

Abstract
Background & Aims
The immunoregulatory cytokine interleukin (IL)-10 is required to maintain immune homeostaisis in the gastrointestinal tract. IL-10-null mice spontaneously develop colitis or are more susceptible to induction of colitis by infections, drugs, and autoimmune reactions. IL-13 regulates inflammatory conditions; its activity might be compromised by the IL-13 decoy receptor (IL-13Rα2).
Methods
We examined the roles of IL-13 and IL-13Rα2 in intestinal inflammation in mice. To study the function of IL-13Rα2, il10−/− mice were crossed with il13rα2−/− to generate il10−/− il13rα2−/− double knockout mice (dKO). Colitis was induced with the gastrointestinal toxin piroxicam or Trichuris muris infection
Results
Induction of colitis by interferon (IFN)-γ or IL-17 in IL-10-null mice requires IL-13Rα2. Following exposure of il10−/− mice to piroxicam or infection with Trichuris muris, production of IL-13Rα2 increased, resulting in decreased IL-13 bioactivity and increased intestinal inflammation in response to IFN-γ or IL-17A. In contrast to il10−/− mice, dKO mice were resistant to piroxicam-induced colitis; they also developed less severe colitis during chronic infection with T. muris infecion. In both models, resistance to IFN-γ and IL-17-mediated intestinal inflammation was associated with increased IL-13 activity. Colitis susceptibility was restored when the dKO mice were injected with monoclonal antibodies against IL-13, confirming its protective role.
Conclusion
Colitis and intestinal inflammation in IL10−/− mice results from IL-13Rα2-mediated attenuation of IL-13 activity. In the absence of IL-13Rα2, IL-13 suppresses pro-inflammatory Th1 and Th17 responses. Reagents that block the IL-13 decoy receptor IL-13Rα2 might be developed for inflammatory bowel disease associated with increased levels of IFN-γ and IL-17.
Keywords: helminth, IBD
Introduction
Studies have suggested that immune homeostasis in the gastrointestinal tract is maintained by a variety of immunoregulatory mechanisms1. Although regulatory T cells play a critical role2, recent studies have suggested that resident non-hematopoietic cells, including epithelial, smooth muscle, and fibroblasts are critically involved in maintaining homeostasis in the gut 3. However, the mechanisms by which these cells regulate immune homeostasis in the intestine remain incompletely defined.
Crohn’s disease (CD) and ulcerative colitis (UC) are believed to be induced by distinct immunological mechanisms4, with mixed Th1/Th17 responses mediating CD5 and persistent Th2-type responses triggering UC6. In the case of Crohn’s disease, a variety of mechanisms have been shown to limit the production of IFN-γ/IL-17A and development of severe disease, including regulatory cell populations7, immunoregulatory cytokines8, and anti-inflammatory proteins9. However, the cytokine IL-10 has emerged as a key suppressive mediator. Indeed, animal studies10 and genetic linkage-association studies have revealed an important protective role for IL-10 in colitis11–13. Although Th2 cytokines, in particular IL-4 and IL-13, can also antagonize Th1/Th17 responses, the mechanisms by which Th2 responses regulate the development of colitis remain much less clear14–15.
Here, using two independent models of chemical and infection induced intestinal inflammation, we show that the development of Th1-Th17-dependent disease in il10−/− mice16 is tightly regulated by the IL-13 decoy receptor (IL-13Rα2). During T. muris infection or following exposure to the non-steroidal anti-inflammatory drug piroxicam (a gastrointestinal toxin)17, production of IL-13Rα2 increased in the absence of IL-10, consistent with our previous studies in the lung and liver21, resulting in decreased IL-13 bioactivity and markedly increased IFNγ/IL-17A-driven intestinal inflammation. As such, these studies reveal a previously unrecognized role for IL-13 and its decoy receptor in the regulation of Th1-Th17 responses in the gut. Because the IL-13Rα2 chain is primarily expressed on epithelial cells, smooth muscle, and fibroblasts, they also illustrate a novel mechanism for cells of non-hematopoietic origin to control IFN-γ/IL-17-mediated intestinal inflammation. Finally, using in vitro polarized CD4+ T cells, we confirm that Th17 cells express a functional IL-13 receptor18, which when activated with IL-13 can directly reduce the frequency of Th17 cells and secretion of IL-17A, thus providing an additional mechanism for IL-13 to limit Th17-dependent pathology in the gastrointestinal tract.
Materials and methods
Animals
Female C57BL/6, BALB/c, BALB/c il13rα2−/−, il13rα1−/−, il10−/− and il10−/−il13rα2−/− 6 – 8 week old mice were obtained from Taconic. Animals were housed under specific pathogen-free conditions at the NIH in an American Association for the Accreditation of Laboratory Animal Care–approved facility. The NIAID animal care and use committee approved all experimental procedures. A minimum of 5 mice per group was used in each experiment, unless indicated.
Piroxicam-induced Colitis
Animals were fed normal animal chow mixed with piroxicam (200 ppm) for 14 consecutive days. Animals were weighed daily and euthanized at day 14 for analysis.
Trichuris muris infection
Mice were infected orally with 200 embryonated T. muris eggs, as described19, 20.
Histopathology
For histopathological analyses, tissues were fixed in 4% phosphate buffered formalin and embedded in paraffin for sectioning. Wright’s Giemsa, hematoxylin and eosin (H&E) or alcian blue periodic acid Schiff (AB-PAS) stains were used. Sub-mucosal inflammation, intramuscular inflammation, mucus and ulcer frequency and severity were scored by a blinded observer on a 1–4+ basis. Eosinophil score was based on % eosinophilia. The same individual scored all histological features and had no knowledge of the experimental groups.
