

Abstract
The title compound, C11H10N4O4S, is a derivative of N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea (AR-A014418), a known glycogen synthase kinase 3β (GSK-3β) inhibitor. All non-H atoms in the molecule are essentially coplanar, with an r.m.s. deviation of 0.045 Å and a maximum deviation of 0.115 (2) Å for the carbonyl O atom. In the crystal structure, molecules are linked via N—H⋯O hydrogen bonds into one-dimensional chains along [101].
Related literature
For background information on the preparation and activity of AR-A014418, see: Bhat et al. (2003 ▶); Inestrosa et al. (2006 ▶). For the radiolabelling procedure of AR-A014418 with carbon-11, see: Vasdev et al. (2005 ▶). For the crystal structure of AR-A014418, see: Vasdev et al. (2007 ▶).
Experimental
Crystal data
C11H10N4O4S
M r = 294.29
Monoclinic,
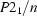
a = 6.8740 (3) Å
b = 12.5840 (7) Å
c = 14.6861 (5) Å
β = 101.622 (3)°
V = 1244.34 (10) Å3
Z = 4
Mo Kα radiation
μ = 0.28 mm−1
T = 150 K
0.28 × 0.24 × 0.22 mm
Data collection
Nonius KappaCCD diffractometer
Absorption correction: multi-scan (SORTAV; Blessing, 1995 ▶) T min = 0.718, T max = 0.948
8106 measured reflections
2825 independent reflections
1979 reflections with I > 2σ(I)
R int = 0.053
Refinement
R[F 2 > 2σ(F 2)] = 0.050
wR(F 2) = 0.145
S = 1.07
2825 reflections
190 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.38 e Å−3
Δρmin = −0.51 e Å−3
Data collection: COLLECT (Nonius, 2002 ▶); cell refinement: DENZO-SMN (Otwinowski & Minor, 1997 ▶); data reduction: DENZO-SMN; program(s) used to solve structure: SIR92 (Altomare et al., 1994 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2009 ▶); software used to prepare material for publication: SHELXTL (Sheldrick, 2008 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810032186/su2205sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810032186/su2205Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H2N⋯O4i | 0.86 (3) | 1.97 (3) | 2.817 (3) | 168 (3) |
| N3—H3N⋯O3i | 0.87 (3) | 2.30 (3) | 3.168 (2) | 174 (2) |
Symmetry code: (i) 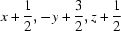 .
.
Acknowledgments
We thank Dr Sylvain Houle for allowing the CAMH PET Centre facilities to be used for this research. Funding was provided by the Ontario Ministry for Research and Innovation (Early Researcher Award to NV).
supplementary crystallographic information
Comment
N-(4-methoxybenzyl)-N'--(5-nitro-1,3-thiazol-2-yl)urea (AR-A014418, Bhat et al., 2003) is a selective glycogen synthase kinase-3β (GSK-3β) inhibitor (Inestrosa et al., 2006). Our initial work on AR-A014418 was to radiolabel this compound with carbon-11 at the methoxy position for positron emission tomography (PET) imaging of brain pathologies (Vasdev et al., 2005). To our surprise, [11C]-AR-A014418 had insignificant brain uptake in rodents despite literature precedence (Bhat et al., 2003). To further understand the role of AR-A014418 and GSK-3β, a single-crystal X-ray structure of AR-A014418 was obtained (Vasdev et al., 2007) and overlaid with the structural determination of the co-crystal of GSK-3β and AR-A014418 (Bhat et al., 2003). For that structure, the benyzl ring was bent out of the binding pocket of the kinase. We now endeavour to explore the binding affinity of an analogous molecule which has reduced flexibility (i.e. the benzyl group is replaced with a phenyl group). Biological assays are underway to determine whether the increased rigidity decreases the binding affinity for GSK-3β. Data from these biological studies as well as the crystal structure determinations will assist in designing future ligands for imaging GSK-3β with PET.
