

Abstract
The title compound C9H9BrO3, was synthesized by the regioselective bromination of 4-methoxyphenylacetic acid using bromine in acetic acid in a 84% yield. In the molecular structure, the methoxy group is almost coplanar with the phenyl ring within 0.06 Å; the acetic acid substituent is tilted by 78.15 (7)° relative to the ring. The C—C—C angles at the OMe, acetyl and Br substituents are 118.2 (2), 118.4 (2) and 121.5 (2)°, respectively, indicating that the Br atom is electron-withdrawing, whereas the other substituents possess electron-donating properties. In the crystal, the molecules form centrosymmetric strongly O—H⋯O hydrogen-bonded dimers of the type R 2 2(8).
Related literature
For the use of the title compound in the synthesis of natural products such as Combretastatin A-4, see: Zou et al. (2008 ▶); for Verongamine, see: Wasserman & Wang (1998 ▶) and for model Vancomycin-type systems, see: Ghosh et al. (2009 ▶). The iodo-analogue featured in the synthesis of (+)-Phleichrome and (+)-Calphostin D, see: Morgan et al. (2010 ▶). For the synthesis of the title compound, see: Coutts et al., (1970 ▶); Morgan et al., (2007 ▶); Zou et al. (2008 ▶); Ghosh et al. (2009 ▶). For background for our program to introduce natural product synthesis, crystal growing techniques and single crystal X-ray diffraction data analysis into the undergraduate curriculum, see: Findlater et al., (2010 ▶); Guzei et al., (2010a
▶). For a discussion of hydrogen-bonding motif assignment, see: Guzei et al. (2010b
▶). Outlier reflections were omitted based on the statistics test described by Prince & Nicholson (1983 ▶) and Rollett (1988 ▶), and implemented in FCF_filter (Guzei, 2007 ▶).
Experimental
Crystal data
C9H9BrO3
M r = 245.06
Monoclinic,
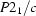
a = 12.5022 (4) Å
b = 8.2690 (2) Å
c = 9.0199 (3) Å
β = 93.573 (1)°
V = 930.67 (5) Å3
Z = 4
Cu Kα radiation
μ = 5.81 mm−1
T = 120 K
0.46 × 0.37 × 0.19 mm
Data collection
Bruker SMART APEXII area-detector diffractometer
Absorption correction: analytical (SADABS; Bruker, 2007 ▶) T min = 0.177, T max = 0.398
12598 measured reflections
1725 independent reflections
1708 reflections with I > 2σ(I)
R int = 0.030
Refinement
R[F 2 > 2σ(F 2)] = 0.026
wR(F 2) = 0.073
S = 1.08
1725 reflections
120 parameters
H-atom parameters constrained
Δρmax = 0.86 e Å−3
Δρmin = −0.47 e Å−3
Data collection: APEX2 (Bruker, 2007 ▶); cell refinement: SAINT-Plus (Bruker, 2007 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: publCIF (Westrip, 2010 ▶) and modiCIFer (Guzei, 2007 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810020143/rk2206sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810020143/rk2206Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Enhanced figure: interactive version of Fig. 1
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O3—H3⋯O2i | 0.84 | 1.82 | 2.661 (2) | 179 |
Symmetry code: (i) 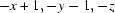 .
.