In vitro cell culture
Lymph node cells were isolated, washed and plated at 5×105 cells per well of a 96-well plate and stimulated with 10μg/ml of T.muris antigen19, 20. For in-vitro Th1 and Th17 differentiation, FACS-purified naïve CD4+CD62LhiCD44lo T cells were stimulated under Th17 (rIL-6 (R&D, 20ng/ml), rhTGFβ (R&D, 5ng/ml), anti-IL-4 (11D11, 10 μg/ml) and anti-IFNγ (XMG1.1, 10μg/ml)) or Th1 (IL-12 (R&D, 10ng/ml) and anti-IL-4 (11D11, 10μg/ml)) conditions with or without rIL-13 at indicated concentrations.
Polymorphonucelar cell (PMN) Analysis
EDTA-treated blood was processed for automated counting using Vista Analyzer (Siemens).
RT-PCR
RNA was isolated from tissue or cells in 1 ml TRIZOL reagent (Invitrogen) and processed as previously described 21, 22. Real-time RT-PCR was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). mRNA levels for each sample were normalized to hypoxanthine guanine phosphoribosyl transferase (HPRT). Primers were either designed using Primer Express software (version 2.0; Applied Biosystems) or adopted from previously published primer sequences21, 22.
ELISA
Cytokines were measured by ELISA using Immulon 2HB plates (Thermo) and manufacturers guidelines. Paired capture and detection antibodies from R&D for IL-17A, IFNγ, IL-4, IL-10 and IL-13 were used. Plates were washed with 0.05% Tween 20 in PBS (PBST) and blocked with 5% milk in PBST. Recombinant cytokine standards (R&D) were used to assess quantity using a standard curve, with OD acquired at 405 nm in an ELISA reader.
Flow cytometry
Following a 3 hour incubation with phorbol 12-myristate 13-acetate (PMA, 10ng/ml), Ionomycin (1μg/ml) and brefeldin A (BFA, 10μg/ml), cells were stained with antibodies diluted in PBS with 0.5% BSA (Sigma-Aldrich) and 0.05% sodium azide (Sigma-Aldrich) for 20 minutes at 4°C. Surface molecule staining (CD4 (BD), CD25 (eBioscience), CD69 (BioLegend), CD44 (BD), CD62L (BD) followed by fixation and permeabilization (BD Cytofix/Cytoperm™) and intracellular staining (IL-17A (BD), IFNγ (BD), Foxp3 (eBioscience)) were carried out on freshly isolated cells. The expression of surface molecules and intracellular cytokines were analyzed on a BD LSR II flow cytometer using FlowJo v.8 software (Tree Star).
Statistical analysis
Data sets were compared using a Mann Whitney test or Kruskal-Wallis test where appropriate, using Prism software v5. Differences were considered significant (*) at P < 0.05.
Results
IL-17A and IFN-γ-associated colitis is reduced in the absence of IL-13Rα2
IL-10 is a critical immunoregulatory cytokine, which maintains intestinal homeostasis and symbiosis with enteric microflora. Germ-line deletion of IL-10 in mice results in spontaneous colitis after 3–4 months, driven by dysregulated immune responses to colonic flora 16. The onset of colitis can be accelerated and synchronized by feeding mice piroxicam, a non-steroidal anti-inflammatory drug (NSAID)23. It has been demonstrated that piroxicam-induced colitis in il10−/− mice is attenuated following intestinal helminth infection15, with infection-driven Th2 responses correlating with protection. Because the bioactivity of IL-13 is attenuated by the IL-13 decoy receptor, IL-13Rα2 22, 24, we tested the role of IL-13Rα2 in piroxicam-induced colitis by deleting IL-13Rα2 on the colitis-prone il10−/− background (dKO, il10−/−il13Rα2−/−). In contrast to il10−/− mice, il10−/−il13Rα2−/−dKO mice were protected from both sub-mucosal and intramuscular inflammation, with less severe colonic ulcers (Fig. 1A). Intestinal pathology in il10−/− mice correlated with weight loss 14 days post treatment and increased polymorphonuclear cells (PMN) (Fig. 1B). dKO mice however did not lose weight and had significantly reduced circulating PMN’s compared to il10−/− mice, correlating with reduced disease severity. These findings suggest that either IL-13Rα2 directly induces disease25, 26, or that IL-13Rα2 functions as a decoy receptor and suppresses a critical anti-inflammatory function of IL-13.
Figure 1. Deletion of IL-13Rα2 suppresses IL-17A and IFNγ production and protects il10−/− mice from piroxicam-induced colitis.
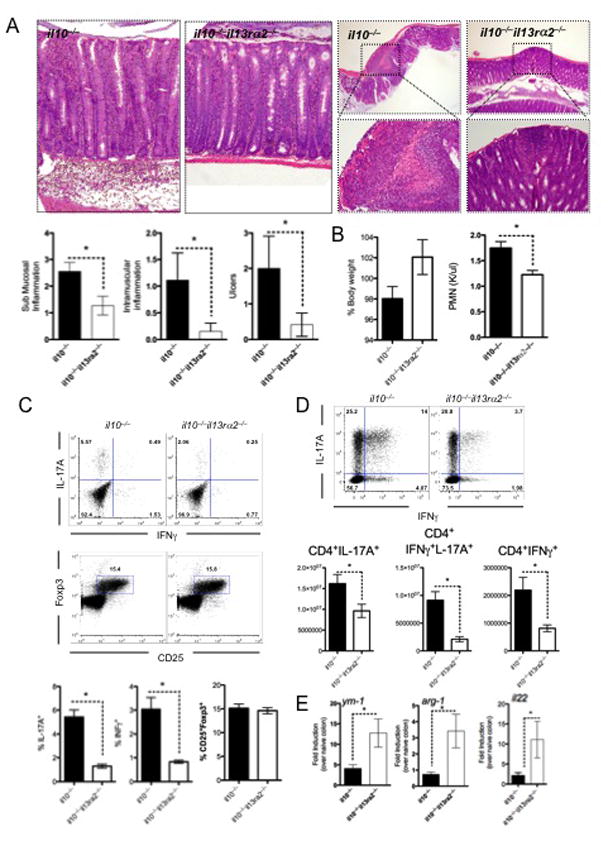
Mice were fed piroxicam infused food for 14 days, with colitis assessed on day 14. Five animals per group were used with 1 of 2 experiments shown. Mean±SEM are shown with * p<0.05 considered statistically significant.
