

Abstract
Background & Aims
Transgenic, insulin–gastrin (INS–GAS) mice have high circulating levels of gastrin. On a FVB/N background, these mice develop spontaneous atrophic gastritis and gastrointestinal intraepithelial neoplasia (GIN) with 80% prevalence 6 months after Helicobacter pylori infection. GIN is associated with gastric atrophy and achlorhydria, predisposing mice to non-helicobacter microbiota overgrowth. We determined if germ-free INS–GAS mice spontaneously develop GIN and if H. pylori accelerates GIN in gnotobiotic INS–GAS mice.
Methods
We compared gastric lesions and levels of mRNA, serum inflammatory mediators, antibodies, and gastrin among germ-free and H. pylori-monoinfected INS-GAS mice. Microbiota composition of specific pathogen-free (SPF) INS-GAS mice was quantified by pyro-sequencing.
Results
Germ-free INS-GAS mice had mild hypergastrinemia but did not develop significant gastric lesions until they were 9 months old; they did not develop GIN through 13 months. H. pylori monoassociation caused progressive gastritis, epithelial defects, oxyntic gland atrophy, marked foveolar hyperplasia and dysplasia, and strong serum and tissue proinflammatory immune responses (particularly in male mice) between 5 and 11 months post infection (P<0.05, compared with germ-free controls). Only 2 of 26 female, whereas 8 of 18 male, H. pylori-infected INS-GAS mice developed low- to high-grade GIN by 11 months post infection. Stomachs of H. pylori-infected SPF male mice had significant reductions in Bacteroidetes and significant increases in Firmicutes.
Conclusions
Gastric lesions take 13 months longer to develop in germ-free INS–GAS mice than male SPF INS-GAS mice. H. pylori-monoassociation accelerated gastritis and GIN but caused less-severe gastric lesions and delayed onset of GIN compared to H. pylori-infected INS-GAS mice with complex gastric microbiota. Changes of gastric microbiota composition might promote GIN in the achlorhydric stomachs of SPF mice.
Keywords: Microbiome, hypergastrinemic mice, gastric adenocarcinoma, enteric microbiota
Introduction
Helicobacter pylori infection, often acquired during childhood, is frequently subclinical but susceptible individuals may develop severe gastritis, peptic ulcer, gastric atrophy and gastric cancer [1]. H. pylori is the most important risk factor for distal stomach cancer and is the fourth most common cancer and second leading cause of cancer deaths [1,2]. H. pylori infection has an overall prevalence from 11% in industrialized to 95% in developing countries [3,4]. Highest infection rates persist among children, disadvantaged and immigrant populations. The mechanisms of H. pylori transmission are poorly understood although data suggest suboptimal sanitation conditions increase risk of H. pylori infection [3].
While H. pylori infection has been unequivocally linked epidemiologically to gastric cancer, other risk factors are important in determining overall gastric cancer risk [1]. Our studies using H. felis and H. pylori infection of C57BL/6 and INS-GAS mice led to the development of mouse models of severe gastritis, gastric atrophy, dysplasia and gastric adenocarcinoma [5–8]. Dysplasia and invasion of the muscularis mucosa and submucosa vasculature by gastric glands in these mice are features compatible with gastric adenocarcinoma in humans [1]. Additionally, intramucosal carcinoma or high-grade gastrointestinal intraepithelial neoplasia (GIN) are compatible with those first described for lower bowel carcinomas in rodents [9].
Transgenic INS-GAS mice over-express human gastrin regulated by a rat insulin promoter. Elevated gastrin is associated with increased risk for gastric glandular atrophy and cancer in humans, making this mouse a unique model of gastric adenocarcinoma [6,10]. Specific pathogen free (SPF) INS-GAS mice exhibit hypergastrinemia that leads to gastric hyperplasia and GIN within 20 months [6,11]. INS-GAS mice infected with H. pylori or H. felis developed gastric carcinomas by 6–7 months of age; analogous to development of gastric cancer after decades of chronic H. pylori gastritis in humans [6–8,12]. Similar to human patients with gastric preneoplasia, INS-GAS mice (uninfected or infected with H. pylori) progress to gastric cancer following development of gastric atrophy and hypochlorhydria, a condition that predisposes the stomach to enteric bacterial overgrowth.
Notably, antibiotic eradication therapy (omeprazole, metronidazole, and clarithromycin) targeting H. pylori infection in male SPF INS-GAS mice resulted in reduced gastric cancer risk; attenuation of gastric lesions was most pronounced when treatment was administered 8 weeks post infection (wpi) compared to mice treated 12 or 22 wpi [13]. Importantly, similar eradication therapy administered to age-matched helicobacter-free male SPF INS-GAS mice also significantly delayed onset of GIN [13,14]. This result suggested antimicrobial therapy may eradicate not only H. pylori but other micro-organisms that may colonize the atrophic gastric epithelium, suggesting enteric bacteria colonizing the atrophic stomach further promote gastritis, genotoxic damage and the progression to neoplasia [13,14].
Because INS-GAS mice progress to GIN following the development of gastric atrophy and metaplasia, which creates a niche for intestinal microbiota, this study determined if germfree (GF) INS-GAS mice developed gastritis or GIN and whether H. pylori monoassociation reproducibly induced GIN in gnotobiotic INS-GAS mice lacking lower bowel microbiota.
Methods
Mice
The animal protocol was approved by the Massachusetts Institute of Technology Committee on Animal Care. GF INS-GAS mice on a FVB/N background (Tg (Ins1-GAS) 1Sbr) [11] were maintained in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. GF mice were housed in sterile isolators on autoclaved hardwood bedding in solid-bottomed polycarbonate cages and fed autoclaved rodent diet (Prolab RMH 3000, PMI Nutrition International) and sterile water ad libitum. Isolators were surveyed weekly and confirmed negative for microbial contaminants. The isolator housing H. pylori-infected mice was contaminated after 8 weeks with Staphylococcus sp; however, no confounding effects were observed and the control mice housed in a separate isolator remained GF. Historically, Staphylococcus sp. have had no measurable impact on the colonization or antibody response in H. pylori-monoassociated mice [15].
Experimental Design
Eighty-seven, 6 week old male and female INS-GAS mice were raised in GF isolators; 46 mice were infected by oral gavage with 200 μl of 109 colony forming units/mL of H. pylori (SS1 strain) on alternate days for a total of 3 doses [16]. At 1 month post-infection (mpi), two mice were euthanized and their stomachs cultured on selective medium to confirm infection. At 5, 7, 9, and 11 mpi, mice were euthanized and necropsied. An additional 5 male SPF INS-GAS mice, 3 infected as described above and 2 uninfected controls, were euthanized and necropsied at 22 weeks of age (15 wpi) for analysis of the gut flora composition.
