

Abstract
In the title compound, [CdBr2(C5H12N2S)2], the CdII atom lies on a twofold rotation axis. It exhibits a distorted tetrahedral coordination environment defined by two S atoms of two tetramethylthiourea (tmtu) ligands and two bromide ions. The crystal structure is consolidated by C—H⋯N and C—H⋯S hydrogen bonds.
Related literature
For crystallographic and spectroscopic studies of thiourea complexes, see: Al-Arfaj et al. (1998 ▶); Ali et al. (2009 ▶); Isab et al. (2009 ▶); Lobana et al. (2008 ▶); Marcos et al. (1998 ▶); Moloto et al. (2003 ▶). The structure of the title compound is isotypic with [Cd(tmtu)2I2] (Nawaz et al., 2010a
▶) and [Hg(tmtu)2Cl2] (Nawaz et al., 2010b
▶).
Experimental
Crystal data
[CdBr2(C5H12N2S)2]
M r = 536.67
Monoclinic,
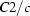
a = 18.6133 (17) Å
b = 10.0690 (9) Å
c = 13.4600 (12) Å
β = 130.834 (1)°
V = 1908.6 (3) Å3
Z = 4
Mo Kα radiation
μ = 5.54 mm−1
T = 292 K
0.24 × 0.23 × 0.20 mm
Data collection
Bruker SMART APEX area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.350, T max = 0.404
12678 measured reflections
2379 independent reflections
2114 reflections with I > 2σ(I)
R int = 0.028
Refinement
R[F 2 > 2σ(F 2)] = 0.021
wR(F 2) = 0.052
S = 1.05
2379 reflections
92 parameters
H-atom parameters constrained
Δρmax = 0.44 e Å−3
Δρmin = −0.45 e Å−3
Data collection: SMART (Bruker, 2008 ▶); cell refinement: SAINT (Bruker, 2008 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810028102/wm2371sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810028102/wm2371Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected bond lengths (Å).
| Cd1—S1 | 2.5580 (6) |
| Cd1—Br1 | 2.5735 (3) |
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2A⋯N2 | 0.96 | 2.53 | 2.855 (4) | 100 |
| C5—H5A⋯S1 | 0.96 | 2.65 | 3.026 (3) | 104 |
Acknowledgments
We gratefully acknowledge King Fahd University of Petroleum and Minerals, Dhahran, Saudi Arabia, for providing the X-ray facility.
supplementary crystallographic information
Comment
The coordination chemistry of thioureas with metal ions has been the subject of several recent investigations because of their variable binding modes and because of the relevance of their binding sites to those in living systems. Crystallographic reports about d10 metal complexes of thioureas established that these ligands are coordinated via the sulfur atom (Al-Arfaj et al., 1998; Moloto et al., 2003). Spectroscopic data is also consistent with this finding (Isab et al., 2009; Ali et al., 2009). Herein, we report the crystal structure of a cadmium bromide complex with tetramethylthiourea (tmtu), [Cd(C5H12N2S2)2Br2], (I).
The crystal structure of (I) consists of discrete molecular species in which the cadmium atom is located on a twofold rotation axis (Fig. 1). It exhibits a distorted tetrahedral coordination environment defined by two tetramethylthiourea (tmtu) ligands and two bromide ions. The tmtu ligand is terminally bound to the CdII atom via coordination of the S1 atom. The Cd—S and Cd—Br bond lengths are 2.5580 (6) and 2.5735 (3) Å, respectively. These values are in agreement with those reported for related compounds, e.g. (Al-Arfaj et al., 1998; Lobana et al. 2008; Marcos et al., 1998; Moloto et al., 2003). The bond angles around Cd are indicative of a slight tetrahedral distortion, with the S—Cd—S angle showing the largest deviation (117.70 (3)°) from the ideal value. The SCN2— moiety of the tmtu ligand is essentially planar, the maximum deviation from the mean plane being 0.007 (2) Å for the carbon atom. The fragments N1—C1—C2—C3 and N2—C1—C4—C5 are also close to planarity. The maximum deviations from the mean planes are 0.072 (2) Å and 0.065 (2) Å for N1 and N2, respectively. These values are consistent with a significant C—N double bond character and electron delocalization in the SCN2— moiety. The steric effect of the two adjacent 1,3-methyl groups imposes a dihedral angle of 47.4 (2) ° for the two mean planes and therefore a tilted conformation. The latter is likely stabilized by non-classical intramolecular hydrogen bonding interactions involving methyl H atoms with sulfur and nitrogen atoms (C2—H2A ···N2 and C5—H5A···S1). The molecules pack to form columns approximately parallel to [110] direction (Fig. 2).
