

Abstract
The structure of the title compound, C12H9BrN4, prepared by the reaction of 2-bromo-1-(6-bromo-3-pyridyl)ethanone with 2-amino-3-methylpyrazine indicates that the compound with the bromopyridyl substituent at position 2 of the imidazopyrazine fused-ring system represents the major product of this reaction. The plane of the pyridine ring forms a dihedral angle of 16.2 (2)° with the essentially planar (r.m.s. deviation = 0.006 Å) imidazopyrazine system. In the crystal, molecules are linked by weak C—H⋯N interactions.
Related literature
For the structure of the related imidazo(1,2-a)pyrazine deivative, see: Lumma & Springer (1981 ▶).
Experimental
Crystal data
C12H9BrN4
M r = 289.14
Monoclinic,
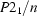
a = 3.9007 (14) Å
b = 13.545 (5) Å
c = 20.673 (8) Å
β = 93.059 (5)°
V = 1090.7 (7) Å3
Z = 4
Mo Kα radiation
μ = 3.75 mm−1
T = 100 K
0.27 × 0.11 × 0.05 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2001 ▶) T min = 0.431, T max = 0.835
19939 measured reflections
2668 independent reflections
1887 reflections with I > 2σ(I)
R int = 0.085
Refinement
R[F 2 > 2σ(F 2)] = 0.057
wR(F 2) = 0.154
S = 1.05
2668 reflections
155 parameters
H-atom parameters constrained
Δρmax = 1.23 e Å−3
Δρmin = −1.30 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810022993/hb5478sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810022993/hb5478Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C7—H7⋯N1i | 0.95 | 2.52 | 3.438 (6) | 163 |
| C10—H10⋯N2ii | 0.95 | 2.60 | 3.484 (7) | 156 |
Symmetry codes: (i) 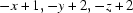 ; (ii)
; (ii) 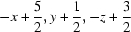 .
.
supplementary crystallographic information
Comment
The reaction of 2-bromo-1-(6-bromo3-pyridyl)ethanone with 2-amino-3-methylpyrazine may potentially produce either 2- or 3-(6-bromo3-pyridyl)-8-methylimidazo[1,2-a]pyrazine. The present study shows that the compound with bromopyridyl substituent in position 2 of imidazopyrazine represents the major product of this reaction (Fig. 1).
The plane of the pyridine ring N1, C1—C5 forms the dihedral angle of 16.2 (2)° with the essentially planar imidazopyrazine system N2, N3, N4, C6—C11. Strange though it may seem, only one purely organic structure with non-protontated non-fused imidazo(1,2 - a)pyrazine system with only carbon substituents has been published heretofore (Lumma & Springer, 1981). The geometry of the bicyclic fragment in this structure is in good agreement with that of the title compound.
Experimental
A mixture of 2-bromo-1-(6-bromo-3-pyridyl)-ethanone (2.70 g, 9.68 mmol), 2-amino-3-methylpyrazine (1.06 g, 9.68 mmol), and sodium bicarbonate (1.22 g, 14.5 mmol) in 40 ml of 2-propanol was heated at 80°C overnight. After cooling down to rt, the reaction mixture was concentrated to dryness. The resulting residue was partitioned between ethyl acetate (100 ml) and water (100 ml). The organic phase was washed with brine (1 × 100 ml), dried over sodium sulfate, concentrated to dryness, and purified by column chromatography with 0 --> 5% MeOH/EA to afford the desired product as a solid (1.25 g, 44.7% yield).
Colourless needles of (I) were grown by slow evaporation of an ethanol/dichloroethane solution.
Refinement
All H atoms were placed in geometrically calculated positions (C—H 0.95 Å for aromatic and 0.98 Å for methyl H atoms, respectively) and included in the refinement in riding motion approximation. The Uiso(H) were set to 1.2Ueq of the carrying atom (1.5Ueq for methyl H atoms). The maximum residual density peak 1.23 e/Å3 is located at a distance of 0.99 Å from the Br1 atom; the deepest hole -1.30 e/Å3 is at a distance of 0.78 Å from the Br1 atom.
Figures
Fig. 1.

Molecular structure of the title compound, showing 50% probability displacement ellipsoids. H atoms are drawn as circles of arbitrary small radius.