Sample Collection
Immediately following CO2 euthanasia, blood was collected via cardiac puncture; serum separated and stored at −70°C. The stomach and proximal duodenum were aseptically removed and incised along the greater curvature. Four individual linear gastric strips from the lesser curvature were sectioned for culture, flash frozen for RNA analysis, stored at −70°C for DNA extraction or preserved in 10% neutral-buffered formalin for histopathologic evaluation. Details of the Histological Analysis, Confirmation of H. pylori Colonization, Gastrin Radioimmunoassay, Analysis of Microbiota Composition, ELISA for Serum IgG2a and IgG1 Response, Cytokine and Chemokine Serum Levels and Gastric mRNA Levels are reported in Supplementary Materials and Methods.
Statistical analysis
Gastric lesion scores and microbiota compositions were compared by Mann-Whitney U test. Antibody, inflammatory mediator, and serum gastrin responses were compared using Student’s t test. Statistical analysis was performed using GraphPad Prism 4.0 (GraphPad Software, Inc., La Jolla, CA) with values of p<0.05 considered significant.
Results
H. pylori colonization was confirmed in all monoassociated INS-GAS mice
H. pylori infection of all dosed mice was confirmed at necropsy by culture of gastric tissue on selective medium. Randomly selected control mice across all time points were negative for infection.
Elevated serum gastrin in germfree INS-GAS mice was further increased by H. pylori infection
GF INS-GAS mice had developed mild hypergastrinemia when first evaluated at 7 months of age, equivalent to the 5 mpi time point, compared to previously published values of 43 ± 13 pM for age-matched wildtype FVB mice [6](Figure 1). The gastrin levels in the GF INS-GAS mice at 7 mpi were comparable to levels in SPF INS-GAS mice[6]. Serum gastrin levels were significantly elevated in male and female H. pylori-monoassociated mice from 7 to 11 mpi compared to age-matched GF controls (all p values <0.05), but did not rise in GF INS-GAS mice. H. pylori-infected males had significantly greater serum gastrin concentrations than infected females at 7 mpi (p < 0.05) but there was no significant difference in gastrin concentrations between infected males and females at 9 or 11 mpi.
Figure 1.
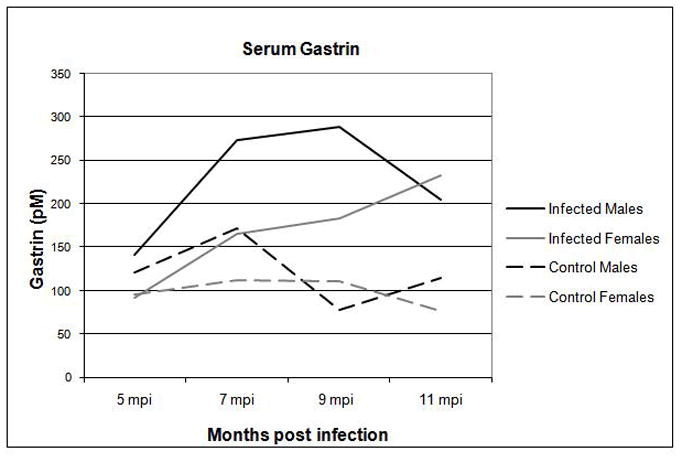
Serum gastrin levels were higher in H. pylori-infected male and female INS-GAS mice compared to GF controls at 5, 7, 9, and 11 mpi.
* p < 0.05 – significant differences between H. pylori-monoassociated and GF control mice.
# p < 0.05 – significant differences between H. pylori-monoassociated male and female mice.
Germfree INS-GAS mice had delayed onset of gastritis and did not develop GIN through the 11 mpi timepoint
Control GF INS-GAS mice had minimal to no gastric lesions when necropsied at 7 months of age. Background lesions attributable to overexpression of gastrin were evident as progressive hyperplasia, dysplasia and atrophy in GF controls between 9 and 11 months of age (p<0.001), correlating to 7 and 9 mpi for the H. pylori-infected mice (Table 1, Figure 2 and 3). No GIN was observed in GF controls through 13 months of age (equivalent to 11 mpi for H. pylori group).
Table 1.
Categorical description of gastric lesions observed in H. pylori-monoassociated and GF control INS-GAS mice at 5, 7, 9, and 11 mpi.
| mpi | M/F | H. pylori | N | Pathology Subfeatures, median (range) | |||||
|---|---|---|---|---|---|---|---|---|---|
| I | ED | A | H | PM | D | ||||
| 5 | C | − | 11 | 0.5 (0–1) | 0.5 (0–1) | 0 (0-0) | 0 (0–0.5) | 0 (0-0) | 0 (0-0) |
| M | + | 5 | 2 (1–2.5)** | 2 (1–2)* | 1.5 (1.5–2)* | 1.5 (0–2)** | 1.5 (1.5–2.5)** | 1 (0–1.5)* | |
| F | + | 6 | 1.5 (1–2)* | 1.5 (1–2)* | 1 (0.5–1.5)* | 0.75 (0.5–1)** | 0.5 (0–0.5)* | 0.5 (0–0.5)* | |
| 7 | C | − | 11 | 0.5 (0–1) | 0.5 (0–1) | 0 (0-0) | 0 (0–0.5) | 0 (0-0) | 0 (0-0) |
| M | + | 5 | 2.5 (2–2.5)* | 2.5 (2.5–3)* | 2.5 (2–3)* | 3.5 (3–4)* | 2.5 (2–3)* | 2.5 (2–3)* | |
| F | + | 7 | 2 (2–3)* | 2.5 (2–3)* | 2 (1.5–2)* | 2.5 (2–3.5)* | 1.5 (1.5–2)* | 1.5 (1.5–2)* | |
| 9 | C | − | 10 | 0.75 (0.5–1) | 1 (0.5–1.5) | 0.5 (0–1) | 1.25 (1–2) | 0.5 (0–1) | 0.25 (0–0.5) |
| M | + | 4 | 2.5 (2–3)** | 3.25 (3–3.5)* | 3 (2–3.5)** | 3.25 (3–4)** | 3 (2–3)* | 3.25 (3–3.5)** | |
| F | + | 6 | 2.5 (2–2.5)* | 2.75 (2–3.5)* | 2.75 (1.5–3)* | 3 (2–3.5)* | 2.5 (2–3)* | 2.5 (2–3)* | |
| 11 | C | − | 11 | 0.5 (0–1) | 0.5 (0–1) | 0.5 (0–1) | 1 (0.5–2) | 0.5 (0–1) | 0.5 (0–1.5) |
| M | + | 4 | 2.75 (2–3)** | 3 (2.5–3.5)* | 3.25 (2–3.5)** | 3.5 (2.5–4)** | 3 (2–3.5)* | 3.5 (2–4)** | |
| F | + | 7 | 2.5 (2–3)* | 2.5 (2.5–3)* | 2.5 (2–4)* | 2.5 (2.5–4)* | 2.5 (2–3)* | 2.5 (1.5–3)* | |
Abbreviations: mpi: months post infection, M. males, F. females, C. combined male and female, I. inflammation, ED. Epithelial defects, A. oxyntic atrophy, H. foveolar hyperplasia, PM. pseudopyloric metaplasia, D. dysplasia.
p<0.01
p<0.05
Control male and female data were combined.