The molecular structure of the title compound is shown in Fig. 1. All non-hydrogen atoms in the molecule are essentially co-planar with a r.m.s. deviation of 0.045 Å and a maximum deviation of 0.115 (2)Å for atom O1.
In the crystal symmetry related molecules are linked via N—H···O hydrogen bonds, to form one-dimensional chains propagating along [101] (Table 1, Fig. 2).
Experimental
The title compound, C11H10N4O4S, was obtained by heating equimolar amounts of 2-amino-5-nitrothiazole (0.65 mmol) and 4-methoxyphenyl isocyanate (0.65 mmol) in dry N,N-dimethyl formamide (5 ml) in a Biotage Initiator Microwave for 1 h at 403 K under nitrogen. Upon cooling, the reaction mixture was partitioned between ethyl acetate and water and the aqueous layer was further extracted with more ethyl acetate (15 ml). The combined organic layers were washed with brine (3 × 30 ml), dried (MgSO4), filtered, and concentrated prior to purification by silica chromatography (Biotage Isolera Flash system, 98% dichloromethane and 2% methanol). C11H10N4O4S was obtained as a red solid in 39% yield (not optimized). X-ray quality crystals were obtained by slow evaporation of a solution of the title compound in ethyl acetate/hexane (50/50) containing 5% ethanol. 1H NMR (DMSO d6, 400 MHz) δ 11.75 (s, 1 H, NH), 9.28 (s, 1H, NH), 8.53 (s, 1H, thiazole), 7.41 (d, J = 8.9 Hz, 2H, Ar), 6.91 (d, J = 8.9 Hz, 2H, Ar), 3.73 (s, 3H, CH3). HRMS (ESI) m/z calcd for C11H11N4O4S, 295.0495; found 295.0483 (M++H), m.p. = 454–456 K.
Refinement
H atoms bonded to C atoms were placed in calculated positions with C—H = 0.95Å or 0.98Å for methyl groups and included in the refinement with Uiso(H) = 1.2Ueq(C) or 1.2Ueq(Cmethyl). H atoms bonded to N atoms were refined independently with isotropic displacement parameters.
Figures
Fig. 1.

The molecular structure of the title compound with probability ellipsoids drawn at the 30% level.
Fig. 2.

Part of the crystal structure of the title compound with hydrogen bonds drawn as dashed lines.
Crystal data
| C11H10N4O4S | F(000) = 608 |
| Mr = 294.29 | Dx = 1.571 Mg m−3 |
| Monoclinic, P21/n | Melting point: 454 K |
| Hall symbol: -P 2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.8740 (3) Å | Cell parameters from 3592 reflections |
| b = 12.5840 (7) Å | θ = 2.6–27.5° |
| c = 14.6861 (5) Å | µ = 0.28 mm−1 |
| β = 101.622 (3)° | T = 150 K |
| V = 1244.34 (10) Å3 | Block, red |
| Z = 4 | 0.28 × 0.24 × 0.22 mm |
Data collection
| Nonius KappaCCD diffractometer | 2825 independent reflections |