Crystal data
| C12H9BrN4 | F(000) = 576 |
| Mr = 289.14 | Dx = 1.761 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 6098 reflections |
| a = 3.9007 (14) Å | θ = 3.3–27.2° |
| b = 13.545 (5) Å | µ = 3.75 mm−1 |
| c = 20.673 (8) Å | T = 100 K |
| β = 93.059 (5)° | Needle, colorless |
| V = 1090.7 (7) Å3 | 0.27 × 0.11 × 0.05 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 2668 independent reflections |
| Radiation source: fine-focus sealed tube | 1887 reflections with I > 2σ(I) |
| graphite | Rint = 0.085 |
| φ and ω scans | θmax = 28.7°, θmin = 1.8° |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | h = −5→5 |
| Tmin = 0.431, Tmax = 0.835 | k = −17→17 |
| 19939 measured reflections | l = −27→26 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.057 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.154 | H-atom parameters constrained |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0701P)2 + 3.9758P] where P = (Fo2 + 2Fc2)/3 |
| 2668 reflections | (Δ/σ)max = 0.001 |
| 155 parameters | Δρmax = 1.23 e Å−3 |
| 0 restraints | Δρmin = −1.30 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.27478 (13) | 0.63452 (4) | 1.03763 (3) | 0.0279 (2) | |
| N1 | 0.5377 (11) | 0.8165 (3) | 1.0087 (2) | 0.0247 (9) | |
| N2 | 1.0290 (10) | 0.9281 (3) | 0.8097 (2) | 0.0195 (8) | |
| N3 | 1.0640 (10) | 1.0898 (3) | 0.83304 (19) | 0.0193 (8) | |
| N4 | 1.4045 (11) | 1.1347 (3) | 0.7232 (2) | 0.0221 (9) | |
| C1 | 0.4653 (12) | 0.7301 (4) | 0.9823 (2) | 0.0215 (10) | |
| C2 | 0.5223 (13) | 0.7047 (4) | 0.9189 (2) | 0.0241 (11) | |
| H2 | 0.4687 | 0.6407 | 0.9026 | 0.029* | |
| C3 | 0.6590 (12) | 0.7755 (4) | 0.8807 (2) | 0.0214 (10) | |
| H3 | 0.7011 | 0.7611 | 0.8369 | 0.026* | |
| C4 | 0.7370 (12) | 0.8692 (3) | 0.9062 (2) | 0.0190 (10) | |
| C5 | 0.6736 (12) | 0.8850 (4) | 0.9704 (2) | 0.0220 (10) | |
| H5 | 0.7290 | 0.9478 | 0.9887 | 0.026* | |
| C6 | 0.8818 (12) | 0.9470 (3) | 0.8672 (2) | 0.0185 (10) | |
| C7 | 0.8997 (12) | 1.0472 (3) | 0.8822 (2) | 0.0207 (10) | |
| H7 | 0.8150 | 1.0790 | 0.9191 | 0.025* | |
| C8 | 1.1395 (12) | 1.0161 (3) | 0.7898 (2) | 0.0174 (9) | |
| C9 | 1.3167 (12) | 1.0417 (4) | 0.7337 (2) | 0.0207 (10) | |
| C10 | 1.3238 (13) | 1.2048 (4) | 0.7677 (3) | 0.0256 (11) | |
| H10 | 1.3897 | 1.2710 | 0.7598 | 0.031* | |
| C11 | 1.1591 (13) | 1.1865 (3) | 0.8214 (3) | 0.0224 (10) | |
| H11 | 1.1088 | 1.2380 | 0.8507 | 0.027* | |
| C12 | 1.4005 (13) | 0.9642 (4) | 0.6864 (2) | 0.0241 (11) | |
| H12A | 1.5153 | 0.9944 | 0.6503 | 0.036* | |
| H12B | 1.1887 | 0.9319 | 0.6700 | 0.036* | |
| H12C | 1.5530 | 0.9152 | 0.7077 | 0.036* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.0245 (3) | 0.0267 (3) | 0.0331 (3) | −0.0011 (2) | 0.0081 (2) | 0.0102 (2) |
| N1 | 0.027 (2) | 0.024 (2) | 0.023 (2) | 0.0019 (18) | 0.0048 (17) | 0.0007 (18) |
| N2 | 0.018 (2) | 0.0173 (19) | 0.024 (2) | 0.0029 (16) | 0.0018 (16) | −0.0005 (16) |
| N3 | 0.018 (2) | 0.016 (2) | 0.025 (2) | 0.0009 (16) | 0.0031 (16) | 0.0015 (16) |
| N4 | 0.021 (2) | 0.018 (2) | 0.027 (2) | −0.0009 (17) | 0.0047 (16) | 0.0024 (17) |
