

Abstract
N-methyl-d-aspartate receptors (NMDARs) play an important role in cell survival versus cell death decisions during neuronal development, ischemia, trauma, and epilepsy. Coupling of neurons by electrical synapses (gap junctions) is high or increases in neuronal networks during all these conditions. In the developing CNS, neuronal gap junctions are critical for two different types of NMDAR-dependent cell death. However, whether neuronal gap junctions play a role in NMDAR-dependent neuronal death in the mature CNS was not known. Using Fluoro-Jade B staining, we show that a single intraperitoneal administration of NMDA (100 mg/kg) to adult wild-type mice induces neurodegeneration in three forebrain regions, including rostral dentate gyrus. However, the NMDAR-mediated neuronal death is prevented by pharmacological blockade of neuronal gap junctions (with mefloquine, 30 mg/kg) and does not occur in mice lacking neuronal gap junction protein, connexin 36. Using Western blots, electrophysiology, calcium imaging, and gas chromatography–mass spectrometry in wild-type and connexin 36 knockout mice, we show that the reduced level of neuronal death in knockout animals is not caused by the reduced expression of NMDARs, activity of NMDARs, or permeability of the blood–brain barrier to NMDA. In wild-type animals, this neuronal death is not caused by upregulation of connexin 36 by NMDA. Finally, pharmacological and genetic inactivation of neuronal gap junctions in mice also dramatically reduces neuronal death caused by photothrombotic focal cerebral ischemia. The results indicate that neuronal gap junctions are required for NMDAR-dependent excitotoxicity and play a critical role in ischemic neuronal death.
INTRODUCTION
In the mammalian CNS, direct intercellular communication between neighboring cells occurs through specialized structures known as gap junctions. A gap junction is a channel between two cells with a pore ∼2 nm in diameter that allows the direct diffusion of ions and small molecules (Bennett and Zukin 2004). The channels are made of proteins known as connexins, which are encoded by a large multigene family with ≥21 genes in mammals (Sohl et al. 2005). Among identified connexins, connexin 36 (Cx36) seems to be an exclusive neuron-specific connexin type (Belluardo et al. 2000; Condorelli et al. 2003). The expression of Cx36 and the coupling of neurons by gap junctions increase during embryonic and/or early postnatal development, then decrease, but increase again in the mature CNS during neuronal injuries such as ischemia, traumatic brain injury, inflammation, and epilepsy (Chang et al. 2000; de Pina-Benabou et al. 2005; Frantseva et al. 2002; Nemani and Binder 2005; Oguro et al. 2001; Perez-Velazquez et al. 1994; Thalakoti et al. 2007). Death of neurons occurs during development and injuries. However, whether gap junctions contribute to neuronal death or survival is still controversial (Decrock et al. 2009; Perez Velazquez et al. 2003).
The N-methyl-d-aspartate receptor (NMDAR), a glutamate-gated ion channel, plays a critical role in cell survival versus cell death decisions during neuronal development, ischemia, trauma, and epilepsy (Arundine and Tymianski 2004; Goodman and Shatz 1993; Hardingham and Bading 2003; Scheetz and Constantine-Paton 1994). Recently, using developing hypothalamic neuronal cultures, we showed a critical role during development for neuronal gap junctions in two types of NMDAR-dependent neuronal cell death, i.e., the deaths caused by hyperactivation or inactivation of NMDARs (de Rivero Vaccari et al. 2007). Specifically, we found that the NMDAR-dependent neuronal deaths do not occur in mature cultures, occur only during the peak of developmental increase in neuronal gap junction coupling [i.e., on days in vitro 14–17 (DIV14-17)], and are prevented by pharmacological inactivation of gap junctions or genetic knockout of Cx36. Compared with cultures, where no neuronal gap junction coupling is detected after cell culture maturation (Arumugam et al. 2005), the coupling and Cx36 expression are found in some neurons in the mature CNS in vivo (Belluardo et al. 2000; Hormuzdi et al. 2001; Long et al. 2005; Venance et al. 2000). Therefore in this study, using NMDA administration in adult mice, we tested the hypothesis that neuronal gap junctions are critical for NMDAR-mediated excitotoxicity in the mature CNS. We also used photothrombotic focal cerebral ischemia in mice to determine whether neuronal gap junctions play a role in ischemic neuronal death.
METHODS
Animal care
Experiments were performed on 2-mo-old wild-type (WT; C57bl/6 strain) and Cx36 knockout (C57bl/6 background strain) male mice. The Cx36 knockout was originally created by Dr. David Paul (Harvard Medical School) and showed almost complete (95–99%) loss of neuronal gap junction coupling (Deans et al. 2001). Mice were genotyped as described (de Rivero Vaccari et al. 2007).