The structure of (I) is isotypic with [Cd(tmtu)2I2] (Nawaz et al., 2010a), with an equivalent degree of distortion from the ideal tetrahedral configuration and similar Cd—S and C—N bond lengths. The tmtu bond lengths are also consistent with those found for the likewise isotypic compound [Hg(tmtu)2Cl2] (Nawaz et al., 2010b), in which a significantly higher distortion of the metal ion coordination sphere is observed.
Experimental
0.35 g (1.0 mmol) cadmium(II) bromide tetrahydrate dissolved in 10 ml water were added to two equivalents of tetramethylthiourea in methanol. A white precipitate formed and was filtered off. The filtrate was kept for crystallization. As a result, an off-white crystalline product suitable for single crystal X-ray diffraction was obtained.
Refinement
H atoms were placed in calculated positions with a C—H distance of 0.96 Å and Uiso(H) = 1.5 Ueq(C).
Figures
Fig. 1.

The molecular structure of title compound with the atomic numbering scheme. Displacement ellipsoids are drawn at the 30% probability level. H-atoms are omitted for clarity.
Fig. 2.

Packing diagram of the title complex
Crystal data
| [CdBr2(C5H12N2S)2] | F(000) = 1048 |
| Mr = 536.67 | Dx = 1.868 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 12678 reflections |
| a = 18.6133 (17) Å | θ = 2.5–28.3° |
| b = 10.0690 (9) Å | µ = 5.54 mm−1 |
| c = 13.4600 (12) Å | T = 292 K |
| β = 130.834 (1)° | Block, colorless |
| V = 1908.6 (3) Å3 | 0.24 × 0.23 × 0.20 mm |
| Z = 4 |
Data collection
| Bruker SMART APEX area-detector diffractometer | 2379 independent reflections |
| Radiation source: normal-focus sealed tube | 2114 reflections with I > 2σ(I) |
| graphite | Rint = 0.028 |
| ω scans | θmax = 28.3°, θmin = 2.5° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −24→24 |
| Tmin = 0.350, Tmax = 0.404 | k = −13→13 |
| 12678 measured reflections | l = −17→17 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.021 | H-atom parameters constrained |
| wR(F2) = 0.052 | w = 1/[σ2(Fo2) + (0.025P)2 + 1.3001P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.05 | (Δ/σ)max = 0.001 |
| 2379 reflections | Δρmax = 0.44 e Å−3 |
| 92 parameters | Δρmin = −0.45 e Å−3 |
| 0 restraints | Extinction correction: SHELXTL (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0068 (2) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cd1 | 1.0000 | 0.70441 (2) | 0.2500 | 0.03969 (8) | |
