

Abstract
Sex-associated differences in hypertension have been repeatedly observed in epidemiological studies. However, the mechanisms conferring vascular protection to females are not totally elucidated. Sex-related differences in intracellular Ca2+ handling or, more specifically, in mechanisms that regulate Ca2+ entry into vascular smooth muscle cells have been identified as players in sex-related differences in hypertension-associated vascular dysfunction. Recently, new signaling components that regulate Ca2+ influx, in conditions of intracellular store depletion, were discovered: stromal interaction molecule 1 (STIM1), which works as an intracellular Ca2+ sensor; and Orai1, which is a component of the Ca+2 release-activated Ca+2 (CRAC) channels. Together, these proteins reconstitute store-operated Ca2+ channel function. Disturbances in STIM1/Orai1 signaling have been implicated in pathophysiological conditions, including hypertension. In this review we analyze evidence for sex-related differences in Ca2+ handling and propose a new hypothesis where sex-related differences in STIM/Orai signaling may contribute to hypertension-associated vascular differences between male and female subjects.
Keywords: STIM, Orai, hypertension, sexual differences
1.0 - Introduction
Defective regulation of intracellular calcium (Ca2+) is a hallmark of hypertension-associated vascular dysfunction and plays a key role in the augmented vascular reactivity, characteristic of clinical and experimental hypertension. The recent discovery of new signaling components linking intracellular Ca+2 stores to plasma membrane Ca2+ entry brought a new insight into the understanding of Ca2+ homeostasis. Stromal interaction molecule 1 (STIM1) is the Ca2+ sensor protein that triggers Ca2+ influx in response to Ca2+ store depletion, whereas Orai is an essential component of CRAC (Ca2+ release-activated Ca2+) channels. Although research on STIM1/Orai signaling is entering an exponential phase of growth, the role of these proteins in vascular dysfunction is unknown.
Mechanisms contributing to hypertension and its associated end-organ dysfunction are differentially regulated in males and females. Critical sex differences are observed in the intrinsic vascular mechanisms that regulate total peripheral resistance, namely that gonadally intact females display less vascular dysfunction associated with experimental hypertension [1–6], compared to gonadally intact males. Although the existence of sex differences in vascular Ca2+ handling is well established [7–10], no studies have addressed differences in vascular STIM1/Orai signaling in male and female hypertensive animals.
Given the importance of STIM1/Orai signaling in intracellular Ca2+ homeostasis, it seems plausible that increased activation of STIM1/Orai contributes to increased vascular contraction in the vasculature of hypertensive animals.
Here we will review evidence supporting these hypotheses. First, we will address the participation of STIM1 and Orai1 on Ca+2 handling mechanisms during physiological condition as well as in hypertension. Then, we will focus on how alteration of STIM1/Orai signaling contributes to differences in Ca+2 handling, and how this impairment may contribute to sex-related differences in vascular function in hypertension.
2.0 - STIM1-Orai1 pathway represents a key Ca+2 handling mechanism
Nearly all cell types depend on cytoplasmatic Ca+2 signals to trigger specific responses [11]. Upon stimulation, most excitable cells display a biphasic increase in cytosolic Ca2+ concentrations. The initial transient increase is due to Ca2+ release from intracellular stores, e.g. inositol trisphosphate (IP3)-mediated release of ER Ca2+, whereas the subsequent prolonged increase requires extracellular Ca2+ influx through various pathways. Upon depletion of Ca2+ from the ER, Ca2+ channels are activated in the plasma-membrane to refill internal Ca2+ stores. This mechanism, by which the ER acts as a capacitor, lead to the term store-operated Ca2+ entry (SOCE) [12, 13]. SOCE carries a highly Ca2+–selective, non-voltage-gated, inwardly rectifying current termed CRAC (Ca2+ release-activated Ca2+) current, or ICRAC [13, 14].
