

Abstract
A major tripartite multidrug efflux pump of Escherichia coli, AcrAB-TolC, confers resistance to a wide variety of compounds. The drug molecule is captured by AcrB probably from the periplasm or the periplasm/inner membrane interface, and is passed through AcrB and then TolC to the medium. Currently there exist numerous crystallographic and mutation data concerning the regions of AcrB and its homologs that may interact with substrates. Starting with these data, we devised fluorescence assays in whole cells to determine the entire substrate path through AcrB. We tested 48 residues in AcrB along the predicted substrate path and 25 gave positive results, based on the covalent labeling of cysteine residues by a lipophilic dye-maleimide and the blocking of Nile Red efflux by covalent labeling with bulky maleimide reagents. These residues are all located in the periplasmic domain, in regions we designate as the lower part of the large external cleft, the cleft itself, the crystallographically defined binding pocket, and the gate between the pocket and the funnel. Our observations suggest that the substrate is captured in the lower cleft region of AcrB, then transported through the binding pocket, the gate, and finally to the AcrB funnel that connects AcrB to TolC.
Keywords: RND transporter, cysteine modification, maleimides, path blockage, Nile Red
Introduction
The major multidrug efflux pump in Escherichia coli consists of three proteins: AcrB, AcrA, and TolC. AcrB is the inner membrane pump (Dinh et al., 1994) that is energized by the proton-motive force (Ma et al., 1993; Ma et al., 1995). AcrA is an inner membrane lipoprotein with its protein domain in the periplasm (Zgurskaya and Nikaido, 1999). TolC (Fralick, 1996) is an outer membrane channel that is continued as a long periplasmic tunnel (Koronakis et al., 2000). AcrB captures the drug molecules and pumps them out through TolC into the medium. AcrA is believed to stabilize the AcrB-TolC interactions. All three proteins are required for AcrAB-TolC-mediated efflux (Nikaido, 1996; Nikaido, 1998).
AcrB is 1050 amino acid long and is almost equally divided into a transmembrane-segment (TMS) domain and a large periplasmic domain. It forms a homotrimer. Each AcrB protomer has 12 TMS helices that contain the proton-relay-network residues (Su et al., 2006; Takatsuka and Nikaido, 2006). The proton translocation from periplasm to cytoplasm is coupled to a large conformational change in the periplasmic domain (Murakami et al., 2006; Seeger et al., 2006; Sennhauser et al., 2007). The substrate specificity is determined by the periplasmic domain (for example see Elkins and Nikaido, 2002), and it is likely that the substrate becomes captured, bound, and then transported through this domain.
Recent asymmetric structures of AcrB trimer (Murakami et al., 2006; Seeger et al., 2006; Sennhauser et al., 2007) show each protomer in a different conformation: Access, Binding, and Extrusion. Importantly, Murakami and associates found substrates, minocycline and doxorubicin, bound to the binding pocket located deep in the periplasmic domain of only the Binding conformer (Murakami et al., 2006)(Fig. 1). The hydrophobic binding pocket is surrounded by many Phe residues, such as Phe178, Phe610, Phe615, and Phe617, and the affinity of ligands to this pocket was recently confirmed by a model docking study (Takatsuka et al., 2010). Furthermore, site-directed mutations of some residues composing this pocket make the bacteria more susceptible to antibiotics (Bohnert et al., 2005; Bohnert et al., 2008; Wehmeier et al., 2009). However, pathways for the entry (or capture) of substrates to the binding pocket and their exit from it into the funnel on top of AcrB (connected to the TolC channel) have not been established experimentally. Once in the funnel, the only path for the substrate to the medium is through the central channel of TolC.
Figure 1. AcrB residues examined in this study.

Structure of the binding protomer of AcrB (from PDB file 2DRD) is shown in a grey diagram. The top half of the molecule is the periplasmic domain (with the TolC-binding subdomain [Tamura et al., 2005] on top), and the bottom half represents the transmembrane domain embedded in the inner membrane. The view is from outside of the trimer. The bound minocycline is shown as a red stick model. The light tan-colored surface diagram shows the connection of this binding pocket to the outside, through three wide channels visualized by the program CAVER (Petrek et al., 2006). One channel exits closest to the reader in the large external cleft; the second one exits to the left into the central cavity, and the third one exits to the right into a vestibule. (These features are similar to those described already [Murakami et al., 2006; Sennhauser et al., 2007]). Residues chosen for conversion to Cys are shown in color-coded spheres. They are from the gate (deep blue), binding pocket (light blue), cleft (orange), bottom of the cleft (mauve), and central cavity (black). Green spheres indicate the residues located outside the probable drug pathway and used as controls. For details, see text.