Figure 2.

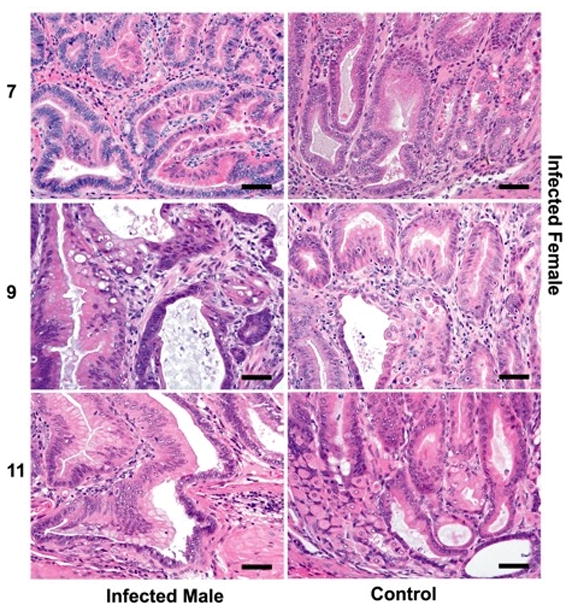
(a) Gastric lesions in H. pylori-monoassociated (male and female) and GF control (male) mice at 5, 7, 9 and 11 mpi. Control GF INS-GAS mice did not develop significant gastric lesions until the 7 mpi timepoint and no GIN was observed through 11 mpi. In contrast H. pylori monoassociation caused progressive gastritis, epithelial defects, oxyntic gland atrophy with metaplasia, marked foveolar hyperplasia and dysplasia, particularly in male INS-GAS mice, between 5 and 11 mpi (p<0.05). Two of 26 female, whereas 8 of 18 male, H. pylori-monoassociated INS-GAS mice developed low to high grade GIN during the 11 mpi observation period. Bars = 200μ for 5 and 7 mpi; 400μ for 9 and 11 mpi. (b) Corresponding higher magnification images (a) highlighting gastric metaplastic and dysplastic changes in H. pylori-monoassociated and control INS-GAS. (All Bars = 40μM). Top left, Male, 7 mpi: Loss of oxyntic glands from glandular metaplasia (pseudopyloric), mild to moderate dysplasia characterized by glandular architectural distortion, loss of columnar orientation, papillary infolding, nuclear stratification and mild nuclear atypia. Top right, Female, 7 mpi: Oxyntic loss, glandular metaplasia globoid mucous change, mild dysplasia characterized by glandular elongation and coiling. Center left, Male, 9 mpi: Glandular metaplasia, moderate to severe dysplasia characterized by severe architectural distortion, glandular ectasia, infolding, disorganized nuclear stratification, cytoplasmic mucous globules and nuclear margination (globoid dysplasia), sloughing of cells and mucous into the glandular lumen. Additionally, loss of basement membranes from dysplastic glands with rafts, finger–like extensions or single polygonal cells in the lamina propria and mild reactive fibrosis indicative of lamina propria invasion. Right center, Female, 9 mpi: Glandular metaplasia, loss of columnar orientation, architectural distortion, infolding, nuclear stratification and nuclear atypia. Bottom left, Male, 11 mpi: Glandular metaplasia, prominent globoid dysplasia, breaching of muscularis mucosa and herniation of dysplastic gland into submucosa. Bottom right, Uninfected Male, 11 mpi: Metaplasia of some oxyntic glands, occasional mild glandular distortion, luminal distension and loss of columnar orientation.
Figure 3.
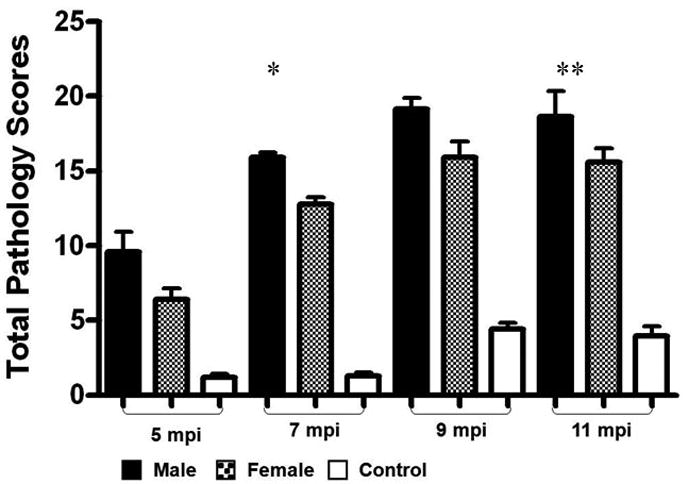
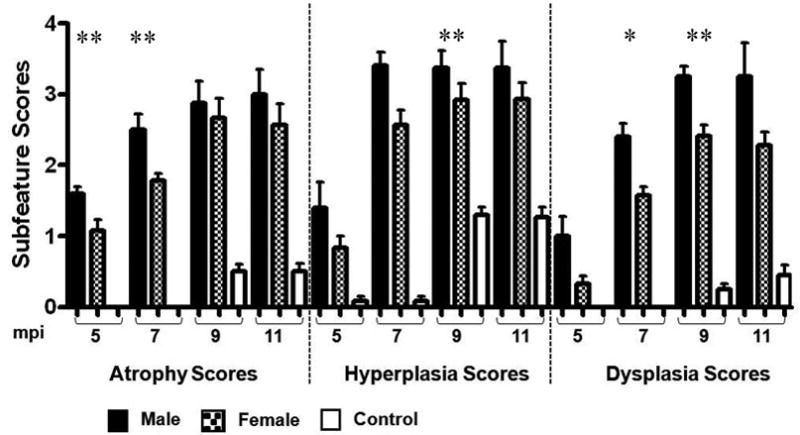
Total pathology scores and subfeature scores of categorical lesions were significantly increased in H. pylori-monoassociated mice compared to GF controls (p values ranging from <0.05 to <0.01) and were more severe in H. pylori-monoassociated INS-GAS male mice compared to females at 5, 7, 9, and 11 mpi.
* p < 0.01 – Significant differences between H. pylori- monoassociated male and female mice.