| Br1 | 1.149352 (17) | 0.56647 (3) | 0.34643 (3) | 0.05549 (9) | |
| S1 | 1.03619 (4) | 0.83583 (7) | 0.44089 (5) | 0.04794 (14) | |
| N1 | 0.91758 (14) | 0.76405 (19) | 0.4771 (2) | 0.0474 (4) | |
| N2 | 0.85096 (13) | 0.8900 (2) | 0.29223 (18) | 0.0471 (4) | |
| C1 | 0.92634 (14) | 0.82923 (19) | 0.39910 (19) | 0.0366 (4) | |
| C2 | 0.8527 (2) | 0.8077 (3) | 0.4968 (3) | 0.0672 (7) | |
| H2A | 0.8267 | 0.8928 | 0.4562 | 0.101* | |
| H2B | 0.8867 | 0.8145 | 0.5891 | 0.101* | |
| H2C | 0.8022 | 0.7444 | 0.4580 | 0.101* | |
| C3 | 0.9861 (2) | 0.6637 (3) | 0.5706 (3) | 0.0724 (8) | |
| H3A | 1.0111 | 0.6189 | 0.5359 | 0.109* | |
| H3B | 0.9552 | 0.6004 | 0.5851 | 0.109* | |
| H3C | 1.0369 | 0.7056 | 0.6522 | 0.109* | |
| C4 | 0.75337 (18) | 0.8418 (3) | 0.2186 (3) | 0.0694 (8) | |
| H4A | 0.7550 | 0.7542 | 0.2480 | 0.104* | |
| H4B | 0.7211 | 0.8394 | 0.1266 | 0.104* | |
| H4C | 0.7203 | 0.9005 | 0.2330 | 0.104* | |
| C5 | 0.8609 (2) | 0.9921 (3) | 0.2254 (3) | 0.0697 (8) | |
| H5A | 0.9218 | 1.0340 | 0.2870 | 0.105* | |
| H5B | 0.8118 | 1.0573 | 0.1892 | 0.105* | |
| H5C | 0.8557 | 0.9524 | 0.1561 | 0.105* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cd1 | 0.04286 (13) | 0.04283 (13) | 0.04311 (13) | 0.000 | 0.03237 (11) | 0.000 |
| Br1 | 0.05068 (15) | 0.05784 (16) | 0.05843 (16) | 0.01407 (10) | 0.03588 (13) | 0.00638 (11) |
| S1 | 0.0391 (3) | 0.0665 (4) | 0.0425 (3) | −0.0082 (2) | 0.0286 (2) | −0.0132 (2) |
| N1 | 0.0572 (11) | 0.0471 (10) | 0.0559 (11) | 0.0041 (9) | 0.0448 (10) | 0.0036 (8) |
| N2 | 0.0448 (10) | 0.0540 (11) | 0.0427 (10) | 0.0038 (8) | 0.0287 (9) | −0.0020 (8) |
| C1 | 0.0406 (10) | 0.0363 (10) | 0.0371 (10) | −0.0014 (8) | 0.0272 (9) | −0.0061 (8) |
| C2 | 0.0799 (19) | 0.0800 (19) | 0.0815 (19) | −0.0012 (15) | 0.0702 (18) | −0.0047 (15) |
| C3 | 0.090 (2) | 0.0600 (16) | 0.082 (2) | 0.0174 (15) | 0.0627 (19) | 0.0238 (15) |
| C4 | 0.0393 (13) | 0.101 (2) | 0.0573 (16) | 0.0026 (14) | 0.0268 (12) | −0.0123 (15) |
| C5 | 0.0804 (19) | 0.0721 (18) | 0.0571 (15) | 0.0165 (15) | 0.0451 (15) | 0.0201 (14) |
Geometric parameters (Å, °)
| Cd1—S1 | 2.5580 (6) | C2—H2B | 0.9600 |
| Cd1—S1i | 2.5580 (6) | C2—H2C | 0.9600 |
| Cd1—Br1i | 2.5735 (3) | C3—H3A | 0.9600 |
| Cd1—Br1 | 2.5735 (3) | C3—H3B | 0.9600 |
| S1—C1 | 1.731 (2) | C3—H3C | 0.9600 |
| N1—C1 | 1.335 (3) | C4—H4A | 0.9600 |
| N1—C3 | 1.460 (3) | C4—H4B | 0.9600 |
| N1—C2 | 1.463 (3) | C4—H4C | 0.9600 |
| N2—C1 | 1.331 (3) | C5—H5A | 0.9600 |