The discovery of new signaling components linking intracellular Ca+2 stores to plasma membrane Ca2+ entry brought a new insight into the understanding of Ca2+ homeostasis. Stromal interaction molecule 1 (STIM1) was identified as a Ca2+ sensor essential for Ca2+–store depletion-triggered Ca2+ influx [15, 16]. Roos and colleagues (2005) showed that knockdown of STIM in Drosophila S2 cells significantly reduced thapsigargin-dependent Ca2+ entry and completely suppressed ICRAC [15]. In addition to being an ER Ca2+ sensor, STIM1 functions within the plasma membrane to control operation of the Ca2+ entry channel itself [17] and STIM1 migrates from the Ca2+ store to the plasma membrane, in conditions of store depletion [18].
It was later demonstrated that Orai1 is an essential pore subunit of the CRAC channel [19]. Accordingly, upon depletion of ER Ca2+ stores, STIM1 and Orai move in a coordinated fashion to form closely apposed clusters in the ER and plasma membrane [20], creating the elementary unit of SOCE [21]. In addition, the interaction between STIM1 and Orai1 is greatly enhanced after thapsigargin treatment, which acts as selective inhibitor of the ER Ca2+ ATPase resulting in depletion of ER Ca2+ stores [22].
2.1 - STIM1-Orai1 pathway alterations and hypertension
Since a rise in intracellular free Ca2+ concentration is the principal process that initiates contraction of vascular smooth muscle cells (VSMCs) [23], the maintenance of the steady-state Ca2+ is critically important to maintain vascular tone and, consequently, total peripheral resistance [24]. Defective regulation of intracellular Ca2+ plays a major role in the augmented vascular reactivity [25]. Augmented Ca2+ levels in VSMCs from hypertensive animals can be attributed to various mechanisms, including increased Ca2+ release from intracellular stores [26]; reduced Ca2+ uptake by the SR [27]; impaired function of Ca2+-binding proteins [28]; decreased Ca2+ extrusion mechanisms in the plasma membrane [26]; and increased Ca2+ influx [29, 30].
Here we will focus on how increased Ca2+ influx through STIM1 and Orai1 pathway may contribute to augmented vascular reactivity in hypertension. We first demonstrated that aortas from stroke-prone spontaneously hypertensive rats (SHRSP), displayed increased force-development during Ca2+ loading, upon depletion of intracellular Ca2+ stores [31]. The SR Ca2+ store is larger in aortas from SHRSP due to an enhanced influx of Ca2+ across the sarcolemma rather than an impaired recycling of the cation by the SR Ca2+-ATPase [32].
It was shown that depletion of ER Ca2+ stores induces greater SOCE activation in vascular myocytes from SHRSP, compared to that in control Wistar Kyoto (WKY - Figure 1) rats [30]. This is associated with augmented vascular contractile responses to Ca2+, which is blocked by molecular (neutralizing antibodies against STIM1 and Orai1) and pharmacological (2-APB and Gd3+) inhibition of STIM1/Orai signaling. In addition, vascular expression of STIM1 (Figure 2) and Orai proteins is increased in SHRSP versus WKY. Thus, augmented STIM1/Orai signaling may represent a mechanism leading to impaired control of intracellular Ca+2 in hypertension.
Fig.1. SHRSP aortas display augmented spontaneous tone after depletion of Ca2+ stores.

Inhibition of the SR Ca2+-ATPase with thapsigargin augmented contractile responses to Ca2+ with greater effects in SHRSP. Representative traces.
Fig. 2. SHRSP displays increased vascular expression of STIM-1.
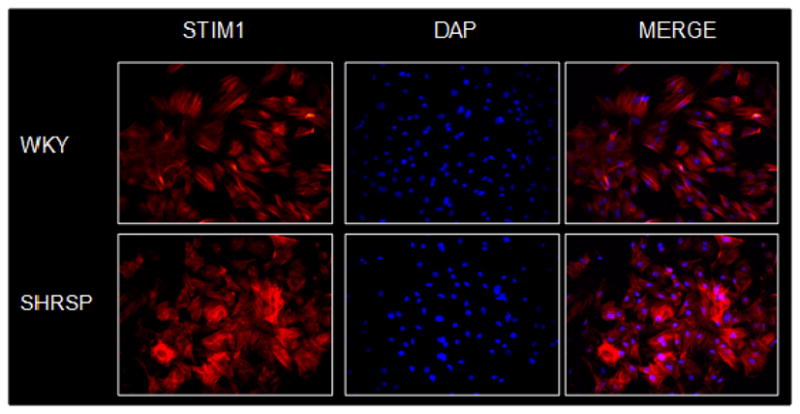
Immunocytochemistry performed in rat aorta vascular smooth muscle cells. Cells were incubated with primary antibodies rabbit anti-STIM1 and counterstained with a Cy3-conjugated anti-mouse IgG secondary antibody. Cells were incubated with 4′,6-diamidino-2-phenyindole (DAPI, D-9564) to detect nuclei.