Crystallographic studies so far suggested three regions of AcrB as the entry points of substrate into AcrB: the central cavity, the vestibule, and the cleft. The central cavity is a large cavity at the bottom of periplasmic domain and at the top of TMS domain, formed by the residues of all three AcrB protomers and presumably the phopholipids of the outer leaflet of inner membrane. The earlier AcrB crystal structure, solved by Murakami et al. (2002), was symmetrical and Yu et al. (2003) found that substrates were bound to the central cavity of similar structures. The central cavity of AcrB is connected to the periplasm via the vestibules, which are located at the bottom of periplasmic domain, and are formed by the residues of two adjacent AcrB protomers (Murakami et al., 2002). Finally the cleft is a large external indentation of the periplasmic domain, formed by two subdomains, PC1 and PC2, of a single AcrB protomer. Drug-AcrB cocrystals with drugs binding to the cleft and the MIC of site-directed mutants suggested that the cleft is directly involved in substrate transport (Yu et al., 2005). The cleft was found with a bound drug also in another crystal structure (Drew et al., 2008). In addition, genetic studies on AcrB and its homologs suggested similar and additional sites that may be important for substrate binding or transport (Mao et al., 2002; Middlemiss and Poole 2004; Murakami et al., 2004; Hearn et al., 2006).
The substrate must eventually leave the binding site in order for its efflux to occur. The asymmetric crystal structure suggests that the gate connects the binding pocket to the funnel in AcrB, and the funnel is connected to TolC. However, there is no experimental evidence for the role of the gate in the efflux process.
We have attempted to elucidate the pathways of the substrates within the AcrB protein by utilizing mutants in which single or double cysteine residue(s) were introduced by site-directed mutagenesis and by covalently modifying these residues with cysteine-specific reagents. Our study suggests that the substrate enters AcrB from the lower part of the cleft before binding to the binding pocket. Our study further confirms the importance of binding pocket and also implicates the role of residues that have not been suspected to participate in substrate capture. Finally, we provide evidence for the importance of gate in substrate transport from the binding pocket to the funnel.
Results
Selection of Residues to be Studied
We wanted to convert into cysteine those AcrB residues that potentially form the pathway of drug uptake, binding, and extrusion, and then study the behavior of these mutants by the covalent modification of these Cys residues with dye-maleimides. We started from the cysteineless AcrBHis (CL-AcrBHis)(Takatsuka and Nikaido, 2007). We selected 48 residues, which can be divided into several groups. Their location in the Binding conformer is shown in Fig. 1. (i) Central cavity (Leu25, Thr98, Phe386 and Phe459) (Yu et al., 2003). (ii) Entrance to the external cleft (Phe664, Phe666, Leu668, Arg717, Leu828). This is where many drugs were found to bind in the symmetric trimer structures (Yu et al., 2005; Drew et al., 2008). (iii) Bottom of the cleft, including residues close to the membrane surface and the vestibules, some of which (Asp566, Val571, Glu673, Thr676, and Gln865) were identified previously as a part of the peripheral binding site (Yu et al., 2005). Gln34, identified as important in a MexD study (Mao et al., 2002) also belongs to this group. (iv) Minocycline/Doxorubicin binding pocket. These residues surround the drugs identified in the asymmetric crystal structure of AcrB (Murakami et al., 2006), and include also the hydrophobic entrance into this site (Ser134, Phe136, Val139, Gln 176, Phe 178, Ser180, Glu273, Asn274, Asp276, Ile277, Phe281, Gly290, Tyr327, Phe617, Arg620, Ile626, Phe628, Ser630). Gln89, identified in the study of MexD (Mao et al., 2002), and its neighbors Thr85 and Thr87, were also included, as well as Met573 that lies in between the binding pocket and the entrance group listed above. Although somewhat farther away from the ligands, Glu607, Phe610, and Val 612 were also tested. (v) Exit from the binding site. This includes Gln124, Gln125, Trp754, Ser757, Tyr758, Lys770, Tyr772, and Arg780. (vi) As negative controls, we selected residues (Gln197 and Lys708) that are not predicted to be in the substrate path but are on the external surface of the periplasmic domain. We also included four residues (Glu346, Leu353, Leu546 and Tyr554) on the hydrophobic face of the transmembrane domain that are unlikely to interact with ligands. The WT-AcrBHis was also tested as a negative control, since the WT-AcrBHis has two native cysteines (Cys493 and Cys887) present in the transmembrane region away from the predicted substrate path.
The functional competence of the mutant proteins was tested by the MIC values of drugs known to be the substrates of the AcrAB-TolC complex. Most mutants exhibited near-normal activity by this criterion. However, the mutants T87C, F178C, E273C, and I626C showed significantly lower novobiocin MIC (data not shown). Also, T87C, F178C, and F628C showed reduced efflux of Nile Red (not shown).