supplementary crystallographic information
Comment
Recently, we have been pursuing simple organic and organometallic compounds as candidates for the introduction of (a) natural product synthesis into the undergraduate teaching laboratory and (b) crystal growing techniques and single crystal X-ray diffraction data analysis into the undergraduate curriculum (Findlater et al., 2010; Guzei at al., 2010a). The 3-bromo-4-methoxyphenylacetic acid I has been employed in the synthesis of natural products such as Combretastatin A-4, (Zou et al., 2008), Verongamine (Wasserman & Wang, 1998) and model Vancomycin-type systems (Ghosh et al., 2009). The iodo-analogue features in the synthesis of the perylenequinones (+)-Phleichrome and (+)-Calphostin D, (Morgan et al., 2010). Our interest in I stems from its role in the synthesis of the antimitotic compound Combretastatin A-4 via a simple Perkin condensation/decarboxylation sequence (Zou et al., 2008). This concise route, employing commercially available starting materials, followed by the facile purification of I to furnish high quality crystals makes it ideal in both regards. Compound I is readily synthesized by the regioselective bromination of 4-methoxyphenylacetic acid using bromine in acetic acid (Coutts et al., 1970; Morgan et al., 2007; Zou et al., 2008; Ghosh et al., 2009). Compound I was isolated and characterized by NMR, mp, and single-crystal X-ray analysis. There are three main structural aspects students should identify. First, the positions of the alkyl substituents on the phenyl ring. The methoxy-group is almost coplanar with the ring, torsion angle C7—O1—C1—C6 is 1.2 (3)°, whereas the acetic acid terminus is nearly perpendicular to the ring with the dihedral angle between the planes defined by atoms C1—C6 and atoms C4,C8,C9,O2,O3 spanning 78.15 (7)°. Secondly, the distortions of the C—C—C angles from 120° at the substituents of the phenyl ring reflect their electronic properties. The stronger the electron-withdrawing power of a substituent, the larger the C—C—C angle. The angles at OMe, Ac and Br are 118.2 (2), 118.4 (2), and 121.5 (2)°, respectively, indicating that the Br atom is electron-withdrawing, whereas the other substituents possess electron-donating properties. Of course, the magnitude of the values is affected by the neighbouring substituents. Thirdly, the molecules of I form centrosymmetric strongly hydrogen-bonded dimers in the lattice. The hydrogen bonding motif is R22(8). A topical discussion of hydrogen bonding motif assignment was published by Guzei et al., 2010b.
Experimental
To a stirred solution of 4-methoxyphenylacetic acid (10 g, 60.2 mmol) in acetic acid (60 ml) was added a solution of bromine (9.62 g, 3.1 ml, 60.2 mmol) in acetic acid (30 ml) slowly dropwise over 30 min. The mixture was stirred at room temperature (60 min) and then poured into 500 ml ice–water. The resultant pale yellow, turbid mixture was stirred (10 min), filtered, rinsed with ice–water (3×10 ml), air-dried (20 min) and recrystallized from hot xylene to give a white crystalline powder. Yield 12.41 g, 84 %. M.p. 386.3–387.2 K.
1H NMR (CDCl3): δ 3.56 (2H, s, CH2); 3.89 (3H, s, OCH3), 6.86 (1H, d), 7.19 (1H, dd), 7.48 (1H, d). 13C(1H) NMR (CDCl3): δ 39.9; 56.5; 111.9; 112.2; 127.0; 129.6; 134.6; 155.5; 178.0.
Refinement
All H-atoms were placed in idealized locations. The C—H distances were 0.98Å for the methyl group, 0.99Å the methylene group, 0.95Å for the sp2-hybridized atoms; the O—H distance was fixed at 0.84Å. All H atoms were refined as riding with thermal displacement coefficients Uiso(H) set to 1.5Ueq(C, O) for the methyl- and hydroxyl-groups and to to 1.2Ueq(C) for the CH- and CH2-groups.
The outlier reflections were omitted based on the statistics test described by Prince & Nicholson, (1983) and Rollett, (1988), and implemented in program FCF_filter (Guzei, 2007). The number of omitted outliers is 4.
Figures
Fig. 1.

Molecular structure of I with the atom numbering scheme. The displacement ellipsoids are shown at 50% probability level.