NMDA and other drug treatments
Mice received a single intraperitoneal administration of sterile saline (100 μl; control), NMDA (Sigma-Aldrich; 100 mg/kg in 100 μl of saline), or mefloquine hydrochloride [a blocker for Cx36-containing gap junctions (Cruikshank et al. 2004; Voss et al. 2009); Sigma-Aldrich; 30 mg/kg in 100 μl of saline]. Some animals received injections of mefloquine (30 mg/kg) or dizocilpine (MK-801; NMDAR blocker; Sigma-Aldrich; 1 mg/kg in 100 μl of saline) that were followed in 30 and 15 min, respectively, by an NMDA injection (100 mg/kg). All solutions also contained Tween80 (1%). After NMDA administration (100 mg/kg), only 1 of 54 WT mice and none of 34 Cx36 knockout mice died during a 24-h period. However, at a higher NMDA dose (150 mg/kg), all tested animals (5 WT and 3 Cx36 knockout) died within 5 min after injection.
Photothrombotic focal cerebral ischemia
Mice were anesthetized (nembutal, 70 mg/kg) and placed in a stereotaxic frame. The skull was exposed by incision of the skin. Rose bengal (1 mg in 100 μl of sterile saline) was injected intraperitoneally. A fiber optic bundle of a cold light source (Zeiss 1500; 1.5 mm aperture; 3,000 K light intensity) was placed in the right hemisphere at anteroposterior (AP) = −1.7 mm and lateral to midline (ML) = +2.0 mm from bregma. To activate Rose bengal, 15 min after its injection, the brain was illuminated through the intact skull for the following 30 min. Furthermore, all animals received a single intraperitoneal saline or mefloquine (30 mg/kg) injection that was given immediately before the Rose bengal injection. In all drug treatment and ischemic experiments, an analgesic was used to reduce possible pain reactions (buprenex, 2 mg/kg).
Fluoro-Jade B and Nissl stainings
Fluoro-Jade B staining is selective and highly sensitive for identification of degenerating neurons (Xu et al. 2004). To conduct the staining, animals were anesthetized and transcardially perfused with PBS (0.01 M; 100 ml) followed by a perfusion with 4% paraformaldehyde (dissolved in 50 ml of PBS; pH = 7.4). The brains were removed. Fifty-micrometer-thick coronal sections were cut with a Leica VT1000S vibratome and mounted on gelatin-coated slides. The sections were hydrated in alcohol, incubated with potassium permanganate (0.006%; 10 min), incubated with Fluoro-Jade B (0.001%; 30 min), rinsed, and air-dried. Staining was visualized using a Nikon Eclipse 80i fluorescent microscope, FITC filter, and Photometrics ES2 digital camera. The staining analysis was done by a person blinded to experimental groups.
In NMDA excitotoxicity experiments, the cell bodies of Fluoro-Jade B–positive neurons were clearly visible. Therefore using OpenLab software (Improvision), the total number of stained neurons was counted in the whole hippocampal region, in both hemispheres, in nine sections in each brain (including −1.0, −1.2, −1.4, −1.6, −1.8, −2.0, −2.2, −2.4, and −2.6 mm from bregma) and was averaged for nine sections.
In ischemic experiments, because of the high amount of staining, the ischemic cortical region was outlined, and the total number of stained pixels was measured in the outlined region using a densitometric thresholding technique implemented with ImageJ software (National Institutes of Health). The threshold was set at a level just above that which would have counted background and nonspecific staining in areas outside the outlined region. The analysis was done in five cortical sections (including −1.5, −1.6, −1.7, −1.8, and −1.9 mm from bregma), and data were averaged for five sections. Because Fluoro-Jade B has an excellent sensitivity and selectivity for identification of degenerating neurons (Xu et al. 2004), no additional co-staining with neuronal markers was necessary.
To conduct Nissl staining, sections were incubated with 0.1% cresyl violet dissolved in PBS, differentiated in 95% ethanol containing acetic acid, and dehydrated.
Western blots
Experiments were conducted as described (Arumugam et al. 2005). Briefly, the hippocampus was dissected and homogenized in a lysis buffer, and total protein was determined. Fifty micrograms of protein was loaded in each lane, transferred to 0.45-μm polyvinylidene difluoride membranes, and processed with a blocking solution and antibodies. Mouse anti-NR1 (1:2,000; cat. 05-432, Upstate), rabbit anti-NR2A (1:10,000; cat. 05-901, Upstate), rabbit anti-NR2B (1:2,000; cat. M265, Sigma-Aldrich), rabbit anti-Cx36 (1:500; cat. 51-6300, Invitrogen), and mouse anti-tubulin (1:10,000; cat. T4026, Sigma-Aldrich) were used as the primary antibodies and were visualized with horseradish peroxidase-conjugated anti-rabbit antibody (1:10,000; cat. G21234, Zymed) or anti-mouse IgG antibody (1:10,000; cat. M5899, Sigma-Aldrich). Band optical density was determined using Quantity One software (Bio-Rad). Optical density signals were normalized relative to tubulin, and normalized values were compared with controls (set at 1.0). Tubulin levels per unit of total protein did not vary significantly among samples.