3.0 - Sex differences in hypertension
Blood pressure is higher in men than in age-matched women and there is a lower incidence of hypertension in pre-menopausal women than men [33–36]. Although gender-associated differences during hypertension have been repeatedly observed in epidemiological studies, the mechanisms for gender differences in blood pressure control are not totally elucidated.
Additionally, because Ca+2 triggers VSMC function and its regulation is highly controlled, differences in Ca+2 handling mechanisms have been proposed to explain sex-related differences in vascular function during hypertension [37] as will be discussed.
3.1 - Sex differences in hypertension and Ca2+ handling by vascular myocytes
Although sex-associated differences in hypertension are well established, with important differences in the neural, renal and vascular mechanisms associated with blood pressure homeostasis [49–53], the mechanisms that determine differences in blood pressure control in males and females are not totally elucidated. Considering that sex-related differences in mechanisms that regulate Ca2+ entry and storage in VSMCs have been identified, differential Ca+2 handling in VSMCs from males and females may explain sex-related differences in vascular function in hypertension [37]. Accordingly, in aorta from both normotensive and female SHR, Ca2+ influx upon contractile stimuli is decreased, compared to that in male SHR [10]. In addition, VSMCs from female rats display reduced Ca2+ entry and reduced depolarization-induced Ca2+ levels, compared to those in male [8]. Differences in intracellular Ca2+ increases in VSMC from male and female SHR are abolished in the absence of extracellular Ca2+ [37–39].
Since augmented STIM1/Orai function may represent one mechanism that contributes to abnormal Ca+2 in VSMC, we hypothesize that vascular protection in hypertensive females reflects an attenuated signaling through STIM1/Orai in vascular myocytes from hypertensive females (figure 3).
Fig. 3. Putative mechanism by which augmented STIM-1/Orai1 signaling contributes to abnormal Ca2+ homeostasis in vascular myocytes.

Compared to female, arteries from male subjects display increased activation of STIM1/Orai1. In conditions of depletion or decrease of intracellular Ca2+ stores, STIM1 is activated, resulting in the dimerization of Orai1, and the formation of CRAC channels. Ca2+ influx, which is increased in VSMCs from hypertensive males compared to hypertensive females, is stimulated via STIM1 and CRAC channels (Orai1). Therefore, arteries from hypertensive male subjects display increased Ca2+ influx. SR Ca2+ ATPase is activated and the intracellular Ca2+stores are refilled. However, the SR in VSMCs of female and male hypertensive displays similar buffering capacity. Consequently, arteries from hypertensive male subjects show increased free cytosolic Ca2+, which can lead to increased vascular contraction.
In agreement with this hypothesis, we showed that upon store depletion, force development during Ca2+ loading period is augmented in aortas from male SHRSP, compared to female SHRSP. Interestingly, pharmacological blockade of CRAC channel is able to abolish sex-differences in spontaneous contractions during Ca2+ loading period [40]. Additionally, after store depletion, neutralizing antibodies against STIM1 and Orai1 abolish sex-differences in spontaneous contractions.
It is possible that augmented STIM1/Orai signaling represents a mechanism for increased activation of Ca2+-dependent signaling pathways in arteries from hypertensive animals. Identification of mechanisms leading to sex differences in hypertension may uncover a regulatory mechanism that may be used to confer cardiovascular protection.