Crystal data
| C9H9BrO3 | F(000) = 488 |
| Mr = 245.06 | Dx = 1.749 Mg m−3 |
| Monoclinic, P21/c | Melting point = 386.3–387.2 K |
| Hall symbol: -P 2ybc | Cu Kα radiation, λ = 1.54178 Å |
| a = 12.5022 (4) Å | Cell parameters from 9563 reflections |
| b = 8.2690 (2) Å | θ = 3.5–69.5° |
| c = 9.0199 (3) Å | µ = 5.81 mm−1 |
| β = 93.573 (1)° | T = 120 K |
| V = 930.67 (5) Å3 | Block, colourless |
| Z = 4 | 0.46 × 0.37 × 0.19 mm |
Data collection
| Bruker SMART APEXII area-detector diffractometer | 1725 independent reflections |
| Radiation source: fine-focus sealed tube | 1708 reflections with I > 2σ(I) |
| graphite | Rint = 0.030 |
| 0.5° ω and 0.5° φ scans | θmax = 69.5°, θmin = 3.5° |
| Absorption correction: analytical (SADABS; Bruker, 2007) | h = −14→15 |
| Tmin = 0.177, Tmax = 0.398 | k = −9→10 |
| 12598 measured reflections | l = −10→10 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.026 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.073 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0442P)2 + 1.1574P] where P = (Fo2 + 2Fc2)/3 |
| 1725 reflections | (Δ/σ)max < 0.001 |
| 120 parameters | Δρmax = 0.86 e Å−3 |
| 0 restraints | Δρmin = −0.47 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.253622 (18) | 0.20274 (3) | 0.38506 (3) | 0.02281 (12) | |
| O1 | 0.05071 (12) | 0.0202 (2) | 0.32075 (17) | 0.0186 (3) | |
| O2 | 0.40281 (14) | −0.3902 (2) | 0.05910 (19) | 0.0234 (4) | |
| O3 | 0.50409 (14) | −0.3283 (2) | −0.12886 (19) | 0.0229 (4) | |
| H3 | 0.5326 | −0.4178 | −0.1071 | 0.034* | |
| C1 | 0.12214 (17) | −0.0244 (3) | 0.2209 (2) | 0.0151 (4) | |
| C2 | 0.22340 (18) | 0.0476 (3) | 0.2339 (2) | 0.0148 (4) | |
| C3 | 0.30189 (17) | 0.0087 (3) | 0.1381 (2) | 0.0160 (4) | |
| H3A | 0.3701 | 0.0593 | 0.1493 | 0.019* | |
| C4 | 0.28118 (18) | −0.1041 (3) | 0.0255 (2) | 0.0172 (5) | |
| C5 | 0.1810 (2) | −0.1765 (3) | 0.0129 (3) | 0.0201 (5) | |
| H5 | 0.1661 | −0.2542 | −0.0631 | 0.024* | |
| C6 | 0.10183 (18) | −0.1383 (3) | 0.1087 (3) | 0.0181 (5) | |
| H6 | 0.0339 | −0.1898 | 0.0977 | 0.022* | |
| C7 | −0.05187 (18) | −0.0591 (3) | 0.3100 (3) | 0.0220 (5) | |
| H7A | −0.0887 | −0.0346 | 0.2135 | 0.033* | |
| H7C | −0.0952 | −0.0204 | 0.3896 | 0.033* | |
| H7B | −0.0416 | −0.1762 | 0.3195 | 0.033* | |
| C8 | 0.3654 (2) | −0.1464 (3) | −0.0810 (3) | 0.0209 (5) | |
| H8A | 0.4175 | −0.0564 | −0.0831 | 0.025* | |
| H8B | 0.3306 | −0.1576 | −0.1821 | 0.025* | |