| Radiation source: fine-focus sealed tube | 1979 reflections with I > 2σ(I) |
| graphite | Rint = 0.053 |
| Detector resolution: 9 pixels mm-1 | θmax = 27.5°, θmin = 2.8° |
| φ scans and ω scans with κ offsets | h = −8→8 |
| Absorption correction: multi-scan (SORTAV; Blessing, 1995) | k = −14→16 |
| Tmin = 0.718, Tmax = 0.948 | l = −17→19 |
| 8106 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.050 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.145 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0755P)2 + 0.3053P] where P = (Fo2 + 2Fc2)/3 |
| 2825 reflections | (Δ/σ)max < 0.001 |
| 190 parameters | Δρmax = 0.38 e Å−3 |
| 0 restraints | Δρmin = −0.51 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expr ession of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.12900 (8) | 0.68571 (5) | 0.41433 (3) | 0.0246 (2) | |
| O1 | 0.2254 (2) | 0.48461 (14) | 0.47569 (10) | 0.0306 (4) | |
| O2 | 0.5006 (3) | 0.00530 (13) | 0.62455 (11) | 0.0340 (4) | |
| O3 | −0.0126 (3) | 0.97318 (14) | 0.33409 (11) | 0.0347 (4) | |
| O4 | 0.0063 (3) | 0.82488 (15) | 0.25900 (11) | 0.0367 (5) | |
| N1 | 0.2026 (3) | 0.79137 (16) | 0.57084 (12) | 0.0277 (5) | |
| N2 | 0.2668 (3) | 0.61085 (16) | 0.58904 (13) | 0.0268 (5) | |
| H2N | 0.327 (4) | 0.627 (2) | 0.6449 (19) | 0.042 (8)* | |
| N3 | 0.3514 (3) | 0.44014 (16) | 0.62860 (13) | 0.0253 (5) | |
| H3N | 0.380 (4) | 0.466 (2) | 0.6845 (19) | 0.037 (8)* | |
| N4 | 0.0257 (3) | 0.87648 (16) | 0.33258 (13) | 0.0270 (5) | |
| C1 | 0.0930 (3) | 0.82155 (19) | 0.41566 (14) | 0.0232 (5) | |
| C2 | 0.1399 (3) | 0.8632 (2) | 0.50308 (15) | 0.0267 (5) | |
| H2A | 0.1292 | 0.9369 | 0.5153 | 0.032* | |
| C3 | 0.2033 (3) | 0.69504 (18) | 0.53334 (15) | 0.0224 (5) | |
| C4 | 0.2790 (3) | 0.50759 (19) | 0.55815 (15) | 0.0243 (5) | |
| C5 | 0.3832 (3) | 0.32879 (18) | 0.62462 (15) | 0.0223 (5) | |
| C6 | 0.3583 (3) | 0.2717 (2) | 0.54193 (15) | 0.0245 (5) | |
| H6A | 0.3153 | 0.3064 | 0.4839 | 0.029* | |
| C7 | 0.3970 (3) | 0.1640 (2) | 0.54544 (15) | 0.0258 (5) | |
| H7A | 0.3791 | 0.1245 | 0.4892 | 0.031* | |
| C8 | 0.4620 (3) | 0.11186 (19) | 0.63008 (15) | 0.0253 (5) | |
| C9 | 0.4842 (4) | 0.1688 (2) | 0.71214 (16) | 0.0281 (6) | |
| H9A | 0.5265 | 0.1342 | 0.7702 | 0.034* | |
| C10 | 0.4439 (4) | 0.2773 (2) | 0.70889 (16) | 0.0277 (5) | |
| H10A | 0.4583 | 0.3164 | 0.7651 | 0.033* | |
| C11 | 0.5519 (4) | −0.0525 (2) | 0.70992 (17) | 0.0360 (6) | |
| H11A | 0.5715 | −0.1276 | 0.6965 | 0.054* | |
| H11B | 0.4446 | −0.0460 | 0.7445 | 0.054* | |