References
- 1.Kahonen M, Tolvanen JP, Sallinen K, Wu X, Porsti I. Influence of gender on control of arterial tone in experimental hypertension. Am J Physiol. 1998;275(1 Pt 2):H15–22. doi: 10.1152/ajpheart.1998.275.1.H15. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol. 2008;294(4):R1220–1226. doi: 10.1152/ajpregu.00864.2007. [DOI] [PubMed] [Google Scholar]
- 3.Tostes RC, David FL, Carvalho MH, Nigro D, Scivoletto R, Fortes ZB. Gender differences in vascular reactivity to endothelin-1 in deoxycorticosterone-salt hypertensive rats. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S99–101. doi: 10.1097/00005344-200036051-00032. [DOI] [PubMed] [Google Scholar]
- 4.Tostes RC, Nigro D, Fortes ZB, Carvalho MH. Effects of estrogen on the vascular system. Braz J Med Biol Res. 2003;36(9):1143–1158. doi: 10.1590/s0100-879x2003000900002. [DOI] [PubMed] [Google Scholar]
- 5.Chappell MC, Westwood BM, Yamaleyeva LM. Differential effects of sex steroids in young and aged female mRen2.Lewis rats: a model of estrogen and salt-sensitive hypertension. Gend Med. 2008;5(Suppl A):S65–75. doi: 10.1016/j.genm.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McIntyre M, Hamilton CA, Rees DD, Reid JL, Dominiczak AF. Sex differences in the abundance of endothelial nitric oxide in a model of genetic hypertension. Hypertension. 1997;30(6):1517–1524. doi: 10.1161/01.hyp.30.6.1517. [DOI] [PubMed] [Google Scholar]
- 7.Khalil RA. Sex hormones as potential modulators of vascular function in hypertension. Hypertension. 2005;46(2):249–254. doi: 10.1161/01.HYP.0000172945.06681.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy JG, Khalil RA. Gender-specific reduction in contractility and [Ca(2+)](i) in vascular smooth muscle cells of female rat. Am J Physiol Cell Physiol. 2000;278(4):C834–844. doi: 10.1152/ajpcell.2000.278.4.C834. [DOI] [PubMed] [Google Scholar]
- 9.Crews JK, Khalil RA. Gender-specific inhibition of Ca2+ entry mechanisms of arterial vasoconstriction by sex hormones. Clin Exp Pharmacol Physiol. 1999;26(9):707–715. doi: 10.1046/j.1440-1681.1999.03110.x. [DOI] [PubMed] [Google Scholar]
- 10.Crews JK, Murphy JG, Khalil RA. Gender differences in Ca(2+) entry mechanisms of vasoconstriction in Wistar-Kyoto and spontaneously hypertensive rats. Hypertension. 1999;34(4 Pt 2):931–936. doi: 10.1161/01.hyp.34.4.931. [DOI] [PubMed] [Google Scholar]
- 11.Putney JW, Jr, McKay RR. Capacitative calcium entry channels. Bioessays. 1999;21(1):38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 12.Putney JW., Jr Pharmacology of capacitative calcium entry. Mol Interv. 2001;1(2):84–94. [PubMed] [Google Scholar]
- 13.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 14.Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. The cellular and molecular basis of store-operated calcium entry. Nat Cell Biol. 2002;4(11):E263–272. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- 15.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci U S A. 2006;103(11):4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443(7108):230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 20.Wu MM, Luik RM, Lewis RS. Some assembly required: constructing the elementary units of store-operated Ca2+ entry. Cell Calcium. 2007;42(2):163–172. doi: 10.1016/j.ceca.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174(6):815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung FP, Yung LM, Yao X, Laher I, Huang Y. Store-operated calcium entry in vascular smooth muscle. Br J Pharmacol. 2008;153(5):846–857. doi: 10.1038/sj.bjp.0707455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rembold CM, Murphy RA. Myoplasmic calcium, myosin phosphorylation, and regulation of the crossbridge cycle in swine arterial smooth muscle. Circ Res. 1986;58(6):803–815. doi: 10.1161/01.res.58.6.803. [DOI] [PubMed] [Google Scholar]