| C1 | 0.017 (2) | 0.023 (2) | 0.025 (2) | 0.0025 (19) | 0.0039 (18) | 0.008 (2) |
| C2 | 0.024 (3) | 0.019 (2) | 0.029 (3) | −0.001 (2) | 0.005 (2) | −0.002 (2) |
| C3 | 0.019 (2) | 0.022 (2) | 0.023 (2) | −0.0002 (19) | 0.0032 (19) | −0.001 (2) |
| C4 | 0.017 (2) | 0.020 (2) | 0.020 (2) | 0.0028 (19) | 0.0033 (17) | 0.0016 (19) |
| C5 | 0.023 (2) | 0.022 (2) | 0.022 (2) | 0.0055 (19) | −0.0005 (19) | −0.0042 (19) |
| C6 | 0.015 (2) | 0.020 (2) | 0.021 (2) | 0.0061 (18) | 0.0003 (18) | 0.0002 (19) |
| C7 | 0.025 (3) | 0.016 (2) | 0.022 (2) | 0.0009 (19) | 0.0065 (19) | −0.0035 (19) |
| C8 | 0.016 (2) | 0.015 (2) | 0.021 (2) | 0.0013 (17) | 0.0026 (18) | 0.0006 (18) |
| C9 | 0.017 (2) | 0.019 (2) | 0.026 (2) | 0.0029 (19) | 0.0026 (19) | 0.003 (2) |
| C10 | 0.027 (3) | 0.019 (2) | 0.031 (3) | −0.001 (2) | 0.001 (2) | 0.001 (2) |
| C11 | 0.020 (2) | 0.013 (2) | 0.034 (3) | 0.0008 (18) | 0.002 (2) | −0.003 (2) |
| C12 | 0.024 (3) | 0.027 (3) | 0.021 (2) | 0.002 (2) | 0.0069 (19) | 0.000 (2) |
Geometric parameters (Å, °)
| Br1—C1 | 1.905 (5) | C3—H3 | 0.9500 |
| N1—C1 | 1.316 (7) | C4—C5 | 1.381 (7) |
| N1—C5 | 1.347 (7) | C4—C6 | 1.458 (7) |
| N2—C8 | 1.340 (6) | C5—H5 | 0.9500 |
| N2—C6 | 1.371 (6) | C6—C7 | 1.393 (7) |
| N3—C7 | 1.359 (6) | C7—H7 | 0.9500 |
| N3—C8 | 1.382 (6) | C8—C9 | 1.424 (7) |
| N3—C11 | 1.386 (6) | C9—C12 | 1.482 (7) |
| N4—C9 | 1.327 (6) | C10—C11 | 1.335 (7) |
| N4—C10 | 1.371 (7) | C10—H10 | 0.9500 |
| C1—C2 | 1.384 (7) | C11—H11 | 0.9500 |
| C2—C3 | 1.369 (7) | C12—H12A | 0.9800 |
| C2—H2 | 0.9500 | C12—H12B | 0.9800 |
| C3—C4 | 1.401 (7) | C12—H12C | 0.9800 |
| C1—N1—C5 | 116.8 (4) | C7—C6—C4 | 126.6 (4) |
| C8—N2—C6 | 104.9 (4) | N3—C7—C6 | 105.4 (4) |
| C7—N3—C8 | 107.6 (4) | N3—C7—H7 | 127.3 |
| C7—N3—C11 | 132.2 (4) | C6—C7—H7 | 127.3 |
| C8—N3—C11 | 120.2 (4) | N2—C8—N3 | 111.1 (4) |
| C9—N4—C10 | 118.5 (4) | N2—C8—C9 | 130.2 (4) |
| N1—C1—C2 | 125.0 (5) | N3—C8—C9 | 118.7 (4) |
| N1—C1—Br1 | 115.9 (4) | N4—C9—C8 | 120.4 (4) |
| C2—C1—Br1 | 119.1 (4) | N4—C9—C12 | 119.8 (5) |
| C3—C2—C1 | 117.3 (5) | C8—C9—C12 | 119.9 (4) |
| C3—C2—H2 | 121.3 | C11—C10—N4 | 124.6 (5) |
| C1—C2—H2 | 121.3 | C11—C10—H10 | 117.7 |
| C2—C3—C4 | 120.2 (5) | N4—C10—H10 | 117.7 |
| C2—C3—H3 | 119.9 | C10—C11—N3 | 117.6 (5) |
| C4—C3—H3 | 119.9 | C10—C11—H11 | 121.2 |
| C5—C4—C3 | 117.0 (4) | N3—C11—H11 | 121.2 |
| C5—C4—C6 | 121.0 (4) | C9—C12—H12A | 109.5 |
| C3—C4—C6 | 122.0 (4) | C9—C12—H12B | 109.5 |
| N1—C5—C4 | 123.8 (5) | H12A—C12—H12B | 109.5 |
| N1—C5—H5 | 118.1 | C9—C12—H12C | 109.5 |
| C4—C5—H5 | 118.1 | H12A—C12—H12C | 109.5 |
| N2—C6—C7 | 110.9 (4) | H12B—C12—H12C | 109.5 |
| N2—C6—C4 | 122.4 (4) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C7—H7···N1i | 0.95 | 2.52 | 3.438 (6) | 163 |
| C10—H10···N2ii | 0.95 | 2.60 | 3.484 (7) | 156 |
Symmetry codes: (i) −x+1, −y+2, −z+2; (ii) −x+5/2, y+1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB5478).
References
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810022993/hb5478sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810022993/hb5478Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report