: etiology, genetics, classification, diagnostic approach, and general aspects. J Pediatr Orthop Part B. 1997;6:159–166. [PubMed] [Google Scholar]
- 26.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokayama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 27.Harper PA, Morton AG. Neuroaxonal dystrophy in Merino sheep. Aust Vet J. 1991;68:152–153. doi: 10.1111/j.1751-0813.1991.tb03162.x. [DOI] [PubMed] [Google Scholar]
- 28.Haynes RL, Borenstein NS, Desilva TM, Folkerth RD, Liu LG, Volpe JJ, Kinney HC. Axonal development in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2005;484:156–167. doi: 10.1002/cne.20453. [DOI] [PubMed] [Google Scholar]
- 29.He Q, Fyfe JC, Schäffer AA, Kilkenney A, Werner P, Kirkness EF, Henthorn PS. Canine Imerslund-Gräsbeck syndrome maps to a region orthologous to HSA14q. Mamm Genome. 2003;14:758–764. doi: 10.1007/s00335-003-2280-1. [DOI] [PubMed] [Google Scholar]
- 30.Hunter AGW, Jimenez CL, Carpenter BF, MacDonald I. Neuroaxonal dystrophy presenting with neonatal dysmorphic features, early onset of peripheral gangrene, and a rapidly lethal course. Am J Med Genet. 1987;28:171–180. doi: 10.1002/ajmg.1320280124. [DOI] [PubMed] [Google Scholar]
- 31.Hwang IK, Choi JH, Li H, Yoo KY, Kim DW, Lee CH, Yi SS, Seong JK, Lee IS, Yoon YS, Won MH. Changes in glial fibrillary protein immunoreactivity in the dentate gyrus and hippocampus proper of adult and aged dogs. J Vet Med Sci. 2008;70:965–969. doi: 10.1292/jvms.70.965. [DOI] [PubMed] [Google Scholar]
- 32.Jäderlund KH, Örvind E, Johnsson E, Matiasek K, Hahn CN, Malm S, Hedhammar Å. A neurologic syndrome in golden retrievers presenting as a sensory ataxic neuropathy. J Vet Intern Med. 2007;21:1307–1315. doi: 10.1892/07-005.1. [DOI] [PubMed] [Google Scholar]
- 33.Janota I. Neuroaxonal dystrophy in the neonate. Acta Neuropathol. 1979;46:151–154. doi: 10.1007/BF00684817. [DOI] [PubMed] [Google Scholar]
- 34.Jennekens FGI, Barth PG, Fleury P, Veldman H, Keuning JF, Westdorp J. Axonal dystrophy in a case of connatal thalamic and brain stem degeneration. Acta Neuropathol. 1984;64:68–71. doi: 10.1007/BF00695608. [DOI] [PubMed] [Google Scholar]
- 35.Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K, Birk OS. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. 2006;79:942–948. doi: 10.1086/508572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 38.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominani E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Nat Acad Sci, USA. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R, Philip SG, Hendriksz C, Moton JEV, Kingston HM, Rosser EM, Wassmer E, Gissen P, Maher ER. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN) Neurology. 70:1623–1629. doi: 10.1212/01.wnl.0000310986.48286.8e. [DOI] [PubMed] [Google Scholar]
- 40.Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, Kotzbauer PT. Disrupted membrane homeostatis and accumulation of ubiquinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J pathol. 2008;172:406–416. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marangos PJ, Zomzely-Neurath C, Luk DC, York C. Isolation and characterization of the nervous system-specific protein 14-3-2 from rat brain. Purification, subunit composition, comparison to the beef brain protein. J Biol Chem. 1975;250:1884–1891. [PubMed] [Google Scholar]