A- 5μm sections of paraffin-embedded colon were stained with H&E and assessed for pathological abnormalities.
B- Animal weights and circulating PMN’s measured in whole blood after 14 days of piroxicam-infused food.
C- Mesenteric lymph node (MLN) cells stimulated with PMA and ionomycin in the presence of BFA and stained with anti-mouse CD4, CD25, IL-17A and IFNγ and Foxp3.
D- Lamina propria lymphocytes isolated and quantified at day 14 were stimulated with PMA and ionomycin in the presence of BFA and stained with anti-mouse CD4, IL-17A and IFNγ.
E- RNA was extracted from the colon of mice, 14 days post piroxicam exposure with ym-1, arg1 and il22 gene transcripts quantified and expressed relative to HPRT.
IL-17A and IFNγ are elevated in IBD patients 27, 28 and both are thought to mediate colitis following piroxicam treatment 29, similar to other models of colitis 5, 30. As expected, significant numbers of IL-17A and IFNγ-producing T cells were observed in the MLN (Fig. 1C) and to a greater extent in the lamina propria of the colon (Fig. 1D) of il10−/− mice following piroxicam treatment. Strikingly however, the frequency and total number of Th1−/−Th17 cells were reduced in dKO mice compared to il10−/− mice, while the number of CD25+Foxp3+ Treg cells was not significantly different. Consistent with this, several Th1-Th17 associated cytokines and chemokines, measured in colon-derived mRNA, were also significantly reduced in the absence of IL-13Rα2 (Sup. Fig. 1), providing an explanation for the reduced circulating PMN’s. Expression of two IL-13/STAT-6-regulated genes, Chi3l3 (Ym1) and Arg1, were markedly increased in the dKO mice (Fig. 1E) compared to il10−/− mice, indicating that IL-13Rα2 was attenuating IL-13 activity in the disease prone il10−/− mice. Furthermore, IL-22, a cytokine which can facilitate mucosal wound healing and protect mice from IBD31, was also significantly elevated in the colon of dKO mice (Fig. 1E). Together these data demonstrate that enhanced IL-13 bioactivity, uncovered in the absence of IL-13Rα2, significantly suppresses Th1/Th17-mediated inflammation and immunopathology. Consequently, these data suggest that the IL-13 decoy receptor (IL-13Rα2) like IL-10, controls susceptibility to Th1/Th17-driven inflammation in the gastrointestinal tract.
Lethal gastrointestinal helminth infection is reversed in the absence of IL-13Rα2
Following infection of genetically resistant mice with the gastrointestinal helminth parasite T. muris, immunity is mediated by a CD4+ Th2 response32, with IL-13 playing a dominant role in resistance33. We showed that IL-10 is critically involved in generating a polarized and protective IL-13 response during T. muris infection20. In the absence of IL-10, parasites are not expelled and the mice develop a lethal intestinal inflammatory response (Fig 2A). The intestinal pathology in il10−/− mice is characterized by reductions in goblet cells, Gob5 mRNA expression, eosinophils, and mucus secretion (Fig. 2B and 2C) and correspondingly increased production of IFN-γ and IL-17A relative to WT mice (Fig 2D). Prior studies conducted with il10−/−il12p40−/− double KO mice suggested that the IFN-γ/IL-17A axis plays a critical pathogenic role in this model 20. Following the observation that IL-13 decoy receptor critically controls susceptibility to piroxicam-induced colitis, we performed a similar series of experiments with T. muris infected il10−/− mice and examined whether the development of lethal Th1/Th17-driven intestinal inflammation during infection is controlled by IL-13 and/or IL-13Rα2.
Figure 2. Lethal intestinal helminth infection in il10−/− mice is reversed by deleting IL-13Rα2.
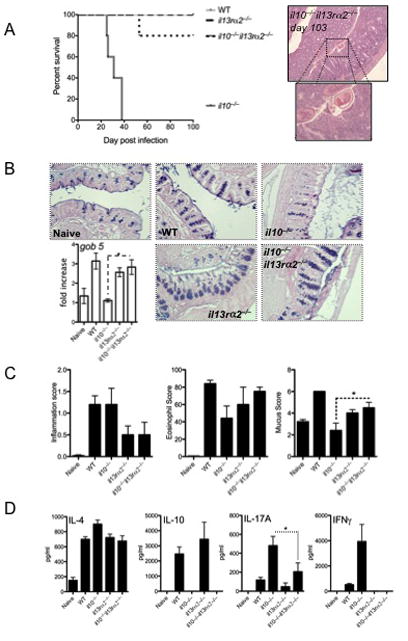
Mice were infected with 200 T. muris eggs and monitored for survival or euthanized and assessed on day-15. Five animals per group were used with 1 of 3 experiments shown. Mean±SEM are shown with p<0.05 considered statistically significant.
A- Survival of mice with evidence of infection at day 103.
B- 5μm sections of paraffin-embedded caecum were stained with AB-PAS and assessed for goblet cell frequency and gob5 gene expression.
C- Inflammation, Eosinophilia and mucus-producing cells was scored from 5μm sections of paraffin-embedded caecum.
D- Mesenteric lymph node cells re-stimulated with 10μg of T. muris antigen were cultured for 4 days. IL-4, IL-10, IL-17A and IFNγ production were measured by ELISA.