Brain slice preparation and electrophysiology
Mice were anesthetized, and coronal 400-μm-thick hippocampal slices were prepared (at 2–4°C) and tested in electrophysiological experiments (at 20–22°C) using artificial cerebrospinal fluid (ACSF) and a pipette solution as described (Arumugam et al. 2005). To test NMDA responses, whole cell voltage-clamp recording was performed at a −60 mV holding potential using a Mutliclamp 700B amplifier and pCLAMP10 software (Molecular Devices). NMDA (10 μM) was applied in Mg2+-free ACSF using bath application. To prevent secondary responses, all media contained TTX (2 μM; a voltage-gated sodium channel blocker), bicuculline (20 μM; a GABAA receptor antagonist), and CNQX (10 μM; a non-NMDA receptor antagonist) (all drugs were from Sigma-Aldrich). The amplitude of NMDA responses was measured and analyzed using Igor Pro (WaveMetrics). To test the effects of mefloquine on electrotonic coupling, dual current-clamp recordings from pairs of randomly chosen neurons in the rostral dentate gyrus acute slices were conducted.
Neuronal cell culture preparation and digital Ca2+ imaging
Pregnant mice were anesthetized, and neuronal cultures were prepared as described (Belousov et al. 2001) from embryonic day 18–19 mouse hippocampus. Neurons were plated on glass coverslips and raised in Neurobasal medium (Invitrogen) with supplements (B27 and glutamine), gentamycin (50 mg/l), and cytosine β-d-arabinofuranoside (5 μM). Each coverslip contained the material obtained from a single embryo. The culture medium was changed twice a week. Fura-2 Ca2+ imaging was performed in mature cultures (1–1.5 mo old) using ACSF as described (Belousov et al. 2001). Cells were loaded with fura-2 AM (5 μM, Invitrogen) for 30 min and examined using a Nikon TE2000s microscope. Conventional dual wavelength ratios (at 340 and 380 nm excitation) were obtained using a Sutter Lambda10-2 filter changer (Sutter Instruments) and Imaging Workbench software (INDEC BioSystems). Ca2+ standards from Invitrogen were used to calibrate the imaging system. NMDA (10 μM) was applied using a flow pipe perfusion system (Belousov et al. 2001) in Mg2+-free, but TTX-, bicuculline-, and CNQX-containing, ACSF (in concentrations as above). The intracellular Ca2+ increases were measured in neuronal cell bodies and were analyzed using Igor Pro software.
Gas chromatography–mass spectrometry assay
Animals were anesthetized and killed by cardiac puncture before (control) or 5, 15, 30, and 120 min after NMDA administration (100 mg/kg). Blood samples were collected and used later to determine the levels of NMDA in the blood plasma. Animals were perfused transcardially with PBS for 5 min to remove blood plasma. The brain tissue (including hippocampus and hypothalamus) was dissected and soaked in PBS to ensure complete removal of the blood from the tissue. The measurements of NMDA in the brain tissue (100 mg) or blood plasma (100 μl) were conducted as described previously in detail (D'Aniello et al. 2000).
Statistical analysis
Data were analyzed using the two-tailed Student's t-test or ANOVA with post hoc Tukey and InStat software (GraphPad Software). Data are reported as mean ± SE for the number of samples indicated.
RESULTS
NMDAR-mediated neuronal death
A single intraperitoneal administration of NMDA to WT mice (100 mg/kg) induced dramatic neuronal death in the hippocampus. The neuronal death was evident 24 h after NMDA injection in brain sections stained with Fluoro-Jade B (Fig. 1, A, B, and E) and 3 wk after NMDA injection in brain sections stained with Nissl (Supplemental Fig. S1A).1 In addition, in Fluoro-Jade B staining experiments, dramatic neuronal death was detected in the hypothalamus (Supplemental Figs. S2, A, B, and E, and S3, C and D) and modest neuronal death in the medial habenula (data not shown). The rest of the forebrain regions did not contain or contained single and very sparse Fluoro-Jade B–positive cells.
Fig. 1.

Inactivation of neuronal gap junctions prevents N-methyl-d-aspartate receptor (NMDAR)-mediated neuronal death. A–D: representative images of Fluoro-Jade B staining in brain sections from control WT (A), NMDA-treated WT (B), NMDA plus mefloquine-treated WT (C), and NMDA-treated Cx36 knockout (Cx36 KO; D) mice are shown. Administration of NMDA induces neuronal death in the rostral dentate gyrus (B) that is prevented by cotreatment with mefloquine (C) or Cx36 knockout (D). E: graph presents statistical analysis of the number of Fluoro-Jade B–positive neurons in the hippocampus (ANOVA; n = 4–7 mice per group; ***P < 0.001; NS, nonsignificant; mean ± SE). The analysis was done 24 h after intraperitoneal (ip) saline or drug administrations (ip). CA3, CA3 hippocampus; cc, corpus callosum; D3V, dorsal 3rd ventricle; GrDG, granule cell layer of dentate gyrus.