| C9 | 0.42502 (19) | −0.3007 (3) | −0.0408 (3) | 0.0170 (5) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.02043 (17) | 0.02308 (18) | 0.02445 (17) | −0.00116 (9) | −0.00227 (11) | −0.00996 (9) |
| O1 | 0.0164 (7) | 0.0206 (8) | 0.0193 (8) | −0.0018 (6) | 0.0047 (6) | −0.0044 (6) |
| O2 | 0.0309 (9) | 0.0194 (8) | 0.0214 (9) | 0.0053 (7) | 0.0119 (7) | 0.0032 (7) |
| O3 | 0.0222 (9) | 0.0241 (9) | 0.0236 (9) | 0.0068 (7) | 0.0096 (7) | 0.0059 (7) |
| C1 | 0.0169 (10) | 0.0131 (10) | 0.0152 (10) | 0.0020 (8) | 0.0009 (8) | 0.0024 (8) |
| C2 | 0.0193 (10) | 0.0110 (10) | 0.0137 (10) | 0.0010 (8) | −0.0029 (8) | −0.0009 (8) |
| C3 | 0.0163 (10) | 0.0137 (10) | 0.0180 (10) | 0.0004 (8) | 0.0005 (8) | 0.0035 (8) |
| C4 | 0.0210 (11) | 0.0152 (11) | 0.0157 (11) | 0.0040 (9) | 0.0041 (8) | 0.0040 (8) |
| C5 | 0.0252 (12) | 0.0181 (11) | 0.0169 (11) | 0.0004 (9) | 0.0013 (9) | −0.0035 (9) |
| C6 | 0.0189 (11) | 0.0165 (11) | 0.0188 (11) | −0.0030 (9) | 0.0005 (9) | −0.0020 (9) |
| C7 | 0.0160 (11) | 0.0241 (12) | 0.0262 (12) | −0.0026 (9) | 0.0028 (9) | −0.0010 (10) |
| C8 | 0.0246 (12) | 0.0211 (12) | 0.0176 (11) | 0.0032 (10) | 0.0072 (9) | 0.0016 (9) |
| C9 | 0.0178 (11) | 0.0188 (12) | 0.0145 (11) | −0.0014 (8) | 0.0019 (9) | −0.0039 (8) |
Geometric parameters (Å, °)
| Br1—C2 | 1.893 (2) | C4—C5 | 1.386 (3) |
| O1—C1 | 1.359 (3) | C4—C8 | 1.510 (3) |
| O1—C7 | 1.438 (3) | C5—C6 | 1.390 (3) |
| O2—C9 | 1.212 (3) | C5—H5 | 0.9500 |
| O3—C9 | 1.326 (3) | C6—H6 | 0.9500 |
| O3—H3 | 0.8400 | C7—H7A | 0.9800 |
| C1—C6 | 1.394 (3) | C7—H7C | 0.9800 |
| C1—C2 | 1.397 (3) | C7—H7B | 0.9800 |
| C2—C3 | 1.385 (3) | C8—C9 | 1.510 (3) |
| C3—C4 | 1.392 (3) | C8—H8A | 0.9900 |
| C3—H3A | 0.9500 | C8—H8B | 0.9900 |
| C1—O1—C7 | 116.85 (18) | C5—C6—H6 | 120.0 |
| C9—O3—H3 | 109.5 | C1—C6—H6 | 120.0 |
| O1—C1—C6 | 124.6 (2) | O1—C7—H7A | 109.5 |
| O1—C1—C2 | 117.3 (2) | O1—C7—H7C | 109.5 |
| C6—C1—C2 | 118.2 (2) | H7A—C7—H7C | 109.5 |
| C3—C2—C1 | 121.5 (2) | O1—C7—H7B | 109.5 |
| C3—C2—Br1 | 119.28 (17) | H7A—C7—H7B | 109.5 |
| C1—C2—Br1 | 119.23 (17) | H7C—C7—H7B | 109.5 |
| C2—C3—C4 | 120.3 (2) | C4—C8—C9 | 113.34 (19) |
| C2—C3—H3A | 119.9 | C4—C8—H8A | 108.9 |
| C4—C3—H3A | 119.9 | C9—C8—H8A | 108.9 |
| C5—C4—C3 | 118.4 (2) | C4—C8—H8B | 108.9 |
| C5—C4—C8 | 120.7 (2) | C9—C8—H8B | 108.9 |
| C3—C4—C8 | 120.9 (2) | H8A—C8—H8B | 107.7 |
| C4—C5—C6 | 121.7 (2) | O2—C9—O3 | 123.7 (2) |
| C4—C5—H5 | 119.2 | O2—C9—C8 | 124.2 (2) |
| C6—C5—H5 | 119.2 | O3—C9—C8 | 112.16 (19) |