Strikingly, although 100% of the il10−/− mice succumbed within 40 days of infection, 80% of the dKO mice survived greater than 100 days (Fig. 2A), despite the fact that they still harbored parasites (Fig. 2A, photomicrographs). Although the quantity of IL-13 in the caecum was similar for all genotypes (Sup. Fig 2), the dKO mice displayed enhanced IL-13 effector function compared to il10−/− mice, confirmed by increased gob5 mRNA and AB-PAS staining (Fig. 2B), increased ccl11 (eotaxin) and tissue eosinophilia (Fig. 2B, C) and increased retlna (FIZZ-1) mRNA responses in the caecum (Sup. Fig 2). As with piroxicam, T. muris-infected il10−/− mice developed significant IL-17A and IFN-γ responses. In contrast, dKO mice displayed minimal IFN-γ/IL-17 responses (Fig. 2D), which resulted in reduced intestinal inflammation compared to il10−/− mice (Fig. 2C). IFNγ-mediated cxcl10 (IP-10) production was also decreased in the dKO (Sup. Fig 2). Thus, although the dKO mice remained infected (>100 days), the enhanced IL-13 effector response revealed in the absence of IL-13Rα2, led to the suppression of IFN-γ/IL-17 responses (Fig 2D), resulting in markedly decreased morbidity and prevention of mortality.
IL-13 negatively regulates Th17 cell differentiation and cytokine secretion in vitro
Deletion of IL-13Rα2 on the il10−/− background resulted in increased IL-13 bioactivity and reduced IFN-γ/IL-17A responses in both models. This is consistent with a recent study that reported exaggerated IL-17A responses in the absence of IL-4 and IL-1334. Although IL-13 has potent anti-inflammatory activity in vivo35 the mechanisms by which IL-13Rα2 controls pro-inflammatory Th17 responses remains unclear. Given the dramatic effect IL-13Rα2 deficiency had on the development of the pathogenic Th1/Th17 responses, we investigated if IL-13 signaling was responsible for the altered response. FACS-purified naïve CD4+ T cells were polarized under Th1, Th2 or Th17 conditions and examined for cytokine production and cytokine receptor expression. As expected, IL-4R mRNA was up-regulated during Th2 polarizing conditions, with only marginal expression detected on Th17 cells (Fig. 3A). Unexpectedly however, IL-13Rα1 was significantly up-regulated on both Th1 and Th17 cells (Fig 3A), confirming a previous observation18. To investigate the functional significance of type II IL-4 receptor expression (IL-4Rα and IL-13Rα1), we polarized naïve CD4+ T cells under Th1 and Th17 conditions in the presence of recombinant IL-13. Although IL-13 had no direct impact on Th1 polarized cells (Sup. Fig 3), the frequency of Th17 cells and secretion of IL-17A was significantly reduced in the presence of IL-13 (Fig 3B), suggesting that IL-13 signaling was negatively regulating the development of Th17 cells, confirming recent observations by Newcomb and colleagues18. Thus, IL-13 likely antagonizes CD4+ Th17 responses by both direct and indirect mechanisms36.
Figure 3. In vitro Th17 cells possess a functional IL-13 receptor that regulates IL-17A production.
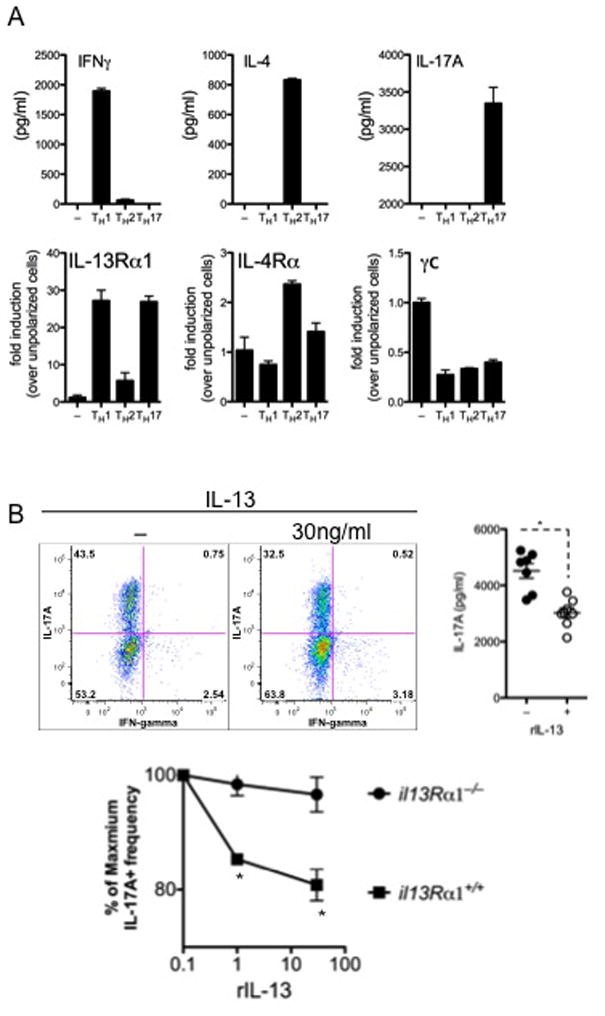
FACS-purified naïve CD4+CD62LhiCD44lo T cells were stimulated under Th1, Th2 or Th17 conditions. One of 4 experiments is shown. Mean±SEM are shown with p<0.05 considered statistically significant.
A- Canonical Th1 (IFNγ), Th2 (IL-4) and Th17 (IL-17A) cytokines were measured in culture supernatants. mRNA transcripts for il13Rα1, il4rα and γC were measured from polarized cells.
B- Th17-polarized cells cultured with rIL-13. Th17 frequency and IL-17A secretion were measured by flow cytometry and ELISA, respectively.