In the hippocampus, neuronal degeneration was observed in the rostral dentate gyrus (Fig. 1B; Supplemental Fig. S3, A and B), primarily in the inner part of the granule cell layer (Supplemental Fig. S1A). However, it was not seen in the caudal dentate gyrus (Supplemental Fig. S4). The NMDAR-mediated cell death was prevented by mefloquine (30 mg/kg; Fig. 1, C and E), which blocks neuronal Cx36-containing gap junctions (Cruikshank et al. 2004; Voss et al. 2009) (Supplemental Fig. S5). In addition, no statistically significant neuronal death was induced by NMDA administration in Cx36 knockout mice (Fig. 1, D and E). Furthermore, in knockout animals, pretreatment with mefloquine that was followed by NMDA injection did not have any additional effect (Fig. 1E). Similar results were obtained in the hypothalamus (Supplemental Fig. S2). As expected, the NMDAR-mediated neuronal death also was prevented by MK-801 (1 mg/kg; NMDAR blocker; Supplemental Fig. S6), confirming that the death is caused by hyperactivation of NMDARs.
Control tests
It is possible that the reduced level of NMDAR-mediated neuronal death in Cx36 knockout mice is caused by the reduced expression of NMDARs, reduced activity of NMDARs, or reduced permeability of the blood–brain barrier (BBB) to NMDA in knockout mice compared with WT mice, and we tested these possibilities. Western blot experiments showed that the expression of NR2A and NR2B NMDAR subunits in the rostral dentate gyrus is not different, and the expression of NR1 subunit is even higher in Cx36 knockout mice compared with WT mice (Fig. 2).
Fig. 2.
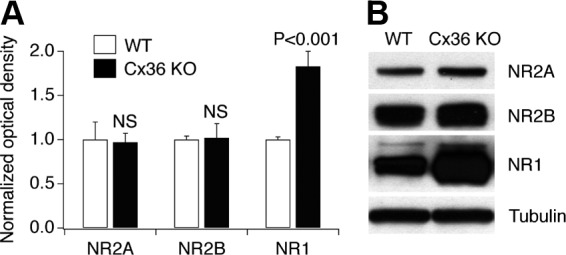
Expression of NMDAR subunits in the rostral dentate gyrus of WT and Cx36 knockout mice. A and B: statistical analysis (A: ANOVA; n = 4 in each group; mean ± SE; shown relative to the corresponding WT group) and representative images (B) from Western blot experiments are shown. Optical density signals are normalized relative to tubulin and compared with the WT group (set at 1.0). The Western blots were done sequentially on 1 membrane.
Patch-clamp recordings from granule cells in acute rostral dentate gyrus slices indicated that the amplitude of NMDA-mediated responses is not significantly different between WT and Cx36 knockout mice (Fig. 3, A–C). Calcium imaging recordings from cultured hippocampal neurons showed that the amplitude of NMDA-mediated Ca2+ increases is even higher in Cx36 knockout than in WT conditions (Fig. 3, D–F). Furthermore, using a gas chromatography–mass spectrometry (GC-MS) assay, the levels of NMDA were measured in the blood and in the brain in mice before and at four time points after NMDA injection and were not statistically different between the Cx36 knockout and WT animals (Fig. 4).
Fig. 3.
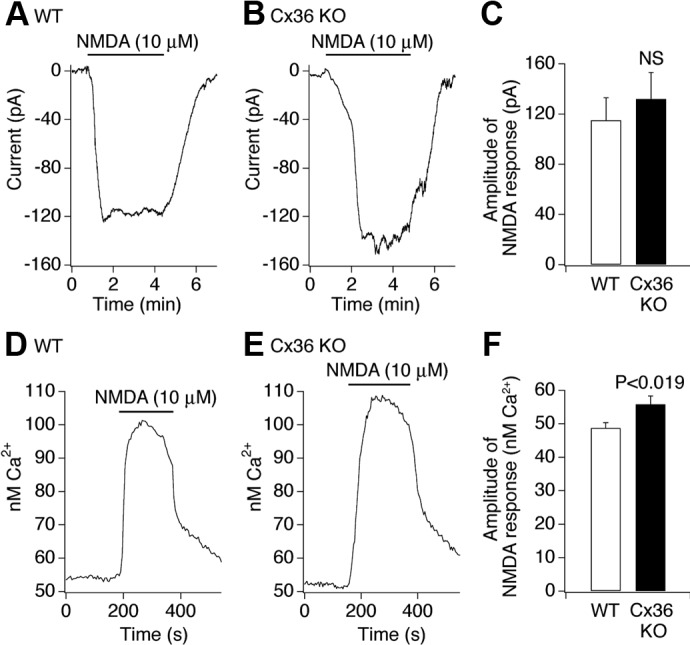
Neuronal NMDA responses in WT and Cx36 knockout mice. A–C: representative patch-clamp recordings from granule cells in acute rostral dentate gyrus slices (A, WT; B, Cx36 knockout) and statistical analysis (C; unpaired Student's t-test; n = 7 neurons in each group; mean ± SE) are shown. D–F: representative Ca2+ imaging recordings from cultured mature hippocampal neurons (D, WT; E, Cx36 knockout) and statistical analysis [F; unpaired Student's t-test; n = 75 (WT) and 78 (Cx36 knockout) neurons; mean ± SE] are shown. All tests were done in a Mg2+-free medium that included TTX (2 μM), bicuculline (20 μM), and CNQX (10 μM).