| N2—C5 | 1.455 (3) | C5—H5B | 0.9600 |
| N2—C4 | 1.471 (3) | C5—H5C | 0.9600 |
| C2—H2A | 0.9600 | ||
| S1—Cd1—S1i | 117.70 (3) | H2A—C2—H2C | 109.5 |
| S1—Cd1—Br1i | 105.899 (14) | H2B—C2—H2C | 109.5 |
| S1i—Cd1—Br1i | 106.524 (15) | N1—C3—H3A | 109.5 |
| S1—Cd1—Br1 | 106.524 (15) | N1—C3—H3B | 109.5 |
| S1i—Cd1—Br1 | 105.899 (14) | H3A—C3—H3B | 109.5 |
| Br1i—Cd1—Br1 | 114.676 (17) | N1—C3—H3C | 109.5 |
| C1—S1—Cd1 | 100.04 (7) | H3A—C3—H3C | 109.5 |
| C1—N1—C3 | 122.1 (2) | H3B—C3—H3C | 109.5 |
| C1—N1—C2 | 122.3 (2) | N2—C4—H4A | 109.5 |
| C3—N1—C2 | 114.2 (2) | N2—C4—H4B | 109.5 |
| C1—N2—C5 | 121.5 (2) | H4A—C4—H4B | 109.5 |
| C1—N2—C4 | 122.8 (2) | N2—C4—H4C | 109.5 |
| C5—N2—C4 | 114.6 (2) | H4A—C4—H4C | 109.5 |
| N2—C1—N1 | 119.41 (19) | H4B—C4—H4C | 109.5 |
| N2—C1—S1 | 121.32 (16) | N2—C5—H5A | 109.5 |
| N1—C1—S1 | 119.26 (16) | N2—C5—H5B | 109.5 |
| N1—C2—H2A | 109.5 | H5A—C5—H5B | 109.5 |
| N1—C2—H2B | 109.5 | N2—C5—H5C | 109.5 |
| H2A—C2—H2B | 109.5 | H5A—C5—H5C | 109.5 |
| N1—C2—H2C | 109.5 | H5B—C5—H5C | 109.5 |
Symmetry codes: (i) −x+2, y, −z+1/2.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2A···N2 | 0.96 | 2.53 | 2.855 (4) | 100 |
| C5—H5A···S1 | 0.96 | 2.65 | 3.026 (3) | 104 |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: WM2371).
References
- Al-Arfaj, A. R., Reibenspies, J. H., Isab, A. A. & Hussain, M. S. (1998). Acta Cryst. C54, 51–53.
- Ali, S., Malik, M. R., Isab, A. A. & Ahmad, S. (2009). J. Coord. Chem.62, 475–480.
- Bruker (2008). SMART and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Isab, A. A., Wazeer, M. I. M. & Ashraf, W. (2009). Spectrochim. Acta Part A, 72, 218–221. [DOI] [PubMed]
- Lobana, T. S., Sharma, R., Sharma, R., Sultana, R. & Butcher, R. J. (2008). Z. Anorg. Allg. Chem.634, 718–723.
- Marcos, C., Alía, J. M., Adovasio, V., Prieto, M. & García-Granda, S. (1998). Acta Cryst. C54, 1225–1229.
- Moloto, M. J., Malik, M. A., O’Brien, P., Motevalli, M. & Kolawole, G. A. (2003). Polyhedron, 22, 595–603.
- Nawaz, S., Sadaf, S., Fettouhi, M., Fazal, A. & Ahmad, S. (2010a). Acta Cryst. E66, m951. [DOI] [PMC free article] [PubMed]
- Nawaz, S., Sadaf, H., Fettouhi, M., Fazal, A. & Ahmad, S. (2010b). Acta Cryst. E66, m952. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810028102/wm2371sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810028102/wm2371Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report