IL-13 suppresses Th1 and Th17 cells in vivo and protects mice from lethal intestinal inflammation
To determine whether IL-13 was responsible for the reduced Th1/Th17 responses in dKO mice, T. muris-infected dKO mice were treated with a neutralizing anti-IL-13 mAb. Efficient neutralization of IL-1337 was confirmed by the marked decrease in gob 5 mRNA (Fig. 4D) and reduced mucus staining observed in the caecum of anti-IL-13-treated WT mice (Fig. 4E). As expected, infected il10−/− mice displayed significant weight loss (Fig. 4A), marked mortality (Fig. 4B), reduced gob5 mRNA expression (Fig. 4D) and decreased mucus producing cells (Fig. 4E). These observations correlated with significant increases in parasite-specific IFN-γ and IL-17A production in the mesenteric lymph nodes (Fig. 4C), increased sub-mucosal inflammation (Fig 4E), and heightened IL-13Rα2 expression in the caecum (Fig. 4D). In contrast, il10−/−il13Rα2−/− dKO mice displayed a completely opposite phenotype, suggesting that, as observed with piroxicam (Fig 2), the absence of the IL-13 decoy receptor was sufficient to restore the protective effects of IL-13. To confirm this hypothesis, additional dKO mice were treated with anti-IL-13 mAb. Similar to il10−/− control group, anti-IL-13 treated dKO animals lost weight, displayed reduced mucus responses (Fig. 4D, 4E), and rapidly succumbed to the infection. Production of the pro-inflammatory cytokines IFN-γ and IL-1A7 was also restored when IL-13 was neutralized (Fig. 4C and 4D), confirming that IL-13Rα2 was functioning as a decoy receptor for IL-13 in the gastrointestinal tract and blocking the protective potent protective effects of IL-13.
Figure 4. IL-13 regulates Th17 and Th1 responses and protects mice from a lethal infection.
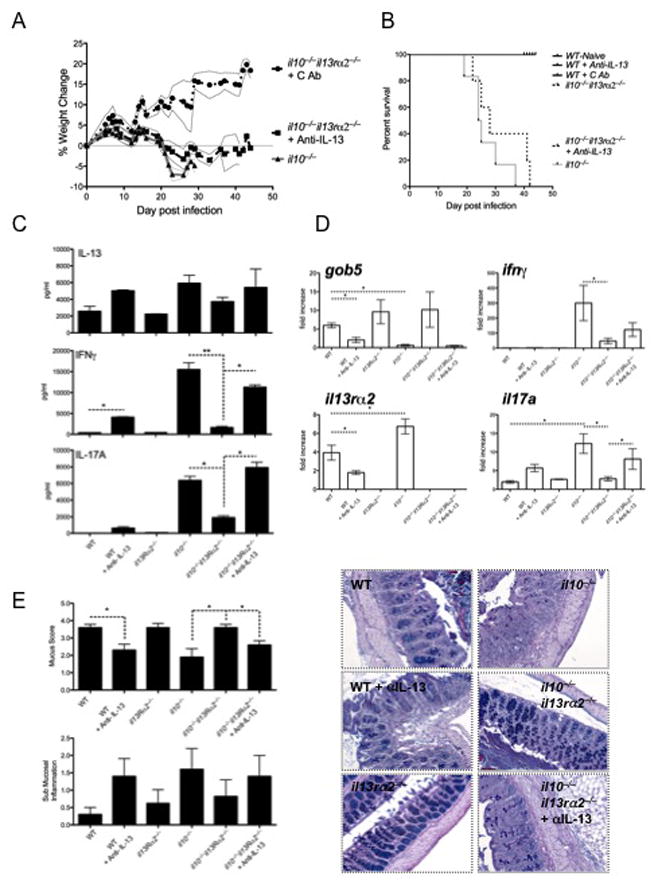
Mice were infected with 200 T. muris eggs and monitored for weight loss and survival or euthanized and assessed at day-21. Animals were treated with 500μg of anti-IL-13 mAb per week starting on day 1. Five animals per group were used with 1 of 2 experiments shown. Mean±SEM are shown with p<0.05 considered statistically significant.
A- Weight monitored daily.
B- Survival of mice.
C- Mesenteric lymph node cells re-stimulated with 10μg of T. muris antigen were cultured for 4 days. IL-13, IFNγ and IL-17A production was measured by ELISA.
D- RNA isolated from the caecum was reverse transcribed and assessed for gob5, ifnγ, il13rα2 and il17a transcripts.
E- 5μm sections of paraffin-embedded caecum were stained with Giemsa and AB-PAS (shown) and assessed for submucosal inflammation and mucus-producing cell frequency.
Discussion
Following piroxicam treatment or infection with T. muris, IFN-γ and IL-17A-driven intestinal inflammation correlated with elevated IL-13Rα2. Deletion of IL-13Rα2 abrogated IFN-γ and IL-17A and significantly attenuated the degree of inflammation in both models. These data suggest that IL-13Rα2 is directly responsible for the development of inflammation, as suggested in a different model of colitis25. Alternatively, these observations suggest that IL-13Rα2 was blocking IL-1322, 24, and that IL-13 functions as a negative regulator of IFN-γ and IL-17A. Following piroxicam treatment or T. muris infection of il10−/−il13Rα2−/− dKO mice, we observed evidence of increased IL-13 activity (increased ym-1 and arg-1 or elevated goblet cell and mucus responses, respectively) and decreased IFN-γ and IL-17A, supporting the latter model. Thus, increased IL-13 activity in the absence of IL-13Rα2 led us to hypothesize that IL-13Rα2 blocks the protective effects IL-13. We tested this hypothesis by neutralizing IL-13 in il10−/−il13Rα2−/− dKO mice, which restored IFN-γ and IL-17A responses, inflammation and mortality following T. muris infection. These observations clearly indicate that the balance between IL-13 and the IFN-γ/IL-17A axis tightly regulates the degree of intestinal inflammation. Furthermore these data also demonstrate that IL-13Rα2 is intricately involved in the regulation of this balance and consequently, the development of IBD.