Fig. 4.
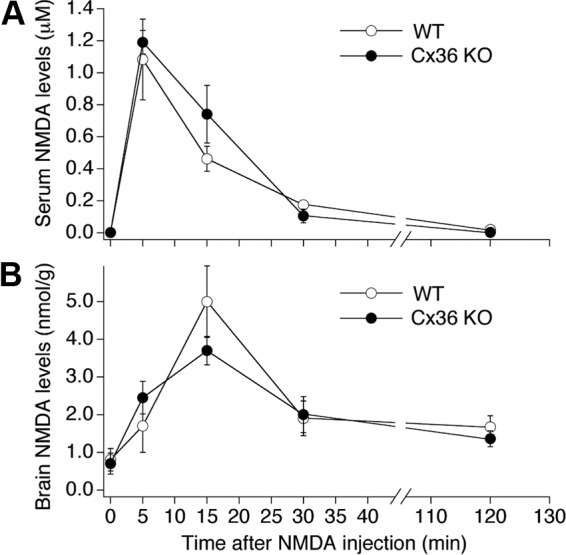
NMDA levels in WT and Cx36 knockout mice after NMDA administration. Data from GC-MS experiments are presented. A and B: the analysis was done in the blood plasma (A) and in the brain (B) before (0) and at 4 time points after administration of NMDA (100 mg/kg; ip). Statistical analysis (ANOVA; n = 3–4 animals per group; mean ± SE) did not show statistical difference between the WT and Cx36 knockout animals.
Finally, using Western blots for Cx36, we tested the possibility that, in WT animals, NMDA administration increases the expression of Cx36-containing gap junctions, thus contributing to neuronal cell death. However, no change in Cx36 expression was detected at four time points after NMDA administration in the rostral dentate gyrus in experiments in vivo (Fig. 5A) and in hippocampal cultures in vitro (Fig. 5B).
Fig. 5.
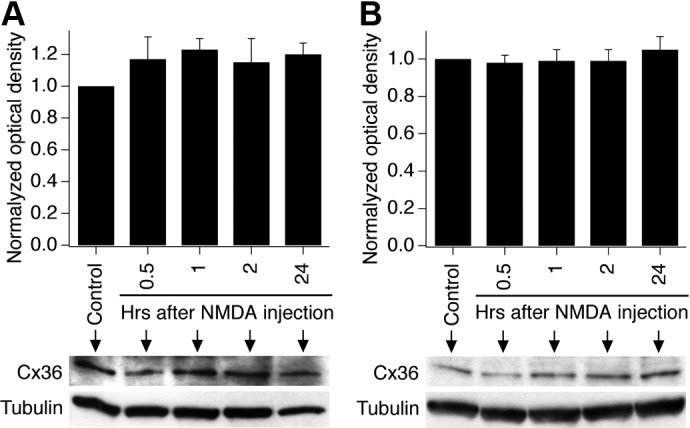
NMDA administration does not affect the expression of Cx36 in WT mice and cultures. A and B: statistical data and representative images from Western blot experiments in the rostral dentate gyrus (A) and in mature hippocampal neuronal cultures (B) are shown. Statistical analysis (ANOVA; n = 5 per group; mean ± SE) did not show statistical difference between the experimental groups.
Ischemia-mediated neuronal death
NMDARs may contribute to neuronal death during ischemia (Arundine and Tymianski 2004), and we tested whether inactivation of neuronal gap junctions also reduces ischemic neuronal death. Photothrombotic focal cerebral ischemia caused substantial neuronal death in the neocortex of WT mice that was detected using Fluoro-Jade B staining in brain sections 48 h after the ischemic procedure (Fig. 6, A–C and F). This neuronal death was dramatically reduced in mefloquine-treated WT (Fig. 6, D and F) and Cx36 knockout (Fig. 6, E and F) mice. However, there was no additional neuroprotective effect of mefloquine in knockout animals (Fig. 6F).
Fig. 6.