** p < 0.05 – Significant differences between H. pylori- monoassociated male and female mice.
H. pylori monoassociation accelerated gastritis and GIN in gnotobiotic INS-GAS mice
H. pylori-infected INS-GAS mice developed significant progressive gastric pathology (p<0.05 to <0.001) between 5 and 9 mpi compared to age-matched uninfected controls (Table 1, Figure 2 and 3). Mucous metaplasia and hyalinosis were similarly noted in mice from each experimental group; thus, these parameters had no association with H. pylori infection (Table 1). H. pylori-infected mice had mild to moderate inflammation, moderate epithelial defects, progressive oxyntic atrophy with pseudopyloric metaplasia, marked foveolar hyperplasia and moderate to severe dysplasia that became progressively more severe with time (Figure 2a, 2b, and 3). Between 5 and 7 mpi there were significant increases in inflammation, metaplasia (p<0.01), dysplasia, oxyntic atrophy, and hyperplasia (p<0.001) contributing to overall higher total pathology scores (p<0.001) in infected mice. Additionally, atrophy, dysplasia, and total pathology scores continued to increase significantly between 7 and 9 mpi (p<0.02). The severity of gastric lesions plateaued in H. pylori-infected mice between 9 and 11 mpi and was restricted primarily to the corpus as noted in previous studies [8,13]. As previously reported [8], H. pylori-infected male mice at 5 mpi had higher atrophy scores than infected females (p<0.05), by 7 mpi had significantly higher atrophy (p<0.05), hyperplasia, dysplasia (p<0.01) and overall total pathology scores (p<0.01) than infected females. Dysplasia and total pathology scores remained significantly higher in infected males compared to infected females at 9 mpi (p<0.04). However, at 11 mpi there were no significant differences in total pathology or subfeature scores between H. pylori-infected males and females (Figure 3).
Development of GIN was characterized by progressive dysplasia including loss of glandular organization, crowding, and atypia with variable globoid dysplasia, a feature characterized by disorderly stratification of proliferating gastric glandular epithelial cells that are expanded by large cytoplasmic mucus vacuoles with nuclear margination, most prominently in infected males[17]. One of 5 H. pylori-infected male mice at 7 mpi and 2 of 4 infected males at 9 mpi had dysplasia scores of 3, consistent with low grade GIN. An additional 2 infected males at 9 mpi had dysplasia scores of 3.5, consistent with high grade GIN. At 11 mpi, 1 of 4 infected males had low grade GIN and 2 of 4 infected males had high grade GIN. Notably, only 1 of 6 infected females developed low grade GIN by 9 mpi and only 1 of 7 infected females had low grade GIN at 11 mpi. In summary, only 2 of 26 female, but 8 of 18 male, H. pylori-infected INS-GAS mice developed low to high grade GIN through 11 mpi attributable to H. pylori monoassociation and hypergastrinemia. Thus, H. pylori-monoassociation accelerated gastritis and GIN compared to GF controls.
Archival slides from a previous study [8,13,14] utilizing H. pylori-infected SPF INS-GAS male mice with a complex microbiota at the same time points was re-scored for overall degree of pathology and dysplasia by our pathologist (S.M.). The raw data generated from the previous study was comparable and there was a high degree of concordance between pathologists. H. pylori-monoassociated INS-GAS male mice had less severe gastric lesions and delayed onset of GIN compared to the SPF INS-GAS males. Median atrophy, metaplasia and dysplasia scores from the previously published study were significantly higher and hyperplasia scores significantly lower in H. pylori-infected SPF mice than in H. pylori-monoassociated male INS-GAS mice at the 7 mpi time point [13] (Figure 4a). SPF control male INS-GAS mice at the same time point had significantly greater inflammation, atrophy, hyperplasia, metaplasia and dysplasia compared to germ free controls (Figure 4a). H. pylori-infected SPF mice had an 100% incidence of GIN at 7mpi, 20% low-grade and 80% high-grade (Figure 4b). Only 10% of H. pyori-monoassociated males developed GIN, all low grade.
Figure 4.
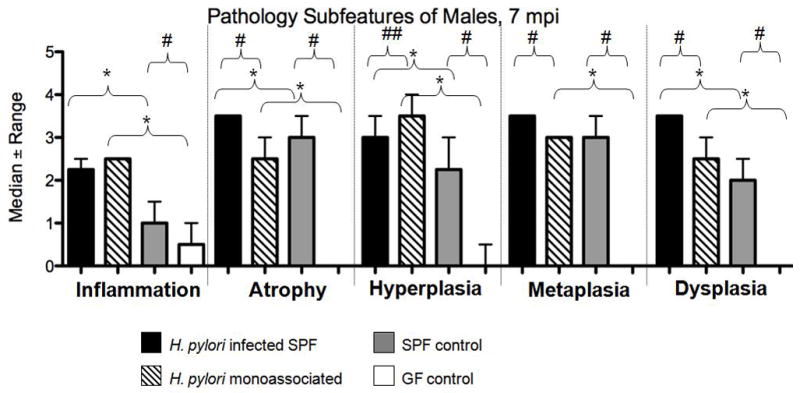
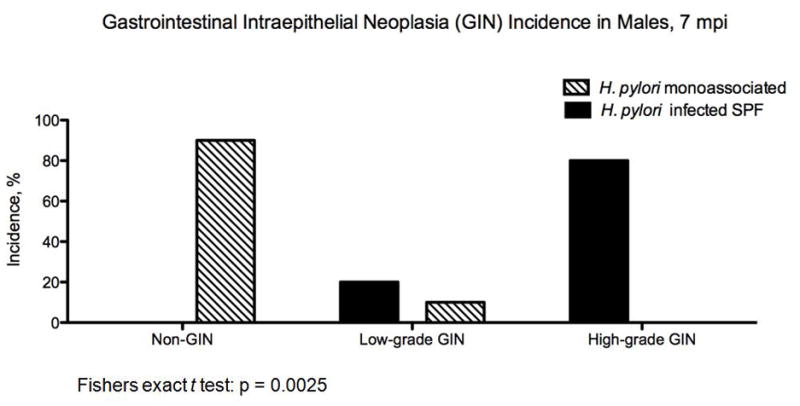
(a) Subfeature pathology scores of categorical lesions and incidence of GIN were significantly increased in H. pylori-infected SPF male mice at 7 mpi compared to H. pylori-monoassociated males at 7 mpi and in the SPF control males compared to the GF control males at 7 mpi (p values ranging from <0.01 <0.05). (b) Development of GIN was significantly increased in H. pylori-infected SPF male mice at 7 mpi compared to H. pylori-monoassociated males at 7mpi (Fishers exact t test: p = 0.0025).
*p ≤ 0.01- Significant differences between H. pylori monoassociated mice and GF control mice or between H. pylori infected SPF mice and SPF control mice.
# p ≤ 0.01, ## p ≤ 0.05 - Significant differences between H. pylori infected SPF mice and H. pylori-monoassociated mice or between SPF control mice and GF control mice.
H. pylori-monoassociated male INS-GAS mice developed a higher IgG2a to IgG1 ratio of H. pylori-specific antibodies than infected females
All H. pylori-infected mice seroconverted by 5 mpi (p<0.01) (data not shown). The Th1-associated IgG2a responses of female and male infected INS-GAS mice were similar (Supplemental Figure 1). While the Th2-associated IgG1 response in H. pylori-infected females remained stable across all time points, the IgG1 response of H. pylori-infected males steadily declined from 5 mpi to 11 mpi. Thus, the H. pylori-infected males had a gradually increasing Th1-associated IgG2a to Th2-associated IgG1 ratio which became significant at 9 mpi (p<0.05). These results reflected a greater pro-inflammatory response to H. pylori in male compared to female mice. Infected females had a significantly higher IgG1 response than males at 11 mpi (p<0.05) indicative of a more robust Th2 response.