Because CD and UC have distinct inflammatory etiologies, with Th1-Th17-associated Crohn’s disease developing in the small and large bowel38 and IL-13-mediated ulcerative colitis occurring primarily in the colon39, the suppression of IL-13 activity by the decoy receptor may be a critical event in the genesis of Th1-Th17 driven inflammation in the gastrointestinal tract. In our studies, the enhanced IL-13 activity observed in the colon of il10−/−il13Rα2−/− mice suppressed the development of Th1-Th17–associated colitis. These data present a new mechanism of IL-13-mediated control of Th1/17 responses. They may also explain why ulcerative colitis and Crohn’s disease are rarely if ever identified in the same individual40, as the key inducer of UC (IL-13) appears to be a negative regulator of Th1-Th17-associated CD.
Interestingly, a recent paper by Shea-Donohue and colleagues41 showed that IL-13 and IL-13Rα2 are expressed much more in the colon than small intestine. Thus, changes in IL-13 and IL-13Rα2 expression might be expected to have a much larger impact on the development of Th1-Th17-mediated inflammation in the colon than in other areas of the gastrointestinal tract. However, the marked absence of IL-13Rα2 in the small bowel might also lead to increased IL-13 bioactivity in this region and thus provide critical protection from Th1-Th17-dependent colitis, while increasing the risk of IL-13-driven UC. Clinical findings support the notion that IL-13 may be protective in Crohn’s disease, as Crohn’s disease patients produce less IL-4 and IL-13 and their PBMC’s are hypo-responsive to IL-1342.
The hypothesis that IL-13 can cross-regulate the development of Th17 cells is supported by the inverse relationship we observed between IL-17A and IL-13. Although IFNγ and IL-17A production was more marked in the absence of IL-10, they also increased when WT mice were treated with anti-IL-13, further supporting a role for IL-13 in the suppression of IFN-γ/IL-17A production in the gut. Together, these observations suggest that in addition to inducing goblet cell hyperplasia43, epithelial cell turnover44, and production of Relm-β (Fizz-2/Retnlb)19, IL-13 also plays a key protective role in the gut by suppressing CD4+ Th1 and Th17 cell development33. When viewed together, these studies reveal a novel protective pathway for IL-13 and its decoy in the control of Th1/Th17-mediated inflammation in the gastrointestinal tract.
In contrast to our findings, two related studies investigating the role of IL-13Rα2 in a model of chronic trinitrobenzene sulfonic acid (TNBS)-induced colitis concluded that IL-13Rα2 functions as a signaling receptor for IL-13 25, 26. Using either a soluble IL-13Rα2-Fc protein or IL-13Rα2-specific small interfering RNA’s to block IL-13Rα2, they concluded that IL-13 signals through IL-13Rα2 and induces IL-13-dependent fibrosis. Although the TNBS model was not employed in our studies, we found no evidence that IL-13Rα2 was functioning as a signaling receptor for IL-13. In fact, our data suggested that IL-13Rα2 primarily blocks IL-13 effector functions, supporting its role as a decoy receptor. We concluded that by blocking IL-13 activity, IL-13Rα2 plays an indispensable role in the genesis of Th1/Th17-associated intestinal pathology in il10−/− mice.
The explanation for the different conclusions is not clear, although the sIL-13Rα2-Fc blocker used in their studies binds IL-13, not IL-13Rα2; therefore it is not capable of distinguishing between IL-13Rα1 and IL-13Rα2-mediated effects. The use of small interfering RNAs in vivo may also have important off-target effects45. Our studies with il13Rα2−/− and il10−/−il13Rα2−/− dKO mice all point to IL-13Rα2 functioning as a decoy receptor for IL-13 in the gastrointestinal tract.
In contrast to the well-known pathogenic properties of IL-13 46, 47, to our knowledge, this is the first study to provide evidence of a tissue-protective role for IL-13 in the GI tract. A similar protective role for IL-13 has been described in the liver following ischemia/reperfusion injury48 and in the CNS following MOG33-55 immunization49. In both cases, pro-inflammatory cytokine production was suppressed, either following rIL-13 treatment or following the induction of IL-13 by IL-25, with hepatic Kupfer cells48 and APC’s49 identified as the key targets of IL-13. It has previously been reported that IL-13 can abrogate Th1 development indirectly 50, by modulating APC function (reviewed in 35, 51); however, this is one of the first studies implicating IL-13 as a negative regulator of Th17 development in vivo.
Previously, using models of pulmonary and hepatic inflammation, we demonstrated that IL-13Rα2 is upregulated by IL-4 and IL-13 and suppressed by IFNγ in vivo52. The soluble form of the IL-13Rα2 was also found in abundance in un-manipulated mice22 and consistent with our findings, was expressed at quite high levels in the colon41. In the lung and liver the decoy IL-13Rα2 restricts the pro-fibrotic and pathogenic activities of IL-13 during allergic inflammation and chronic helminth infection22. Therefore, in some circumstances, disrupting IL-13Rα2 might have unintended consequences such as exacerbating IL-13-driven diseases like allergic asthma or infection induced hepatic fibrosis 21, 46. This report identifies a similar role for IL-13Rα2 in the intestine, however instead of restricting IL-13 and preventing IL-13 related pathology, our data suggest that IL-13Rα2 is key to generating IFNγ/IL-17A-driven inflammation and immunopathology in the bowel. The heightened susceptibility of IL-10-deficient mice to colitis is clearly controlled by this mechanism, as il10−/− mice developed increased IL-13Rα2 responses and reduced IL-13 activity during T. muris infection or following treatment with piroxicam. In both cases this led to the development of IFNγ/IL-17-associated colitis and intestinal inflammation. Consequently, reagents that specifically target and inhibit the IL-13 decoy receptor and enhance IL-13 effector function might offer a new strategy to prevent or reverse inflammatory bowel disease.