Inactivation of neuronal gap junctions reduces ischemia-mediated neuronal death in mice. Representative images of Fluoro-Jade B staining (A–E) and statistical analysis (F) are shown. All images are taken at bregma −1.7 mm (i.e., at the center of ischemic injury). Images in A–C are taken from the same brain section, and the regions that are boxed in A and B are shown at a higher magnification as, respectively, B and C. A–E: photothrombotic focal ischemia induces neuronal death in the cortex of WT mice (A–C) that is reduced by a pretreatment with mefloquine (D) and Cx36 knockout (E). F: number of stained pixels in the ischemic cortical region is analyzed (ANOVA; n = 4–10 mice per group; mean ± SE). Statistical difference is shown relative to the saline-treated WT mice (***P < 0.001 and **P < 0.01) and between saline- and mefloquine-treated Cx36 knockout mice (#NS, nonsignificant). The analysis was done 48 h after ischemia. S1Tr, primary somatosensory, trunk; S1BF, primary somatosensory, barrel field.
DISCUSSION
The level of neuronal gap junction coupling and the expression of Cx36 are high in the developing mammalian CNS and subsequently decrease (Arumugam et al. 2005; Bennett and Zukin 2004). In the mature CNS, the neuronal coupling and Cx36 expression are found in some regions, including the hippocampal dentate gyrus and hypothalamus (Arumugam et al. 2005; Belluardo et al. 2000; Hormuzdi et al. 2001; Long et al. 2005; Venance et al. 2000), and the coupling is eliminated in these regions in Cx36 knockout mice (Hormuzdi et al. 2001; Long et al. 2005). In this study, we showed that a single administration of NMDA to adult WT mice induces massive neuronal death in the rostral dentate gyrus and hypothalamus, and this neuronal death is prevented by pharmacological and genetic inactivation of Cx36-containing gap junctions. Thus the results suggest a critical role for neuronal gap junctions in NMDAR-mediated excitotoxicity in the mature CNS.
Data from control experiments in WT and Cx36 knockout mice indicate that the reduced level of NMDAR-mediated neuronal death in knockout animals is not caused by reduced expression and activity of NMDARs or permeability of the BBB to NMDA. The lack of Cx36 upregulation by NMDA in WT mice suggests that the NMDAR-dependent neuronal death in these animals is caused by a contribution of already existing Cx36-containing gap junctions rather than newly synthesized ones. It is still possible that NMDA administration increases the expression of non–Cx36-comprising neuronal gap junctions or hemichannels. However, given our previous studies showing that activation of NMDARs decreases, rather than increases, neuronal gap junction coupling (Arumugam et al. 2005) and that hemichannels do not contribute to the NMDAR-mediated neuronal death in developing neurons (de Rivero Vaccari et al. 2007), the above possibilities seem unlikely.
Adult Cx36 knockout mice lack obvious anatomical, physiological, and behavioral abnormalities, although showe some mild deficiencies (Deans et al. 2001; Hormuzdi et al. 2001; Long et al. 2005). Therefore it is also possible that abnormal animal development contributes in some way to the lower NMDAR-mediated neuronal death in Cx36-null mice. A strong argument against this suggestion, however, comes from our previous study (de Rivero Vaccari et al. 2007). The study was conducted using NMDA administration in neuronal cultures obtained from Cx36 knockout embryos (i.e., obtained from the animals even before their birth), but showed results similar to those in the adult Cx36 knockout mice reported here. In addition, in this study, we tested the effects of mefloquine on NMDAR- and ischemia-mediated neuronal death. Mefloquine was reported to be a potent blocker of Cx36-containing gap junctions (Cruikshank et al. 2004; Voss et al. 2009). In the used concentrations, mefloquine effectively reduced electrotonic coupling between neurons in brain slices in vitro (Supplemental Fig. S5), prevented NMDAR-mediated neuronal death in vivo (Fig. 1), and reduced ischemic neuronal death in vivo (Fig. 6), and the concentrations of mefloquine in vivo and in vitro were comparable: 30 mg/kg and 21 mg/l (50 μM), respectively. This confirmed that the neuroprotection in Cx36-deficient animals was likely caused by elimination of Cx36.
It should be noted that, in addition to the blockade of Cx36-containing gap junctions, mefloquine also may block Cx50-containing gap junctions (which, however, are restricted to the lens), pannexin 1 hemichannels, and adenosine A2A receptors (Cruikshank et al. 2004; Iglesias et al. 2008; Weiss et al. 2003). Therefore the actions of mefloquine on these channels and/or receptors may potentially be responsible (at least in part) for neuroprotection. In this study, however, mefloquine did not have any additional neuroprotective effects in Cx36-deficient mice in both NMDA excitotoxicity and ischemic models. This suggests that neuroprotective effects of mefloquine were specifically via blockade of Cx36-containing gap junctions.