H. pylori-monoassociated mice had significant pro- and anti-inflammatory systemic and gastric tissue responses
When gastric pathology peaked in severity at 9 mpi, serum levels of pro- and anti-inflammatory cytokines were significantly greater in H. pylori-monoassociated male and female mice compared to GF controls (figure 5a). Additionally, H. pylori-monoassociated female INS-GAS mice had a delayed inflammatory response (figure 5b). Details are reported in Supplementary Materials.
Figure 5.
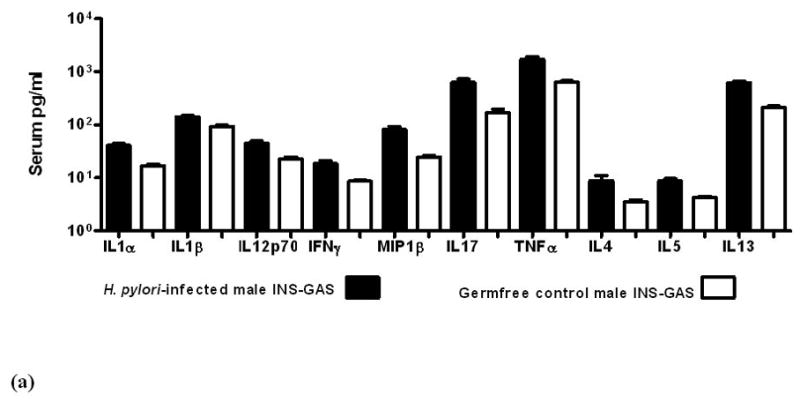
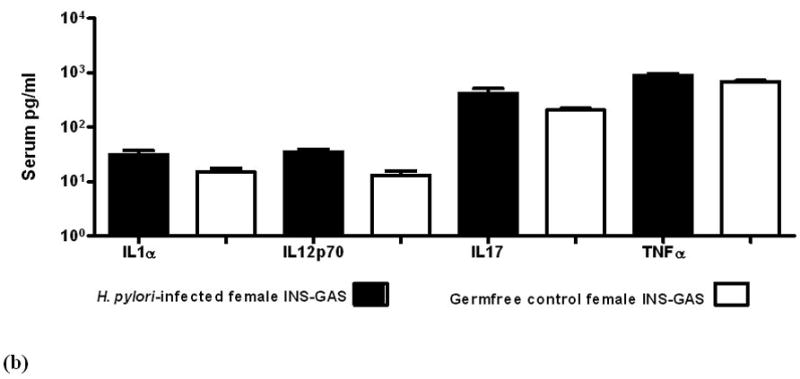
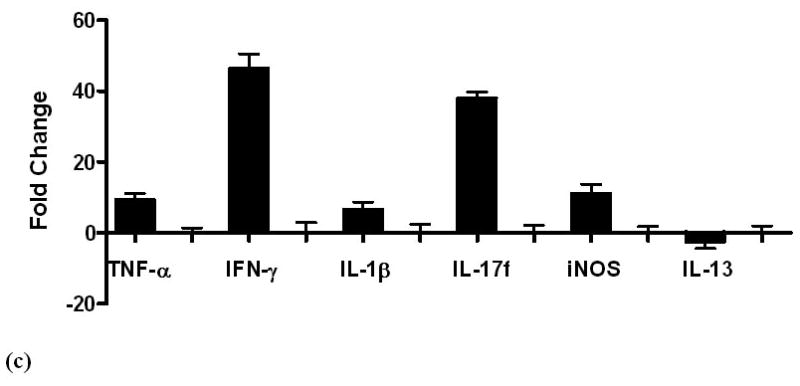
Serum levels of cytokines and chemokines in GF control and H. pylori-monoassociated (a) male INS-GAS mice at 9 mpi and in (b) comparable groups of females at 11 mpi. Within each gender, all differences between infected and GF control mice were significant (males, p<0.02; females p<0.04). (c) Fold-change of mRNA expression levels in gastric tissue from H. pylori-monoassociated INS-GAS mice (gender data combined) at 9 mpi when gastric lesions were most severe. Fold-change represents comparison to baseline levels pre-set for gastric tissues from control GF mice. There were no gender differences (not shown) and all differences between expression levels of H. pylori-monoassociated and GF control mice were significant (p<0.01, all but IL-13; p<0.04 for IL-13). Error bars for panels (a), (b) and (c) represent standard deviation.
Gastric tissue mRNA levels maintained a pattern of predominantly proinflammatory gene expression in all H. pylori-infected mice compared to GF controls across all time points (p<0.01, figure 5c). At 5 mpi, H. pylori-monoassociated females expressed higher levels of some pro-inflammatory genes, however, the gender difference was insignificant at 7 mpi and by 11 mpi, males had higher levels of some pro-inflammatory genes. Details are reported in Supplementary Materials
H. pylori infection and microbiota composition in male SPF INS-GAS mice
Samples from 5 male INS-GAS/FVB mice (three 22 week old (15 wpi) H. pylori-infected mice and two uninfected controls) were subjected to microbiota analysis using 454 sequencing of partial 16S rDNA amplicons. The gastric microbiota composition of infected vs. control mice showed significant differences at the phylum level (figure 6). H. pylori-infected SPF male mice showed a significant reduction in the relative number of Gram-negative Bacteroidetes and a significant increase in the relative number of Firmicutes in the stomach (p<0.05 for both comparisons), while no significant alterations of microbiome composition were detected in cecum or colon. As expected owing to different concentrations of oxygen available in stomach vs. cecum or colon, the composition within the Firmicutes phylum differed significantly between stomach, cecum and colon samples, with a very high proportion of Lactobacillaceae in the gastric samples (84% vs. 2.2% in cecum and colon), and high content in Ruminococcaceae and Lachnospiraceae in the cecum/colon samples. There were no significant differences of Firmicutes composition between infected and control animals. H. pylori-infected SPF INS-GAS/FVB mice showed a significantly increased species richness (ACE and Chao estimators) in all analyzed parts of the GI tract. Details are reported in Supplementary Materials.
Figure 6.
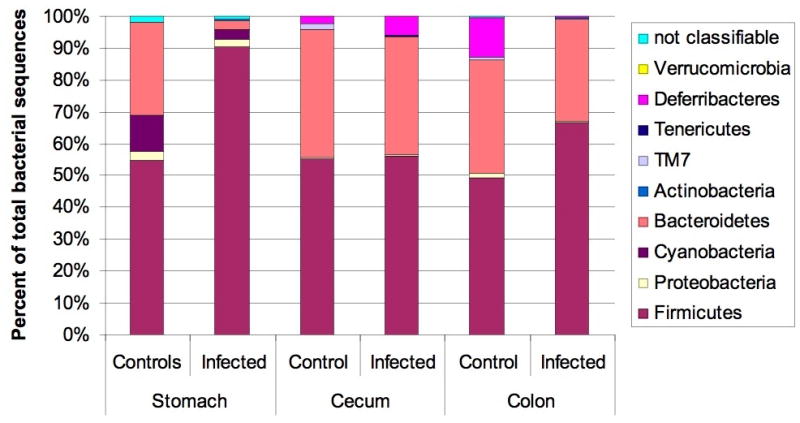
Microbiota composition in stomach, cecum and colon of H. pylori infected male INS-GAS mice (n=3, 15 wpi) vs. uninfected controls (n=2). Note the significant increase in the relative abundance of Firmicutes and decrease of Bacteroidetes in the stomachs of H. pylori infected mice (p<0.05), while no significant changes were observed in the other parts of the gastrointestinal tract.