Supplementary Material
Acknowledgments
The authors acknowledge the meticulous care of all animals by Nicole Mahoney, Joy McFarlane, Jose Encarnacion, Lauren Donato and SoBran staff. We thank Mary Collins and Marion Kasaian at Wyeth LLC for providing IL-13Rα2-deficient mice. We thank Sandra D. White, Robert W. Thompson and NIH Clinical Center staff for additional animal care, technical assistance and CBC processing. We also thank Dr. Luke Barron and Dr. Kevin Vanella for helpful and constructive discussion. This research was supported by the Intramural Research Program of the NIH, NIAID.
Abbreviations
- dKO
double knockout
Footnotes
Wilson – study design, acquisition and analysis of data, co-wrote the paper, Ramalingam – acquisition of data, Rivollier - study design, acquisition and analysis of data, Shenderov - acquisition of data, M. Mentink – acquisition of data, Madala - acquisition of data, Cheever - study design, acquisition and analysis of data, Artis - study design, Kelsall - study design and co-wrote the paper, Wynn - study design, analysis and interpretation of data, and drafting of the manuscript.
Disclosures: i. None of the authors have any financial arrangement to disclose.
ii. This works was supported by the intramural program of the NIH/NIAID.
iii. The manuscript was written exclusively by the authors listed above.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kelsall BL. Innate and adaptive mechanisms to control [corrected] pathological intestinal inflammation. J Pathol. 2008;214:242–59. doi: 10.1002/path.2286. [DOI] [PubMed] [Google Scholar]
- 2.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–38. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 3.Artis D, Grencis RK. The intestinal epithelium: sensors to effectors in nematode infection. Mucosal Immunol. 2008;1:252–64. doi: 10.1038/mi.2008.21. [DOI] [PubMed] [Google Scholar]
- 4.Cucino C, Sonnenberg A. Cause of death in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2001;7:250–5. doi: 10.1097/00054725-200108000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382–8. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 6.Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–38. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 7.Chamouard P, Monneaux F, Richert Z, Voegeli AC, Lavaux T, Gaub MP, Baumann R, Oudet P, Muller S. Diminution of Circulating CD4+CD25 high T cells in naive Crohn’s disease. Dig Dis Sci. 2009;54:2084–93. doi: 10.1007/s10620-008-0590-6. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–8. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stumhofer JS, Silver J, Hunter CA. Negative regulation of Th17 responses. Semin Immunol. 2007;19:394–9. doi: 10.1016/j.smim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leach MW, Davidson NJ, Fort MM, Powrie F, Rennick DM. The role of IL-10 in inflammatory bowel disease: “of mice and men”. Toxicol Pathol. 1999;27:123–33. doi: 10.1177/019262339902700124. [DOI] [PubMed] [Google Scholar]
- 11.Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, Domingues FS, Albrecht M, Nothnagel M, Ellinghaus D, Sina C, Onnie CM, Weersma RK, Stokkers PC, Wijmenga C, Gazouli M, Strachan D, McArdle WL, Vermeire S, Rutgeerts P, Rosenstiel P, Krawczak M, Vatn MH, Mathew CG, Schreiber S. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–23. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 12.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, Hatscher N, Pfeifer D, Sykora KW, Sauer M, Kreipe H, Lacher M, Nustede R, Woellner C, Baumann U, Salzer U, Koletzko S, Shah N, Segal AW, Sauerbrey A, Buderus S, Snapper SB, Grimbacher B, Klein C. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Correa I, Veny M, Esteller M, Pique JM, Yague J, Panes J, Salas A. Defective IL-10 production in severe phenotypes of Crohn’s disease. J Leukoc Biol. 2009;85:896–903. doi: 10.1189/jlb.1108698. [DOI] [PubMed] [Google Scholar]
- 14.Karttunnen R, Breese EJ, Walker-Smith JA, MacDonald TT. Decreased mucosal interleukin-4 (IL-4) production in gut inflammation. J Clin Pathol. 1994;47:1015–8. doi: 10.1136/jcp.47.11.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinstock JV, Summers RW, Elliott DE. Role of helminths in regulating mucosal inflammation. Springer Semin Immunopathol. 2005;27:249–71. doi: 10.1007/s00281-005-0209-3. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 17.Hale LP, Gottfried MR, Swidsinski A. Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and mucosal exposure to intestinal bacteria. Inflamm Bowel Dis. 2005;11:1060–9. doi: 10.1097/01.mib.0000187582.90423.bc. [DOI] [PubMed] [Google Scholar]
- 18.Newcomb DC, Zhou W, Moore ML, Goleniewska K, Hershey GK, Kolls JK, Peebles RS., Jr A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol. 2009;182:5317–21. doi: 10.4049/jimmunol.0803868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, Schad GA, Scott P, Wu GD. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci U S A. 2004;101:13596–600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schopf LR, Hoffmann KF, Cheever AW, Urban JF, Jr, Wynn TA. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J Immunol. 2002;168:2383–92. doi: 10.4049/jimmunol.168.5.2383. [DOI] [PubMed] [Google Scholar]
- 21.Ramalingam TR, Pesce JT, Sheikh F, Cheever AW, Mentink-Kane MM, Wilson MS, Stevens S, Valenzuela DM, Murphy AJ, Yancopoulos GD, Urban JF, Jr, Donnelly RP, Wynn TA. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor alpha1 chain. Nat Immunol. 2008;9:25–33. doi: 10.1038/ni1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson MS, Elnekave E, Mentink-Kane MM, Hodges MG, Pesce JT, Ramalingam TR, Thompson RW, Kamanaka M, Flavell RA, Keane-Myers A, Cheever AW, Wynn TA. IL-13Ralpha2 and IL-10 coordinately suppress airway inflammation, airway-hyperreactivity, and fibrosis in mice. J Clin Invest. 2007;117:2941–51. doi: 10.1172/JCI31546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–20. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mentink-Kane MM, Wynn TA. Opposing roles for IL-13 and IL-13 receptor alpha 2 in health and disease. Immunol Rev. 2004;202:191–202. doi: 10.1111/j.0105-2896.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- 25.Fichtner-Feigl S, Young CA, Kitani A, Geissler EK, Schlitt HJ, Strober W. IL-13 signaling via IL-13R alpha2 induces major downstream fibrogenic factors mediating fibrosis in chronic TNBS colitis. Gastroenterology. 2008;135:2003–13. 2013, e1–7. doi: 10.1053/j.gastro.2008.08.055. [DOI] [PubMed] [Google Scholar]