| C5—C6—C1 | 120.0 (2) | ||
| C7—O1—C1—C6 | 1.2 (3) | C3—C4—C5—C6 | 0.4 (3) |
| C7—O1—C1—C2 | −177.70 (19) | C8—C4—C5—C6 | −179.3 (2) |
| O1—C1—C2—C3 | 179.39 (19) | C4—C5—C6—C1 | 0.1 (4) |
| C6—C1—C2—C3 | 0.4 (3) | O1—C1—C6—C5 | −179.4 (2) |
| O1—C1—C2—Br1 | −1.0 (3) | C2—C1—C6—C5 | −0.5 (3) |
| C6—C1—C2—Br1 | 180.00 (17) | C5—C4—C8—C9 | −81.5 (3) |
| C1—C2—C3—C4 | 0.1 (3) | C3—C4—C8—C9 | 98.8 (3) |
| Br1—C2—C3—C4 | −179.53 (17) | C4—C8—C9—O2 | 5.8 (3) |
| C2—C3—C4—C5 | −0.5 (3) | C4—C8—C9—O3 | −175.0 (2) |
| C2—C3—C4—C8 | 179.2 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O2i | 0.84 | 1.82 | 2.661 (2) | 179 |
Symmetry codes: (i) −x+1, −y−1, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: RK2206).
References
- Bruker (2007). APEX2, SADABS and SAINT-Plus Bruker AXS Inc., Madison, Wisconsin, USA.
- Coutts, I. G. C., Durbin, A. K. & Schofield, K. (1970). Aust. J. Chem 23, 791–800.
- Findlater, M., Hill, N. J. & Cowley, A. H. (2010). J. Chem. Crystallogr 40, 64–66.
- Ghosh, S., Kumar, A. S., Mehta, G. N. & Soundararajan, R. (2009). Synthesis, pp. 3322–3326.
- Guzei, I. A. (2007). In-house Crystallographic Programs: FCF_filter, INSerter and modiCIFer Molecular Structure Laboratory, University of Wisconsin–Madison, Madison, Wisconsin, USA.
- Guzei, I. A., Hill, N. J. & Van Hout, M. R. (2010a). Acta Cryst. E66, o40–o41. [DOI] [PMC free article] [PubMed]
- Guzei, I. A., Spencer, L. C., Ainooson, M. K. & Darkwa, J. (2010b). Acta Cryst. C66, m89–m96. [DOI] [PubMed]
- Morgan, B. J., Mulrooney, C. A., O’Brien, E. M. & Kozlowski, M. C. (2010). J. Org. Chem 75, 30–43. [DOI] [PMC free article] [PubMed]
- Morgan, B. J., Xie, X., Phuan, P.-W. & Kozlowski, M. C. (2007). J. Org. Chem 72, 6171–6182. [DOI] [PubMed]
- Prince, E. & Nicholson, W. L. (1983). Acta Cryst. A39, 407–410.
- Rollett, J. S. (1988). Crystallographic Computing, edited by N. W. Isaacs & M. R. Taylor, Vol. 4, pp. 149–166. Oxford University Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wasserman, H. H. & Wang, J. (1998). J. Org. Chem 63, 5581–5586.
- Westrip, S. P. (2010). J. Appl. Cryst.43 Submitted.
- Zou, Y., Xiao, C.-F., Zhong, R.-Q., Wei, W., Hunag, W.-M. & He, S.-J. (2008). J. Chem. Res. pp. 354–356.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810020143/rk2206sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810020143/rk2206Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Enhanced figure: interactive version of Fig. 1