- 42.Mellersh CS, Langston AA, Acland GM, Fleming MA, Ray K, Wiegand NA, Francisco LV, Gibbs M, Aguirre GD, Ostrander EA. A linkage map of the canine genome. Genomics. 1997;46:326–336. doi: 10.1006/geno.1997.5098. [DOI] [PubMed] [Google Scholar]
- 43.Miller MM, Collatos C. Equine degenerative myeloencephalopathy. Vet Clin North Am Equine Pract. 1997;13:43–52. doi: 10.1016/s0749-0739(17)30254-7. [DOI] [PubMed] [Google Scholar]
- 44.Moessinger AC. Fetal akinesia deformation sequence: an animal model. Pediatrics. 1983;72:857–863. [PubMed] [Google Scholar]
- 45.Muntoni F, Goodwin F, Sewry C, Cox P, Cowan F, Airaksinen E, Patel S, Ignatius J, Dubowitz V. Clinical spectrum and diagnostic difficulties of infantile pontocerebellar hypoplasia type I. Neuropediatrics. 1999;30:243–248. doi: 10.1055/s-2007-973498. [DOI] [PubMed] [Google Scholar]
- 46.Nibe K, Kita C, Morozumi M, Awamura Y, Tamura S, Okuno S, Kobayashi T, Uchida K. Clinicopathological feature of canine neuroaxonal dystrophy and cerebellar cortical abiotrophy in papillion and papillion-related dogs. J Vet Med Sci. 2007;69:1047–1052. doi: 10.1292/jvms.69.1047. [DOI] [PubMed] [Google Scholar]
- 47.Nibe K, Nakyama H, Uchida K. Immunohistochemical features of dystrophic axons in Papillion dogs with neuroaxonal dystrophy. Vet Pathol. 2009;46:474–483. doi: 10.1354/vp.08-VP-0156-U-FL. [DOI] [PubMed] [Google Scholar]
- 48.Porter HJ. Lethal arthrogryposis multiplex congenital (fetal akinesia deformation sequence, FADS) Pediatr Pathol Lab Med. 1995;15:617–637. doi: 10.3109/15513819509026997. [DOI] [PubMed] [Google Scholar]
- 49.Ribera J, Ayala V, Esquerda JE. c-Jun-like immunoreactivity in apoptosis is the result of a crossreaction with neoantigenic sites exposed by caspase-3-mediated proteolysis. J Histochem Cytochem. 2002;50:961–972. doi: 10.1177/002215540205000710. [DOI] [PubMed] [Google Scholar]
- 50.Richman M, Mellersh CS, André C, Galibert F, Ostrander EA. Characterization of a minimal screening set of 172 microsatellite markers for genome-wide screens of the canine genome. J Biochem Biophys Methods. 2001;47:137–149. doi: 10.1016/s0165-022x(00)00160-3. [DOI] [PubMed] [Google Scholar]
- 51.Riemersma S, Vincent A, Beeson D, Newland C, Hawke S, Vernet-der Garabedian B, Eymard B, Newsom-Davis J. Association of arthrogryposis multiplex congenital with maternal antibodies inhibiting fetal acetylcholine receptor function. J Clin Invest. 1996;98:2358–2363. doi: 10.1172/JCI119048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rudnik-Schöneborn S, Sztriha L, Aithala GR, Houge G, Lægreid LM, Seeger J, Huppke M, Wirth B, Zerres K. Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. Am J Med Genet. 2003;117A:10–17. doi: 10.1002/ajmg.a.10863. [DOI] [PubMed] [Google Scholar]
- 53.Ryan MM, Cooke-Yarborough CM, Procopis PG, Ouvrier RA. Anterior horn cell disease and olivopontocerebellar hypoplasia. Pediatr Neurol. 2000;23:180–184. doi: 10.1016/s0887-8994(00)00166-1. [DOI] [PubMed] [Google Scholar]
- 54.Sago K, Tamahara S, Tomihari M, Matsuki N, Asahara Y, Takei A, Bonkobara M, Washizu T, Ono K. In vitro differentiation of canine celiac adipose tissue-derived stromal cells into neuronal cells. J Vet Med Sci. 2008;70:353–357. doi: 10.1292/jvms.70.353. [DOI] [PubMed] [Google Scholar]