- 24.Himpens B, Missiaen L, Casteels R. Ca2+ homeostasis in vascular smooth muscle. J Vasc Res. 1995;32(4):207–219. doi: 10.1159/000159095. [DOI] [PubMed] [Google Scholar]
- 25.Tostes RC, Wilde DW, Bendhack LM, Webb RC. Calcium handling by vascular myocytes in hypertension. Braz J Med Biol Res. 1997;30(3):315–323. doi: 10.1590/s0100-879x1997000300004. [DOI] [PubMed] [Google Scholar]
- 26.Lompre AM. Sarcoplasmic reticulum in vascular cells in hypertension and during proliferation. Clin Exp Pharmacol Physiol. 1999;26(7):553–557. doi: 10.1046/j.1440-1681.1999.03079.x. [DOI] [PubMed] [Google Scholar]
- 27.Levitsky DO, Clergue M, Lambert F, Souponitskaya MV, Le Jemtel TH, Lecarpentier Y, Lompre AM. Sarcoplasmic reticulum calcium transport and Ca(2+)-ATPase gene expression in thoracic and abdominal aortas of normotensive and spontaneously hypertensive rats. J Biol Chem. 1993;268(11):8325–8331. [PubMed] [Google Scholar]
- 28.Niki I, Yokokura H, Sudo T, Kato M, Hidaka H. Ca2+ signaling and intracellular Ca2+ binding proteins. J Biochem. 1996;120(4):685–698. doi: 10.1093/oxfordjournals.jbchem.a021466. [DOI] [PubMed] [Google Scholar]
- 29.Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455(3):465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- 30.Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, Tostes RC, Webb RC. Increased Activation of Stromal Interaction Molecule-1/Orai-1 in Aorta From Hypertensive Rats. A Novel Insight Into Vascular Dysfunction. Hypertension. 2009;53(2):409–416. doi: 10.1161/HYPERTENSIONAHA.108.124404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanagy NL, Ansari MN, Ghosh S, Webb RC. Recycling and buffering of intracellular calcium in vascular smooth muscle from genetically hypertensive rats. J Hypertens. 1994;12(12):1365–1372. [PubMed] [Google Scholar]
- 32.Peel SE, Liu B, Hall IP. ORAI and store-operated calcium influx in human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2008;38(6):744–749. doi: 10.1165/rcmb.2007-0395OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hajjar I, Kotchen JM, Kotchen TA. Hypertension: trends in prevalence, incidence, and control. Annu Rev Public Health. 2006;27:465–490. doi: 10.1146/annurev.publhealth.27.021405.102132. [DOI] [PubMed] [Google Scholar]
- 34.Mosca L, Manson JE, Sutherland SE, Langer RD, Manolio T, Barrett-Connor E. Cardiovascular disease in women: a statement for healthcare professionals from the American Heart Association. Writing Group. Circulation. 1997;96(7):2468–2482. doi: 10.1161/01.cir.96.7.2468. [DOI] [PubMed] [Google Scholar]
- 35.Safar ME, Smulyan H. Hypertension in women. Am J Hypertens. 2004;17(1):82–87. doi: 10.1016/s0895-7061(03)01008-2. [DOI] [PubMed] [Google Scholar]
- 36.Taler SJ. Hypertension in women. Curr Hypertens Rep. 2009;11(1):23–28. doi: 10.1007/s11906-009-0006-9. [DOI] [PubMed] [Google Scholar]
- 37.Devynck MA. Gender and vascular smooth muscle cells: a direct influence on Ca2+ handling? J Hypertens. 2002;20(11):2139–2140. doi: 10.1097/00004872-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Loukotova J, Kunes J, Zicha J. Gender-dependent difference in cell calcium handling in VSMC isolated from SHR: the effect of angiotensin II. J Hypertens. 2002;20(11):2213–2219. doi: 10.1097/00004872-200211000-00021. [DOI] [PubMed] [Google Scholar]
- 39.Loukotova J, Kunes J, Zicha J. Cytosolic free calcium response to angiotensin II in aortic VSMC isolated from male and female SHR. Physiol Res. 1998;47(6):507–510. [PubMed] [Google Scholar]
- 40.Giachini F, Carneiro FS, Lima VV, Carneiro ZN, Dorrance A, Tostes RC, Webb RC. Sex differences in vascular expression and activation of STIM-1/Orai-1 during hypertension: focus on calcium regulation. FASEB J. 2009;23:781.715. Meeting abstracts. [Google Scholar]