Discussion
While the presence of gastric atrophy (loss of parietal cells) predisposes humans to the development of gastric cancer, it is clear that the time course for progression is quite delayed, suggesting that other factors may be required. Acute parietal cell loss (such as in the DMP777 transgenic mouse model) is not sufficient [18]. The presence of chronic inflammation, associated with H. pylori infection, is also presumed to be required. Parietal cell loss has a number of consequences, including the loss of gastric acid secretion which inhibits the growth of most (non-H. pylori) bacteria. Indeed, several studies have suggested a variety of enteric bacteria colonize the hypochlorhydric stomach. Bacterial overgrowth of the stomach leads to increased conversion of dietary nitrates into carcinogens such as N-nitrosamines or nitric oxide, both potent mutagens, thus promoting gastric cancer. Additionally, overgrowth with enteric organisms may enhance inflammatory responses accelerating atrophy, metaplasia and cancer. Thus, there is a strong correlation between H. pylori infection, inflammation leading to gastric atrophy and bacterial overgrowth [13,14,19–22]. Enteric bacterial overgrowth plus H. pylori infection may be the predominant causes of gastric atrophy, rather than strictly H. pylori infection [23,24]. A number of animal studies demonstrated bacterial overgrowth in the setting of achlorhydria, or infection with non-H. pylori pathogens (such as Acinetobacter lwoffii) can lead to chronic gastritis and atrophy [25,26]. Indeed, in late stages of chronic H. pylori infection, H. pylori colonization decreases, and thus the beneficial effects of antibiotic eradication regimens could be due as much to effects on the bacterial overgrowth as they are to specific eradication of H. pylori [13].
Prior reports in H. pylori-monoassociated tox 176 transgenic mice on a FVB/N background and wild type FVB/N mice demonstrated direct causation between gastritis and H. pylori infection [27]. Similarly, H. felis-monoassociated Swiss Webster mice and H. felis-monoassociated Sprague Dawley rats developed chronic active gastritis by 8 weeks post infection [28,29]. However, in the current study, we have attempted to separate out the distinct effects of H. pylori and enteric flora on gastric carcinogenesis. Our findings, for the first time, demonstrate H. pylori-infection alone can cause gastric cancer in INS-GAS mice. In general, the degree of atrophy, metaplasia, dysplasia and development of GIN in H. pylori-monoassociated male mice were markedly lower at 7 mpi than those observed in studies with H. pylori-infected SPF INS-GAS males [6,8,13]. There was no significant difference in inflammation scores between H. pylori- monoassociated and H. pylori-infected SPF male mice at 7mpi. In a H. felis, H. polygyrus co-infection study in C57BL/6 mice there were similarly increased gastric inflammatory scores in the H. felis and co-infected groups; however, the co-infected group developed less gastric atrophy, hyperplasia, and metaplasia [30]. Thus inflammation alone does not correlate with the development of preneoplastic lesions. Significant increases in gastric hyperplasia, atrophy and dysplasia were not apparent until 9 mpi in H. pylori-monoassociated mice. Consistent with these pathological changes, and previously noted in SPF H. pylori-infected mice, pro-inflammatory mediators and Th-1 associated H. pylori serum antibody were also significantly increased in H. pylori-monoassociated mice compared to uninfected controls. At 7 mpi in male H. pylori-monoassociated INS-GAS and previously reported H. pylori-infected SPF INS-GAS mice, levels of tissue-derived TNF-α and IFN-γ mRNA, as well as inflammation-associated iNOS, were significantly upregulated compared to age matched controls [13]. However, mRNA tissue levels of IFNγ, a potent proinflammatory cytokine known to induce reactive oxygen species, were four fold lower in the H. pylori-monoassociated mice compared to historical results from H. pylori-infected SPF mice, further supporting the attenuated pathology noted in the H. pylori-monoassociated mice [13,31]. The differences noted between these two models support our hypothesis that enteric microbiota colonizing the stomach (i.e. bacterial overgrowth) exacerbate H. pylori-initiated disease and thus play an important role in gastric cancer progression. Also consistent with recent studies, a Th17 response, evident by upregulation of IL-17 in gastric tissue and sera, was associated with H. pylori-induced gastric pathology in monoassociated INS-GAS mice [32–35].
Interestingly, GF INS-GAS mice demonstrated delayed development and significantly decreased hypergastrinemia associated lesions compared to age-matched SPF INS-GAS mice [6,13]. GF INS-GAS mice had lower serum gastrin levels than SPF INS-GAS mice at 11 mpi, suggesting a role for gastric colonization with enteric microbiota in the progression and severity of hypergastrinemia-associated gastritis [6]. These data are consistent with earlier published reports that antibiotic therapy of omeprazole-treated animals resulted in lower serum gastrin levels, suggesting that hypergastrinemia in the setting of hypochlorhydria was largely due to bacterial overgrowth of the stomach [26]. Thus colonization of the atrophic stomach with complex microbiota may promote gastric inflammation, premalignancy, and cancer.
We previously described male predominance of H. pylori-induced lesions in INS-GAS mice [7,8]. In this study, H. pylori-infected males consistently demonstrated earlier and more severe gastric pathology. For example, at 9 mpi, 100% of H. pylori-infected males, but only 1 of 6 infected females had low to high grade GIN. Th1-associated antibody responses, serum gastrin and host inflammatory responses were higher in infected males than infected females across several time points. Interestingly, some of these gender differences were lost with age. For example, while serum gastrin levels were significantly greater in infected males compared to females at 5 and 7 mpi, levels were similar at 9 and 11 mpi. Consistent with post-menopausal women having greater risk for developing gastric cancer, it is possible that as infected INS-GAS females passed reproductive age, lower estrogen levels resulted in loss of protection against GIN [36]. Additionally, H. pylori-infected ovariohysterectomized INS-GAS females developed significantly more severe gastritis and increased inflammatory mediator responses compared to infected intact females [12]. This effect was reversed with exogenous estrogen, demonstrating a protective effect of estradiol (E2) in H. pylori-induced gastric cancer in the INS-GAS model [12].