- 55.Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 56.Sacre BJ, Cummings JF, de Lahunta A. Neuroaxonal dystrophy in a Jack Russell terrier pup resembling human infantile neuroaxonal dystrophy. Cornell Vet. 1993;83:133–142. [PubMed] [Google Scholar]
- 57.Saxena S, Caroni P. Mechanisms of axon degeneration: From development to disease. Prog Neurobiol. 2007;23:174–191. doi: 10.1016/j.pneurobio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Schmidt RE, Coleman BD, Nelson JS. Differential effect of chronic vitamin E deficiency on the development of neuroaxonal dystrophy in rat gracile/cuneate nuclei and prevertebral sympathetic ganglia. Neurosci Lett. 1991;123:102–106. doi: 10.1016/0304-3940(91)90168-s. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt RE, Dorsey D, Parvin CA, Beaudet LN, Plurad SB, Roth KA. Dystrophic axonal swellings develop as a function of age and diabetes in human dorsal root ganglia. Neuropathol Exp Neurol. 1997;56:1028–1043. doi: 10.1097/00005072-199709000-00008. [DOI] [PubMed] [Google Scholar]
- 60.Shelton GD, Cardinet GH, Bandman E. Expression of fiber type specific proteins during ontogeny of canine temporalis muscle. Musc & Nerve. 1988;11:124–132. doi: 10.1002/mus.880110207. [DOI] [PubMed] [Google Scholar]
- 61.Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neuroscience. 2008;28:2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sisó S, Hanzlíček D, Fluehmann G, Kathmann I, Tomek A, Papa V, Vandevelde M. Neurodegenerative diseases in domestic animals: a comparative review. Vet J. 2006;171:20–38. doi: 10.1016/j.tvjl.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 63.Sisó S, Tort S, Aparici C, Pérez L, Vidal E, Pumarola M. Abnormal neuronal expression of the calcium-binding proteins, parvalbumin, and calbindin D-28k, in aged dogs. J Comp Path. 2003;128:9–14. doi: 10.1053/jcpa.2002.0597. [DOI] [PubMed] [Google Scholar]
- 64.Summers BA, Cummings JF, de Lahunta A. Veterinary Neuropathology. Mosby; St. Louis: 1995. [Google Scholar]
- 65.Tachibana H, Hayashi T, Kajii T, Takashima S, Sasaki K. Neuroaxonal dystrophy of neonatal onset with unusual clinicopathological findings. Brain Dev. 1986;8:605–609. doi: 10.1016/s0387-7604(86)80007-9. [DOI] [PubMed] [Google Scholar]
- 66.Toma JG, Akhavan M, Fernandes KJ, Barnabe-Heider F, Sadikot A, Kaplan DR, Miller FD. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3:778–784. doi: 10.1038/ncb0901-778. [DOI] [PubMed] [Google Scholar]
- 67.Ulfig N, Nickel J, Bohl J. Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res. 1998;291:433–443. doi: 10.1007/s004410051013. [DOI] [PubMed] [Google Scholar]
- 68.Wang QJ, Ding Y, Kohtz S, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woodard JC, Collins GH, Hessler JR. Feline hereditary neuroaxonal dystrophy. Am J Pathol. 1974;74:551–566. [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y, Xiong H, Chang X, Bao X, Zhang Y, Xiao J, Wu X. Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy. Eur J Neurol. 2009;16:240–245. doi: 10.1111/j.1468-1331.2008.02397.x. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Y, Flandin P, Long JE, Cuesta MD, Westphal H, Rubenstein JL. Distinct molecular pathways for development of telencephalic interneuron subtypes revealed through analysis of Lhx6 mutants. J Comp Neurol. 2008;510:79–99. doi: 10.1002/cne.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]