As discussed above, whether gap junctions contribute to neural cell death or survival is controversial. It has been suggested that contributions of gap junctions to either neuronal death or survival are through propagation of some gap junction–permeable signals, neurodegenerative or neuroprotective, respectively (Decrock et al. 2009; Perez Velazquez et al. 2003). Although speculations on possible candidates exist (e.g., Ca2+, Na+, IP3), the nature of these signals is largely unknown. Presumably, in our experimental conditions, these are NMDAR-dependent neurodegenerative (death) signals. If such death signals were generated in all neurons independently and equally, it would be expected that the affected neurons would die regardless of whether or not they are coupled to other neurons. However, NMDAR-dependent cell death is prevented by inactivation of gap junctions, indicating that the gap junction–coupled neurons have a higher probability of dying than those that are uncoupled. We hypothesize therefore that, for example, in the rostral dentate gyrus, the generation of death signals is unequal in different neurons. This can be caused by unequal distribution of NMDARs among different neurons and/or unequal expression of NMDAR-dependent intracellular signaling pathways and molecules. It is possible that at the NMDA concentration that was used in this study, only a small fraction of neurons generates lethal levels of (presumptive) gap junction–permeable death signals. The blockade of gap junctions rescues the majority of neurons in the rostral dentate gyrus, and only a small (nonsignificant) fraction of neurons die. We propose that these neurons, which produce lethal levels of gap junction–permeable death signals, are those that can be seen in NMDA-treated Cx36 knockout mice and in WT mice co-treated with NMDA and mefloquine. A focus of future studies will be to identify the specific gap junction–permeable signaling molecules that are generated during the NMDAR-mediated excitotoxicity.
In addition, with the systemic administration of NMDA that was used in this study, the NMDAR-mediated neuronal death was detected in the forebrain only in three regions: hippocampus, hypothalamus, and medial habenula. Moreover, in the hippocampus, neuronal death was observed in the rostral dentate gyrus (primarily in the inner part of the granule cell layer) but not in the caudal dentate gyrus. This clearly suggests the region specificity for the induction of NMDAR-mediated neuronal death. Once again, this potentially may be because of the difference in the expression of NMDARs among different brain regions. If this is the case, neuronal death in other brain regions should be expected if NMDA is delivered to those regions in sufficiently high concentrations: in fact, the increased neuronal death is observed in the hippocampal CA1 after direct intra-hippocampal NMDA administration (Brennan et al. 2009). However, another possibility is that the level of neuronal gap junction coupling is higher in these selective regions than in the rest of the forebrain, which makes them more susceptible to NMDAR-mediated excitotoxicity. If this is the case, the level of neuronal gap junction coupling should be expected to be higher in the rostral than in the caudal dentate gyrus. Future experiments will evaluate this prediction.
As discussed above, in this study, we also used photothrombotic focal cerebral ischemia in adult mice to determine the role of neuronal gap junctions in ischemic neuronal death. In general, the photothrombotic stroke model is less sensitive to genetic and pharmacological manipulations than stroke induced by transient occlusion of a major artery, such as the middle cerebral artery. Nevertheless, our study showed that neuronal death caused by photothrombotic ischemia is substantially reduced by genetic and pharmacological inactivation of neuronal gap junctions. Given that NMDARs are essential for induction of the secondary neuronal death during ischemia (Arundine and Tymianski 2004) and that neuronal gap junctions play critical role in the NMDAR-mediated excitotoxicity and ischemic neuronal death (this study), inactivation of neuronal gap junctions may be considered a promising tool for neuroprotection during ischemic injuries.
GRANTS
This work was supported by National Institutes of Health Grants R01 NS-064256, R01 DA-015088, and P20 RR-024214, Kansas IDeA Network of Biomedical Research Excellence, and University of Kansas Medical Center funds to A. B. Belousov. Core support was provided by National Institutes of Health Grant HD-002528.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Marla Feller and David Paul for providing Cx36 knockout mice and Dr. Randolph Nudo for generous help.
Footnotes
The online version of this article contains supplemental data.