| H11C | 0.6749 | −0.0236 | 0.7472 | 0.054* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0269 (3) | 0.0260 (4) | 0.0198 (3) | 0.0001 (2) | 0.0024 (2) | −0.0017 (2) |
| O1 | 0.0405 (10) | 0.0273 (10) | 0.0225 (9) | 0.0016 (8) | 0.0028 (7) | −0.0024 (7) |
| O2 | 0.0482 (11) | 0.0242 (9) | 0.0304 (9) | 0.0047 (8) | 0.0097 (8) | 0.0018 (7) |
| O3 | 0.0418 (11) | 0.0279 (10) | 0.0335 (10) | 0.0046 (8) | 0.0057 (8) | 0.0080 (8) |
| O4 | 0.0450 (11) | 0.0437 (12) | 0.0193 (8) | 0.0073 (9) | 0.0015 (7) | −0.0019 (8) |
| N1 | 0.0368 (12) | 0.0251 (11) | 0.0214 (10) | 0.0020 (9) | 0.0059 (8) | −0.0009 (8) |
| N2 | 0.0352 (12) | 0.0240 (11) | 0.0205 (10) | 0.0011 (9) | 0.0040 (8) | −0.0033 (9) |
| N3 | 0.0315 (11) | 0.0238 (11) | 0.0204 (10) | 0.0003 (9) | 0.0048 (8) | −0.0014 (9) |
| N4 | 0.0239 (11) | 0.0318 (12) | 0.0250 (10) | 0.0008 (9) | 0.0041 (8) | 0.0044 (9) |
| C1 | 0.0226 (12) | 0.0249 (13) | 0.0223 (11) | 0.0005 (10) | 0.0049 (9) | 0.0036 (10) |
| C2 | 0.0310 (13) | 0.0242 (13) | 0.0246 (12) | 0.0003 (10) | 0.0046 (9) | −0.0005 (10) |
| C3 | 0.0241 (12) | 0.0231 (13) | 0.0206 (11) | −0.0022 (9) | 0.0063 (8) | −0.0015 (9) |
| C4 | 0.0243 (12) | 0.0251 (13) | 0.0247 (12) | −0.0007 (10) | 0.0079 (9) | −0.0009 (10) |
| C5 | 0.0206 (11) | 0.0247 (13) | 0.0226 (11) | −0.0003 (9) | 0.0070 (8) | 0.0002 (10) |
| C6 | 0.0233 (12) | 0.0285 (14) | 0.0219 (11) | 0.0006 (10) | 0.0051 (9) | 0.0007 (10) |
| C7 | 0.0253 (12) | 0.0293 (14) | 0.0233 (11) | −0.0025 (10) | 0.0063 (9) | −0.0030 (10) |
| C8 | 0.0257 (12) | 0.0214 (12) | 0.0309 (12) | 0.0006 (10) | 0.0105 (9) | 0.0002 (10) |
| C9 | 0.0310 (13) | 0.0314 (14) | 0.0231 (11) | 0.0006 (11) | 0.0087 (9) | 0.0044 (10) |
| C10 | 0.0338 (14) | 0.0273 (13) | 0.0235 (12) | 0.0006 (11) | 0.0097 (10) | −0.0020 (10) |
| C11 | 0.0477 (16) | 0.0261 (14) | 0.0345 (14) | 0.0045 (12) | 0.0089 (11) | 0.0074 (11) |
Geometric parameters (Å, °)
| S1—C3 | 1.723 (2) | C1—C2 | 1.364 (3) |
| S1—C1 | 1.728 (2) | C2—H2A | 0.9500 |
| O1—C4 | 1.227 (3) | C5—C10 | 1.385 (3) |
| O2—C8 | 1.373 (3) | C5—C6 | 1.391 (3) |
| O2—C11 | 1.431 (3) | C6—C7 | 1.381 (3) |
| O3—N4 | 1.246 (3) | C6—H6A | 0.9500 |
| O4—N4 | 1.245 (2) | C7—C8 | 1.397 (3) |
| N1—C3 | 1.332 (3) | C7—H7A | 0.9500 |
| N1—C2 | 1.349 (3) | C8—C9 | 1.384 (3) |
| N2—C3 | 1.356 (3) | C9—C10 | 1.391 (3) |
| N2—C4 | 1.384 (3) | C9—H9A | 0.9500 |
| N2—H2N | 0.86 (3) | C10—H10A | 0.9500 |
| N3—C4 | 1.353 (3) | C11—H11A | 0.9800 |
| N3—C5 | 1.421 (3) | C11—H11B | 0.9800 |
| N3—H3N | 0.87 (3) | C11—H11C | 0.9800 |
| N4—C1 | 1.398 (3) | ||
| C3—S1—C1 | 86.27 (11) | C10—C5—C6 | 120.0 (2) |