Once a threshold of genetic damage in gastric mucosa from chronic H. pylori-induced gastritis has been achieved and additional microflora have colonized the hypochlorhydric stomach and further promoted inflammation and progression to GIN, H. pylori may no longer be essential for progression to cancer [13,22,37]. Infection and/or host-mediated inflammation can disturb the composition of intestinal microbiota resulting in the elimination of some bacteria and overgrowth of others [38]. In the INS-GAS model, there is a loss of parietal cells, resulting in the elevated gastric pH consistently observed in atrophic gastritis [1,6]. H. pylori-monoassociated INS-GAS mice demonstrated elevated gastric pH at 7 and 9 mpi (unpublished). When the stomach mucosa is colonized with H. pylori, gastritis rapidly ensues and parietal cell loss is accelerated with subsequent colonization of different, non-indigenous microbiota such as enteric bacteria [13]. The link between hypochlorhydria, gastric microbial diversity and density has been established in human clinical trials and in an epidemiological study [39–42]. H. pylori may also increase gastric microbial mass and alter microbiota composition by improving growth conditions for gut bacteria via production of ammonia and bicarbonate substrates [20]. Our analysis by 454 pyrosequencing of the microbiota composition of a small number of H. pylori-infected male SPF INS-GAS vs. control male SPF INS-GAS mice provides evidence for a significant impact of H. pylori infection on the composition of the gastric microbiota in INS-GAS mice. Prominent alterations in the proportions of Gram-negative Bacteroidetes and of Gram-positive Firmicutes were observed that were highly significant despite the small number of animals tested. Interestingly, we also noted a significant increase in the number of species detected in all parts of the GI tract (“richness” as quantified by ACE and Chao estimators) in the H. pylori-infected mice compared to controls. A more extensive study with deeper sampling will therefore be of interest as part of future studies into the mechanisms of cancer induction by H. pylori in this model.
In conclusion, use of axenic animals can be an important component in elucidating the role of commensal and pathogenic microbiota in intestinal inflammation and disease and can be used to delineate the essential factors in gastric cancer progression [43,44]. Our findings indicate that H. pylori alone can induce cancer in INS-GAS mice; however, the absence of gastric colonization with enteric microbes significantly inhibited the development of gastric cancer in the INS-GAS model. Importantly, the INS-GAS mouse is a model of gastric corpus cancer, and our study has not addressed the importance of enteric flora in the development of antral gastric cancer. [45]. Additional gnotobiotic studies utilizing a defined group of or single commensal bacteria will provide further insight into the contributions of specific enteric microbiota in progression of H. pylori-associated gastric disease [46,47]. Immunostimulatory properties of specific bacterial species in the H. pylori-infected INS-GAS model also can be studied. For example, further dissection of Th17 responses in germ-free mice colonized with specific bacterial species and H. pylori can be realized [48,49].
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Grant Support: R01 AI37750 (Fox), R01 CA093405 (Wang, Fox), P30 ES02109 (Fox), P01 CA028842 (Fox), T32 RR07036 (Fox), German Research Foundation (SFB621/B8 and SFB 900/Z1) (Suerbaum).
We extend special acknowledgements to Carlos Umana and Oscar Acevedo for their expert care of our gnotobiotic animals. We are very grateful to Christine Josenhans for critical reading of the manuscript and helpful discussions, to Friederike Kops, Stella Lamprecht and Sabrina Woltemate for excellent technical assistance, and to Markus Uhr for computational support.
Abbreviations
- GIN
gastrointestinal intraepithelial neoplasia
- mpi
months post infection
- SPF
specific pathogen free
- wpi
weeks post infection
- GF
germfree
Footnotes
Disclosure: Authors have no disclosure
Jennifer L. Lofgren (study concept and design, acquisition, analysis and interpretation of data, statistical analysis, drafting and revision of manuscript), Mark T. Whary (study concept and design, analysis and interpretation of data, statistical analysis, drafting and revision of manuscript, study supervision), Zhongming Ge (study concept and design, acquisition, analysis and interpretation of data, revision of manuscript), Sureshkumar Muthupalani (study concept and design, histopathological analysis and interpretation of data, statistical analysis, drafting of manuscript), Nancy S. Taylor (study concept and design, acquisition, analysis and interpretation of data), Melissa Mobley (acquisition and analysis of data, technical), Amanda Potter (acquisition and analysis of data, technical), Andrea Varro (acquisition and analysis of data, technical, material support), Daniel Eibach (acquisition and analysis of data, revision of manuscript), Sebastian Suerbaum (analysis and interpretation of data, revision of manuscript), Timothy C. Wang (study concept and design, material support, revision of manuscript), James G. Fox (study concept and design, analysis and interpretation of data, drafting and revision of manuscript, study supervision)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner H, Rothenbacher D, Arndt V. Epidemiology of stomach cancer. Methods Mol Biol. 2009;472:467–477. doi: 10.1007/978-1-60327-492-0_23. [DOI] [PubMed] [Google Scholar]
- 3.Bruce MG, Maaroos HI. Epidemiology of Helicobacter pylori infection. Helicobacter. 2008;13 (Suppl 1):1–6. doi: 10.1111/j.1523-5378.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 4.Whary MT, Sundina N, Bravo LE, et al. Intestinal helminthiasis in Colombian children promotes a Th2 response to Helicobacter pylori: possible implications for gastric carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2005;14:1464–1469. doi: 10.1158/1055-9965.EPI-05-0095. [DOI] [PubMed] [Google Scholar]
- 5.Fox JG, Li X, Cahill RJ, et al. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology. 1996;110:155–166. doi: 10.1053/gast.1996.v110.pm8536852. [DOI] [PubMed] [Google Scholar]
- 6.Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000;118:36–47. doi: 10.1016/s0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- 7.Fox JG, Wang TC, Rogers AB, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003;124:1879–1890. doi: 10.1016/s0016-5085(03)00406-2. [DOI] [PubMed] [Google Scholar]
- 8.Fox JG, Rogers AB, Ihrig M, et al. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 2003;63:942–950. [PubMed] [Google Scholar]
- 9.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 10.Wang TC, Bonner-Weir S, Oates PS, et al. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest. 1993;92:1349–1356. doi: 10.1172/JCI116708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang TC, Koh TJ, Varro A, et al. Processing and proliferative effects of human progastrin in transgenic mice. J Clin Invest. 1996;98:1918–1929. doi: 10.1172/JCI118993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohtani M, Garcia A, Rogers AB, et al. Protective role of 17 beta -estradiol against the development of Helicobacter pylori-induced gastric cancer in INS-GAS mice. Carcinogenesis. 2007;28:2597–2604. doi: 10.1093/carcin/bgm150. [DOI] [PubMed] [Google Scholar]