REFERENCES
- Arumugam et al., 2005. Arumugam H, Liu X, Colombo PJ, Corriveau RA, Belousov AB. NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci 8: 1720–1726, 2005 [DOI] [PubMed] [Google Scholar]
- Arundine and Tymianski, 2004. Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 61: 657–668, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluardo et al., 2000. Belluardo N, Mudo G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res 865: 121–138, 2000 [DOI] [PubMed] [Google Scholar]
- Belousov et al., 2001. Belousov AB, O'Hara BF, Denisova JV. Acetylcholine becomes the major excitatory neurotransmitter in the hypothalamus in vitro in the absence of glutamate excitation. J Neurosci 21: 2015–2027, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett and Zukin, 2004. Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron 41: 495–511, 2004 [DOI] [PubMed] [Google Scholar]
- Brennan et al., 2009. Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci 12: 857–863, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang et al., 2000. Chang Q, Pereda A, Pinter MJ, Balice-Gordon RJ. Nerve injury induces gap junctional coupling among axotomized adult motor neurons. J Neurosci 20: 674–684, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli et al., 2003. Condorelli DF, Trovato-Salinaro A, Mudo G, Mirone MB, Belluardo N. Cellular expression of connexins in the rat brain: neuronal localization, effects of kainate-induced seizures and expression in apoptotic neuronal cells. Eur J Neurosci 18: 1807–1827, 2003 [DOI] [PubMed] [Google Scholar]
- Cruikshank et al., 2004. Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc Natl Acad Sci USA 101: 12364–12369, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Aniello et al., 2000. D'Aniello A, Di Fiore MM, Fisher GH, Milone A, Seleni A, D'Aniello S, Perna AF, Ingrosso D. Occurrence of D-aspartic acid and N-methyl-D-aspartic acid in rat neuroendocrine tissues and their role in the modulation of luteinizing hormone and growth hormone release. FASEB J 14: 699–714, 2000 [DOI] [PubMed] [Google Scholar]
- de Pina-Benabou et al., 2005. de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff HJ, Stanton PK, Rozental R. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke 36: 2232–2237, 2005 [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari et al., 2007. de Rivero Vaccari JC, Corriveau RA, Belousov AB. Gap junctions are required for NMDA receptor-dependent cell death in developing neurons. J Neurophysiol 98: 2878–2886, 2007 [DOI] [PubMed] [Google Scholar]
- Deans et al., 2001. Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron 31: 477–485, 2001 [DOI] [PubMed] [Google Scholar]
- Decrock et al., 2009. Decrock E, Vinken M, De Vuyst E, Krysko DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V, Leybaert L. Connexin-related signaling in cell death: to live or let die? Cell Death Differ 16: 524–536, 2009 [DOI] [PubMed] [Google Scholar]
- Frantseva et al., 2002. Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci 22: 644–653, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman and Shatz, 1993. Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell 72(Suppl): 77–98, 1993 [DOI] [PubMed] [Google Scholar]
- Hardingham and Bading, 2003. Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci 26: 81–89, 2003 [DOI] [PubMed] [Google Scholar]
- Hormuzdi et al., 2001. Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA, Monyer H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron 31: 487–495, 2001 [DOI] [PubMed] [Google Scholar]
- Iglesias et al., 2008. Iglesias R, Locovei S, Roque A, Alberto AP, Dahl G, Spray DC, Scemes E. P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am J Physiol Cell Physiol 295: C752–C760, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long et al., 2005. Long MA, Jutras MJ, Connors BW, Burwell RD. Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat Neurosci 8: 61–66, 2005 [DOI] [PubMed] [Google Scholar]
- Nemani and Binder, 2005. Nemani VM, Binder DK. Emerging role of gap junctions in epilepsy. Histol Histopathol 20: 253–259, 2005 [DOI] [PubMed] [Google Scholar]
- Oguro et al., 2001. Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, Grooms SY, Bennett MV, Zukin RS. Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci 21: 7534–7542, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Velazquez et al., 2003. Perez Velazquez JL, Frantseva MV, Naus CC. Gap junctions and neuronal injury: protectants or executioners? Neuroscientist 9: 5–9, 2003 [DOI] [PubMed] [Google Scholar]
- Perez-Velazquez et al., 1994. Perez-Velazquez JL, Valiante TA, Carlen PL. Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electrotonic coupling in epileptogenesis. J Neurosci 14: 4308–4317, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheetz and Constantine-Paton, 1994. Scheetz AJ, Constantine-Paton M. Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J 8: 745–752, 1994 [DOI] [PubMed] [Google Scholar]
- Sohl et al., 2005. Sohl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci 6: 191–200, 2005 [DOI] [PubMed] [Google Scholar]
- Thalakoti et al., 2007. Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE, Durham PL. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache 47: 1008–1023, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venance et al., 2000. Venance L, Rozov A, Blatow M, Burnashev N, Feldmeyer D, Monyer H. Connexin expression in electrically coupled postnatal rat brain neurons. Proc Natl Acad Sci USA 97: 10260–10265, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss et al., 2009. Voss LJ, Jacobson G, Sleigh JW, Steyn-Ross A, Steyn-Ross M. Excitatory effects of gap junction blockers on cerebral cortex seizure-like activity in rats and mice. Epilepsia 50: 1971–1978, 2009 [DOI] [PubMed] [Google Scholar]
- Weiss et al., 2003. Weiss SM, Benwell K, Cliffe IA, Gillespie RJ, Knight AR, Lerpiniere J, Misra A, Pratt RM, Revell D, Upton R, Dourish CT. Discovery of nonxanthine adenosine A2A receptor antagonists for the treatment of Parkinson's disease. Neurology 61: S101–S106, 2003 [DOI] [PubMed] [Google Scholar]
- Xu et al., 2004. Xu L, Heinze T, Pogge A, Slikker W, Schmued L. Isolation and characterization of Fluoro-Jade B, a selective histochemical stain for neuronal degeneration. J Liq Chromatogr 27: 1627–1640, 2004 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.