

Abstract
In the title compound, C7H4F2O2, the dihedral angle between the benzene ring and the carboxylate group is 33.70 (14)°. In the crystal structure, inversion dimers linked by pairs of O—H⋯O hydrogren bonds occur, generating R 2 2(8) loops. The dimers are linked into sheets lying parallel to (102) by C—H⋯F hydrogen bonds.
Related literature
For general background to 2,6-diflorobenzylchloride derivatives, see: Beavo (1995 ▶); Beavo & Reifsnyder (1990 ▶); Nicholson et al. (1991 ▶). For the stability of the temperature controller used in the data collection, see: Cosier & Glazer (1986 ▶).
Experimental
Crystal data
C7H4F2O2
M r = 158.10
Monoclinic,
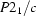
a = 3.6517 (4) Å
b = 14.1214 (15) Å
c = 12.2850 (13) Å
β = 95.651 (3)°
V = 630.42 (12) Å3
Z = 4
Mo Kα radiation
μ = 0.16 mm−1
T = 100 K
0.73 × 0.19 × 0.09 mm
Data collection
Bruker APEXII DUO CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.841, T max = 0.986
6112 measured reflections
2190 independent reflections
1895 reflections with I > 2σ(I)
R int = 0.029
Refinement
R[F 2 > 2σ(F 2)] = 0.049
wR(F 2) = 0.143
S = 1.12
2190 reflections
116 parameters
All H-atom parameters refined
Δρmax = 0.47 e Å−3
Δρmin = −0.31 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810028758/hb5558sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810028758/hb5558Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H1O2⋯O3i | 0.95 (4) | 1.68 (4) | 2.6318 (14) | 174 (4) |
| C3—H3⋯F2ii | 0.98 (2) | 2.54 (2) | 3.3428 (16) | 138.7 (16) |
Symmetry codes: (i) 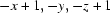 ; (ii)
; (ii) 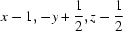 .
.
Acknowledgments
NM gratefully acknowledges funding from Universiti Sains Malaysia (USM) under the University Research Grant (No. 1001/PFARMASI/815025). HKF and CSY thank USM for the Research University Golden Goose Grant (No. 1001/PFIZIK/811012). CSY also thanks USM for the award of a USM Fellowship.
supplementary crystallographic information
Comment
The derivatives of 2,6-diflorobenzylchloride involved in the inhibition of phosphodiesterases (PDEs) are enzymes which catalyze PDEs. These derivatives are classified into seven families, five of which, PDE1–PDE5, have been characterized (Beavo, 1995). The hydrolysis of cyclic nucleotides was evaluated according to the methods in given the references (Beavo & Reifsnyder, 1990; Nicholson et al., 1991).
The molecule of the title compound, (I), (Fig. 1) is not planar with the dihedral angle between the benzene ring and the carboxylate group being 33.70 (14)°. In the crystal structure, the molecules are linked into pairs of centrosymmetric dimers by intermolecular O2—H1O2···O3 hydrogren bonds (Table 1). These dimers are linked into two-dimensional plane by the intermolecular C3—H3A···F2 hydrogen bonds (Fig. 2, Table 1) parallel to (102).
Experimental
2,6-Difluorobenzylchloride (0.01 mol, 1.7 g) was added drop-wise with stirring into a round bottom flask containing 25 ml water and then refluxed for two and half hours. The gum compound precipitate formed was filtered and dissolved in alkaline water. Hydrochloric acid was then added drop-wise with stirring. The white precipitate formed was dissolved in methanol. Colourless needles of (I) were formed at room temperature overnight and filtrated and dried at 333 K.
Refinement
All hydrogen atoms were located in a difference Fourier map and refined freely.
Figures
Fig. 1.

The molecular structure of (I) with 50% probability ellipsoids for non-H atoms.
Fig. 2.

The crystal packing of (I), viewed down the b axis, showing two 2-D planes.