| C8—O2—C11 | 117.49 (19) | C10—C5—N3 | 116.5 (2) |
| C3—N1—C2 | 109.40 (18) | C6—C5—N3 | 123.5 (2) |
| C3—N2—C4 | 124.68 (19) | C7—C6—C5 | 119.0 (2) |
| C3—N2—H2N | 115.2 (19) | C7—C6—H6A | 120.5 |
| C4—N2—H2N | 118.6 (19) | C5—C6—H6A | 120.5 |
| C4—N3—C5 | 128.5 (2) | C6—C7—C8 | 121.3 (2) |
| C4—N3—H3N | 117.8 (18) | C6—C7—H7A | 119.4 |
| C5—N3—H3N | 113.6 (18) | C8—C7—H7A | 119.4 |
| O4—N4—O3 | 122.61 (19) | O2—C8—C9 | 124.7 (2) |
| O4—N4—C1 | 117.3 (2) | O2—C8—C7 | 115.9 (2) |
| O3—N4—C1 | 120.09 (19) | C9—C8—C7 | 119.4 (2) |
| C2—C1—N4 | 127.3 (2) | C8—C9—C10 | 119.5 (2) |
| C2—C1—S1 | 112.58 (17) | C8—C9—H9A | 120.3 |
| N4—C1—S1 | 120.16 (17) | C10—C9—H9A | 120.3 |
| N1—C2—C1 | 114.6 (2) | C5—C10—C9 | 120.8 (2) |
| N1—C2—H2A | 122.7 | C5—C10—H10A | 119.6 |
| C1—C2—H2A | 122.7 | C9—C10—H10A | 119.6 |
| N1—C3—N2 | 119.29 (19) | O2—C11—H11A | 109.5 |
| N1—C3—S1 | 117.15 (17) | O2—C11—H11B | 109.5 |
| N2—C3—S1 | 123.53 (17) | H11A—C11—H11B | 109.5 |
| O1—C4—N3 | 126.7 (2) | O2—C11—H11C | 109.5 |
| O1—C4—N2 | 121.2 (2) | H11A—C11—H11C | 109.5 |
| N3—C4—N2 | 112.05 (19) | H11B—C11—H11C | 109.5 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2N···O4i | 0.86 (3) | 1.97 (3) | 2.817 (3) | 168 (3) |
| N3—H3N···O3i | 0.87 (3) | 2.30 (3) | 3.168 (2) | 174 (2) |
Symmetry codes: (i) x+1/2, −y+3/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: SU2205).
References
- Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994). J. Appl. Cryst.27, 435.
- Bhat, R., Xue, Y. F., Berg, S., Hellberg, S., Ormo, M., Nilsson, Y., Radesater, A. C., Jerning, E., Markgren, P. O., Borgegard, T., Nylof, M., Gimenez-Cassina, A., Hernandez, F., Lucas, J. J., Diaz-Nido, J. & Avila, J. (2003). J. Biol. Chem.278, 45937–45945. [DOI] [PubMed]
- Blessing, R. H. (1995). Acta Cryst. A51, 33–38. [DOI] [PubMed]
- Inestrosa, N. C., Farías, G. & Colombres, M. (2006). Glycogen Synthase Kinase 3 (GSK-3) and Its Inhibitors: Drug Discovery and Development, edited by A. Martinez, A. Castro & M. Medina, pp. 25–43. New Jersey: John Wiley & Sons Inc.
- Nonius (2002). COLLECT Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Vasdev, N., Garcia, A., Stableford, W. T., Young, A. B., Meyer, J. H., Houle, S. & Wilson, A. A. (2005). Bioorg. Med. Chem. Lett.15, 5270–5273. [DOI] [PubMed]
- Vasdev, N., Wilson, A. A., Houle, S. & Lough, A. J. (2007). Acta Cryst. E63, o1653–o1655.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810032186/su2205sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810032186/su2205Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report