- 13.Lee CW, Rickman B, Rogers AB, et al. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008;68:3540–3548. doi: 10.1158/0008-5472.CAN-07-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CW, Rickman B, Rogers AB, et al. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009;69:8166–8174. doi: 10.1158/0008-5472.CAN-08-3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabir AM, Aiba Y, Takagi A, et al. Prevention of Helicobacter pylori infection by lactobacilli in a gnotobiotic murine model. Gut. 1997;41:49–55. doi: 10.1136/gut.41.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CW, Rao VP, Rogers AB, et al. Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2−/− mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect Immun. 2007;75:2699–2707. doi: 10.1128/IAI.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers AB, Taylor NS, Whary MT, et al. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 2005;65:10709–10715. doi: 10.1158/0008-5472.CAN-05-1846. [DOI] [PubMed] [Google Scholar]
- 18.Li Q, Karam SM, Gordon JI. Diphtheria toxin-mediated ablation of parietal cells in the stomach of transgenic mice. J Biol Chem. 1996;271:3671–3676. [PubMed] [Google Scholar]
- 19.Falk PG, Syder AJ, Guruge JL, et al. Theoretical and experimental approaches for studying factors defining the Helicobacter pylori-host relationship. Trends Microbiol. 2000;8:321–329. doi: 10.1016/s0966-842x(00)01780-7. [DOI] [PubMed] [Google Scholar]
- 20.Aebischer T, Fischer A, Walduck A, et al. Vaccination prevents Helicobacter pylori-induced alterations of the gastric flora in mice. FEMS Immunol Med Microbiol. 2006;46:221–229. doi: 10.1111/j.1574-695X.2005.00024.x. [DOI] [PubMed] [Google Scholar]
- 21.Kato S, Fujimura S, Kimura K, et al. Non-Helicobacter bacterial flora rarely develops in the gastric mucosal layer of children. Dig Dis Sci. 2006;51:641–646. doi: 10.1007/s10620-006-3185-0. [DOI] [PubMed] [Google Scholar]
- 22.Wong BC, Lam SK, Wong WM, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–194. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 23.Sanduleanu S, Jonkers D, de Bruine A, Hameeteman W, Stockbrugger RW. Changes in gastric mucosa and luminal environment during acid-suppressive therapy: a review in depth. Dig Liver Dis. 2001;33:707–719. doi: 10.1016/s1590-8658(01)80050-5. [DOI] [PubMed] [Google Scholar]
- 24.Sanduleanu S, Jonkers D, De Bruine A, Hameeteman W, Stockbrugger RW. Non-Helicobacter pylori bacterial flora during acid-suppressive therapy: differential findings in gastric juice and gastric mucosa. Aliment Pharmacol Ther. 2001;15:379–388. doi: 10.1046/j.1365-2036.2001.00888.x. [DOI] [PubMed] [Google Scholar]
- 25.Zavros Y, Rieder G, Ferguson A, Merchant JL. Gastritis and hypergastrinemia due to Acinetobacter lwoffii in mice. Infect Immun. 2002;70:2630–2639. doi: 10.1128/IAI.70.5.2630-2639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zavros Y, Rieder G, Ferguson A, Samuelson LC, Merchant JL. Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology. 2002;122:119–133. doi: 10.1053/gast.2002.30298. [DOI] [PubMed] [Google Scholar]
- 27.Bjorkholm B, Guruge J, Karlsson M, et al. Gnotobiotic transgenic mice reveal that transmission of Helicobacter pylori is facilitated by loss of acid-producing parietal cells in donors and recipients. Microbes Infect. 2004;6:213–220. doi: 10.1016/j.micinf.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Fox JG, Lee A, Otto G, Taylor NS, Murphy JC. Helicobacter felis gastritis in gnotobiotic rats: an animal model of Helicobacter pylori gastritis. Infect Immun. 1991;59:785–791. doi: 10.1128/iai.59.3.785-791.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee A, Fox JG, Otto G, Murphy J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology. 1990;99:1315–1323. doi: 10.1016/0016-5085(90)91156-z. [DOI] [PubMed] [Google Scholar]
- 30.Fox JG, Beck P, Dangler CA, et al. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med. 2000;6:536–542. doi: 10.1038/75015. [DOI] [PubMed] [Google Scholar]
- 31.Yang D, Elner SG, Bian ZM, et al. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85:462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2009;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khamri W, Walker MM, Clark P, et al. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun. 2009;78:845–853. doi: 10.1128/IAI.00524-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otani K, Watanabe T, Tanigawa T, et al. Anti-inflammatory effects of IL-17A on Helicobacter pylori-induced gastritis. Biochem Biophys Res Commun. 2009;382:252–258. doi: 10.1016/j.bbrc.2009.02.107. [DOI] [PubMed] [Google Scholar]
- 35.Shiomi S, Toriie A, Imamura S, et al. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter. 2008;13:518–524. doi: 10.1111/j.1523-5378.2008.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sipponen P, Correa P. Delayed rise in incidence of gastric cancer in females results in unique sex ratio (M/F) pattern: etiologic hypothesis. Gastric Cancer. 2002;5:213–219. doi: 10.1007/s101200200037. [DOI] [PubMed] [Google Scholar]
- 37.Dicksved J, Lindberg M, Rosenquist M, et al. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J Med Microbiol. 2009;58:509–516. doi: 10.1099/jmm.0.007302-0. [DOI] [PubMed] [Google Scholar]
- 38.Lupp C, Robertson ML, Wickham ME, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Mowat C, Williams C, Gillen D, et al. Omeprazole, Helicobacter pylori status, and alterations in the intragastric milieu facilitating bacterial N-nitrosation. Gastroenterology. 2000;119:339–347. doi: 10.1053/gast.2000.9367. [DOI] [PubMed] [Google Scholar]
- 40.Stark CA, Adamsson I, Edlund C, et al. Effects of omeprazole and amoxycillin on the human oral and gastrointestinal microflora in patients with Helicobacter pylori infection. J Antimicrob Chemother. 1996;38:927–939. doi: 10.1093/jac/38.6.927. [DOI] [PubMed] [Google Scholar]
- 41.Adamsson I, Edlund C, Nord CE. Impact of treatment of Helicobacter pylori on the normal gastrointestinal microflora. Clin Microbiol Infect. 2000;6:175–177. doi: 10.1111/j.1469-0691.2000.00028.x. [DOI] [PubMed] [Google Scholar]
- 42.Kato S, Tsukamoto T, Mizoshita T, et al. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int J Cancer. 2006;119:1558–1566. doi: 10.1002/ijc.21810. [DOI] [PubMed] [Google Scholar]
- 43.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Wagner RD. Effects of microbiota on GI health: gnotobiotic research. Adv Exp Med Biol. 2008;635:41–56. doi: 10.1007/978-0-387-09550-9_4. [DOI] [PubMed] [Google Scholar]
- 45.Takaishi S, Tu S, Dubeykovskaya ZA, et al. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. Am J Pathol. 2009;175:365–375. doi: 10.2353/ajpath.2009.081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarma-Rupavtarm RB, Ge Z, Schauer DB, Fox JG, Polz MF. Spatial distribution and stability of the eight microbial species of the altered schaedler flora in the mouse gastrointestinal tract. Appl Environ Microbiol. 2004;70:2791–2800. doi: 10.1128/AEM.70.5.2791-2800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dewhirst FE, Chien CC, Paster BJ, et al. Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol. 1999;65:3287–3292. doi: 10.1128/aem.65.8.3287-3292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivanov, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ivanov, Frutos Rde L, Manel N, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.