Crystal data
| C7H4F2O2 | F(000) = 320 |
| Mr = 158.10 | Dx = 1.666 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 3166 reflections |
| a = 3.6517 (4) Å | θ = 3.3–32.1° |
| b = 14.1214 (15) Å | µ = 0.16 mm−1 |
| c = 12.2850 (13) Å | T = 100 K |
| β = 95.651 (3)° | Needle, colourless |
| V = 630.42 (12) Å3 | 0.73 × 0.19 × 0.09 mm |
| Z = 4 |
Data collection
| Bruker APEXII DUO CCD diffractometer | 2190 independent reflections |
| Radiation source: fine-focus sealed tube | 1895 reflections with I > 2σ(I) |
| graphite | Rint = 0.029 |
| φ and ω scans | θmax = 32.1°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −5→5 |
| Tmin = 0.841, Tmax = 0.986 | k = −20→20 |
| 6112 measured reflections | l = −18→18 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.049 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.143 | All H-atom parameters refined |
| S = 1.12 | w = 1/[σ2(Fo2) + (0.0668P)2 + 0.3079P] where P = (Fo2 + 2Fc2)/3 |
| 2190 reflections | (Δ/σ)max < 0.001 |
| 116 parameters | Δρmax = 0.47 e Å−3 |
| 0 restraints | Δρmin = −0.31 e Å−3 |
Special details
| Experimental. The crystal was placed in the cold stream of an Oxford Cryosystems Cobra open-flow nitrogen cryostat (Cosier & Glazer, 1986) operating at 100.0 (1) K. |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | 0.0410 (3) | −0.01635 (6) | 0.16839 (7) | 0.0312 (2) | |
| F2 | 0.2348 (3) | 0.26707 (6) | 0.36896 (7) | 0.0285 (2) | |
| O2 | 0.2287 (3) | 0.09843 (7) | 0.46658 (7) | 0.0238 (2) | |
| O3 | 0.4751 (3) | −0.00746 (7) | 0.35958 (8) | 0.0222 (2) | |
| C1 | 0.0163 (4) | 0.07830 (9) | 0.17659 (9) | 0.0194 (2) | |
| C2 | −0.1413 (4) | 0.12763 (10) | 0.08679 (10) | 0.0228 (3) | |
| C3 | −0.1734 (4) | 0.22519 (10) | 0.09441 (10) | 0.0234 (3) | |
| C4 | −0.0482 (4) | 0.27250 (10) | 0.19005 (11) | 0.0230 (3) | |
| C5 | 0.1044 (4) | 0.22003 (9) | 0.27793 (9) | 0.0184 (2) | |
| C6 | 0.1398 (3) | 0.12160 (8) | 0.27576 (9) | 0.0160 (2) | |
| C7 | 0.2939 (3) | 0.06665 (8) | 0.37288 (9) | 0.0156 (2) | |
| H2 | −0.221 (7) | 0.0927 (16) | 0.0182 (18) | 0.038 (6)* | |
| H3 | −0.285 (6) | 0.2599 (15) | 0.0302 (17) | 0.035 (5)* | |
| H4 | −0.074 (6) | 0.3406 (16) | 0.1979 (18) | 0.035 (5)* | |
| H1O2 | 0.341 (11) | 0.062 (3) | 0.526 (3) | 0.098 (12)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0518 (6) | 0.0183 (4) | 0.0219 (4) | 0.0019 (4) | −0.0044 (4) | −0.0039 (3) |
| F2 | 0.0459 (6) | 0.0169 (4) | 0.0210 (4) | −0.0010 (4) | −0.0048 (4) | −0.0018 (3) |
| O2 | 0.0340 (6) | 0.0238 (5) | 0.0135 (4) | 0.0038 (4) | 0.0014 (4) | 0.0013 (3) |
| O3 | 0.0269 (5) | 0.0179 (4) | 0.0216 (4) | 0.0056 (4) | 0.0012 (4) | 0.0030 (3) |
| C1 | 0.0238 (6) | 0.0181 (5) | 0.0162 (5) | 0.0001 (4) | 0.0014 (4) | 0.0003 (4) |