- 26.Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK, Schlitt HJ, Kitani A, Strober W. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007;178:5859–70. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- 27.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakuraba A, Sato T, Kamada N, Kitazume M, Sugita A, Hibi T. Th1/Th17 Immune Response is Induced by Mesenteric Lymph Node Dendritic Cells in Crohn’s Disease. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 29.Elliott DE, Metwali A, Leung J, Setiawan T, Blum AM, Ince MN, Bazzone LE, Stadecker MJ, Urban JF, Jr, Weinstock JV. Colonization with Heligmosomoides polygyrus suppresses mucosal IL-17 production. J Immunol. 2008;181:2414–9. doi: 10.4049/jimmunol.181.4.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–57. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cliffe LJ, Grencis RK. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv Parasitol. 2004;57:255–307. doi: 10.1016/S0065-308X(04)57004-5. [DOI] [PubMed] [Google Scholar]
- 33.Bancroft AJ, McKenzie AN, Grencis RK. A critical role for IL-13 in resistance to intestinal nematode infection. J Immunol. 1998;160:3453–61. [PubMed] [Google Scholar]
- 34.He R, Kim HY, Yoon J, Oyoshi MK, MacGinnitie A, Goya S, Freyschmidt EJ, Bryce P, McKenzie AN, Umetsu DT, Oettgen HC, Geha RS. Exaggerated IL-17 response to epicutaneous sensitization mediates airway inflammation in the absence of IL-4 and IL-13. J Allergy Clin Immunol. 2009;124:761–70. e1. doi: 10.1016/j.jaci.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Vries JE. The role of IL-13 and its receptor in allergy and inflammatory responses. J Allergy Clin Immunol. 1998;102:165–9. doi: 10.1016/s0091-6749(98)70080-6. [DOI] [PubMed] [Google Scholar]
- 36.McKenzie AN, Culpepper JA, de Waal Malefyt R, Briere F, Punnonen J, Aversa G, Sato A, Dang W, Cocks BG, Menon S, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci U S A. 1993;90:3735–9. doi: 10.1073/pnas.90.8.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang G, Li L, Volk A, Emmell E, Petley T, Giles-Komar J, Rafferty P, Lakshminarayanan M, Griswold DE, Bugelski PJ, Das AM. Therapeutic dosing with anti-interleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther. 2005;313:8–15. doi: 10.1124/jpet.104.076133. [DOI] [PubMed] [Google Scholar]
- 38.Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58:1152–67. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 39.Fuss IJ, Strober W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol. 2008;1 (Suppl 1):S31–3. doi: 10.1038/mi.2008.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White CL, 3rd, Hamilton SR, Diamond MP, Cameron JL. Crohn’s disease and ulcerative colitis in the same patient. Gut. 1983;24:857–62. doi: 10.1136/gut.24.9.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morimoto M, Morimoto M, Zhao A, Madden KB, Dawson H, Finkelman FD, Mentink-Kane M, Urban JF, Jr, Wynn TA, Shea-Donohue T. Functional importance of regional differences in localized gene expression of receptors for IL-13 in murine gut. J Immunol. 2006;176:491–5. doi: 10.4049/jimmunol.176.1.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kucharzik T, Lugering N, Weigelt H, Adolf M, Domschke W, Stoll R. Immunoregulatory properties of IL-13 in patients with inflammatory bowel disease; comparison with IL-4 and IL-10. Clin Exp Immunol. 1996;104:483–90. doi: 10.1046/j.1365-2249.1996.39750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan WI, Blennerhasset P, Ma C, Matthaei KI, Collins SM. Stat6 dependent goblet cell hyperplasia during intestinal nematode infection. Parasite Immunol. 2001;23:39–42. doi: 10.1046/j.1365-3024.2001.00353.x. [DOI] [PubMed] [Google Scholar]
- 44.Cliffe LJ, Humphreys NE, Lane TE, Potten CS, Booth C, Grencis RK. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science. 2005;308:1463–5. doi: 10.1126/science.1108661. [DOI] [PubMed] [Google Scholar]
- 45.Snove O, Jr, Rossi JJ. Expressing short hairpin RNAs in vivo. Nat Methods. 2006;3:689–95. doi: 10.1038/nmeth927. [DOI] [PubMed] [Google Scholar]
- 46.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–85. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 48.Yoshidome H, Kato A, Miyazaki M, Edwards MJ, Lentsch AB. IL-13 activates STAT6 and inhibits liver injury induced by ischemia/reperfusion. Am J Pathol. 1999;155:1059–64. doi: 10.1016/S0002-9440(10)65208-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA, Cua DJ. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–70. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deepak P, Kumar S, Jr, Kishore D, Acharya A. IL-13 from Th2-type cells suppresses induction of antigen-specific Th1 immunity in a T-cell lymphoma. Int Immunol. 22:53–63. doi: 10.1093/intimm/dxp114. [DOI] [PubMed] [Google Scholar]
- 51.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–56. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 52.Wynn TA, Hesse M, Sandler NG, Kaviratne M, Hoffmann KF, Chiaramonte MG, Reiman R, Cheever AW, Sypek JP, Mentink-Kane MM. P-selectin suppresses hepatic inflammation and fibrosis in mice by regulating interferon gamma and the IL-13 decoy receptor. Hepatology. 2004;39:676–87. doi: 10.1002/hep.20102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.