| C2 | 0.0242 (6) | 0.0289 (6) | 0.0150 (5) | −0.0002 (5) | −0.0006 (4) | 0.0019 (4) |
| C3 | 0.0224 (6) | 0.0289 (6) | 0.0184 (5) | 0.0037 (5) | −0.0001 (4) | 0.0078 (4) |
| C4 | 0.0272 (6) | 0.0193 (6) | 0.0225 (6) | 0.0041 (5) | 0.0016 (5) | 0.0062 (4) |
| C5 | 0.0210 (5) | 0.0174 (5) | 0.0166 (5) | 0.0004 (4) | 0.0012 (4) | 0.0011 (4) |
| C6 | 0.0182 (5) | 0.0160 (5) | 0.0137 (4) | 0.0009 (4) | 0.0014 (4) | 0.0019 (3) |
| C7 | 0.0174 (5) | 0.0148 (5) | 0.0148 (4) | −0.0006 (4) | 0.0020 (4) | 0.0012 (3) |
Geometric parameters (Å, °)
| F1—C1 | 1.3442 (15) | C2—H2 | 0.99 (2) |
| F2—C5 | 1.3467 (14) | C3—C4 | 1.3892 (19) |
| O2—C7 | 1.2794 (14) | C3—H3 | 0.98 (2) |
| O2—H1O2 | 0.96 (4) | C4—C5 | 1.3807 (17) |
| O3—C7 | 1.2574 (15) | C4—H4 | 0.97 (2) |
| C1—C2 | 1.3815 (17) | C5—C6 | 1.3965 (17) |
| C1—C6 | 1.3976 (16) | C6—C7 | 1.4866 (15) |
| C2—C3 | 1.387 (2) | ||
| C7—O2—H1O2 | 113 (2) | C5—C4—H4 | 119.3 (13) |
| F1—C1—C2 | 117.86 (11) | C3—C4—H4 | 122.2 (13) |
| F1—C1—C6 | 118.83 (11) | F2—C5—C4 | 117.84 (11) |
| C2—C1—C6 | 123.29 (12) | F2—C5—C6 | 118.74 (10) |
| C1—C2—C3 | 118.58 (12) | C4—C5—C6 | 123.38 (11) |
| C1—C2—H2 | 119.4 (13) | C5—C6—C1 | 115.44 (10) |
| C3—C2—H2 | 122.0 (13) | C5—C6—C7 | 122.18 (10) |
| C2—C3—C4 | 120.79 (11) | C1—C6—C7 | 122.37 (11) |
| C2—C3—H3 | 118.2 (13) | O3—C7—O2 | 123.76 (11) |
| C4—C3—H3 | 121.0 (13) | O3—C7—C6 | 119.51 (10) |
| C5—C4—C3 | 118.49 (12) | O2—C7—C6 | 116.72 (10) |
| F1—C1—C2—C3 | 179.13 (13) | C4—C5—C6—C7 | −177.92 (12) |
| C6—C1—C2—C3 | 1.2 (2) | F1—C1—C6—C5 | −179.87 (12) |
| C1—C2—C3—C4 | 0.3 (2) | C2—C1—C6—C5 | −1.92 (19) |
| C2—C3—C4—C5 | −0.9 (2) | F1—C1—C6—C7 | −0.65 (19) |
| C3—C4—C5—F2 | 178.01 (12) | C2—C1—C6—C7 | 177.31 (12) |
| C3—C4—C5—C6 | 0.0 (2) | C5—C6—C7—O3 | −147.25 (13) |
| F2—C5—C6—C1 | −176.65 (11) | C1—C6—C7—O3 | 33.57 (18) |
| C4—C5—C6—C1 | 1.31 (19) | C5—C6—C7—O2 | 33.33 (17) |
| F2—C5—C6—C7 | 4.12 (18) | C1—C6—C7—O2 | −145.84 (13) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H1O2···O3i | 0.95 (4) | 1.68 (4) | 2.6318 (14) | 174 (4) |
| C3—H3···F2ii | 0.98 (2) | 2.54 (2) | 3.3428 (16) | 138.7 (16) |
Symmetry codes: (i) −x+1, −y, −z+1; (ii) x−1, −y+1/2, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB5558).
References
- Beavo, J. A. (1995). Physiol. Rev.75, 725–748. [DOI] [PubMed]
- Beavo, J. A. & Reifsnyder, D. H. (1990). Trends Pharmacol. Sci.11, 150–155. [DOI] [PubMed]
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Cosier, J. & Glazer, A. M. (1986). J. Appl. Cryst.19, 105–107.
- Nicholson, C. D., Chaliss, R. A. & Shalid, M. (1991). Trends Pharmacol. Sci.12, 19–27. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810028758/hb5558sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810028758/hb5558Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report