Bodipy labeling
Our first approach for the detection of residues on the substrate path was to use a lipophilic maleimide reagent that may act as a substrate of AcrB. Such a reagent was expected to become accumulated within the path and to have a higher probability of reacting with the Cys residues encountered. If the reagent is used at low concentrations, the labeling may become specific, in the sense that only the residues on the substrate path would be labeled. We tried labeling with Alexa 488 maleimide, Alexa Fluor 594 C5-maleimide, Bodipy-FL-maleimide, and Eosin-5-maleimide at several concentrations. We found that Bodipy-FL-maleimide was most efficient in modifying the AcrB mutants at low concentrations (results not shown).
We therefore used a low concentration (6μM) of Bodipy-FL-maleimide routinely on intact cells. (Although the cells expressed mutant AcrB proteins from a medium-copy-number vector pSPORT1, the cells were grown without inducer and AcrB was overexpressed only to a modest extent [15-fold above normal chromosomal level]. Since 10 to 12 AcrA proteins are present for each AcrB protein in wild-type E. coli [Tikhonova and Zgurskaya, 2004], we expect that there is no severe shortage of AcrA under our conditions based on the recently proposed 1:1 stoichiometry between AcrA and AcrB in the tripartite complex [Symmons et al., 2009]). At this concentration, the “control” Cys residues well out of the presumed substrate pathway (Q197C, K708C, E346C, L353C, L546C, Y554C and C493/C887) were not labeled (Fig. 2). In contrast, residues presumably on the substrate path, such as N274C became strongly labeled (Fig. 2). Since much of the substrate path is expected to have a relatively hydrophobic surface, it is important that even residues on the hydrophobic surface of the TMS domain (E346C, L353C, L546C, and Y553C) were not labeled; this shows that the generally hydrophobic environment of the tested residue is not sufficient to produce the positive labeling. In order to further confirm our hypothesis that the labeling requires the passage of Bodipy-FL-maleimide through the normal substrate path, we tested the labeling of several strongly labeled residues in a non-functional mutant of AcrB. For this purpose, we inactivated AcrB by introducing additional D407A mutation, which renders AcrB inactive apparently by abolishing the proton translocation (Su et al., 2006; Takatsuka and Nikaido, 2006). In such mutants, the “functionally rotating” conformational changes of the protomers (Murakami et al., 2006) cannot occur (Takatsuka and Nikaido, 2009), and only one protomer out of three would have the binding conformation that would allow a full access to substrates; in contrast, in the functional AcrB trimer, all protomers will become accessible to substrates over time. Indeed, AcrBN274C/D407A, AcrBD276C/D407A, and AcrBF666C/D407A were stained less strongly by Bodipy-FL-maleimide, compared to the corresponding AcrB mutants without the D407A alteration (61, 42, and 47% of the control, respectively), a result supporting the notion that substrate transport is required to stain these residues fully. (On the other hand there was no difference in the staining of AcrBT676C and AcrBT676C/D407A; this finding will be interpreted in Discussion).
Figure 2. Examples of the staining of CL-AcrBHis mutants by Bodipy-FL-maleimide.

The overnight-grown whole cells at OD660 3.5 were stained with 6 μM Bodipy-FL-maleimide for 1 h. The CL-AcrBHis was isolated from 5 ml of cells and was eluted with 100 μl of elution buffer. A portion (7.5 μl) of the eluate was resolved on SDS-PAGE. The resolved AcrB bands were observed for staining by Bodipy-FL-maleimide using Typhoon phosphorimager, and the same gel was then stained using the Coomassie stain. The WT-AcrBHis contains two native cysteines, Cys493 and Cys887. The other alterations are made in CL-AcrBHis, which does not have native cysteines.
Having convinced ourselves that 6μM Bodipy-FL-maleimide likely modifies only the residues in the substrate path, we examined the labeling in whole cells of the entire 48 mutant set. We found that AcrB mutants with following alterations were modified: Q89C, S134C, F136C, Q176C, F178C, S180C, E273C, N274C, D276C, I277C, G290C, Y327C, D566C, F617C, R620C, F664C, F666C, L668C, E673C, T676C, R717C, and L828C (Fig. 3). We also tested double mutants Q124C/Y758C, Q125C/K770C, Q125C/Y758C and K770C/Y758C, but the labeling was not increased significantly in comparison with the levels obtained with single mutants (Fig. 3). Among the double mutants only AcrBQ124C/Y758C had a significantly low novobiocin MIC and showed a reduced efflux of Nile Red (see below).
Figure 3. Covalent labeling of AcrB mutants in whole cells with 6 μM Bodipy-FL-maleimide.
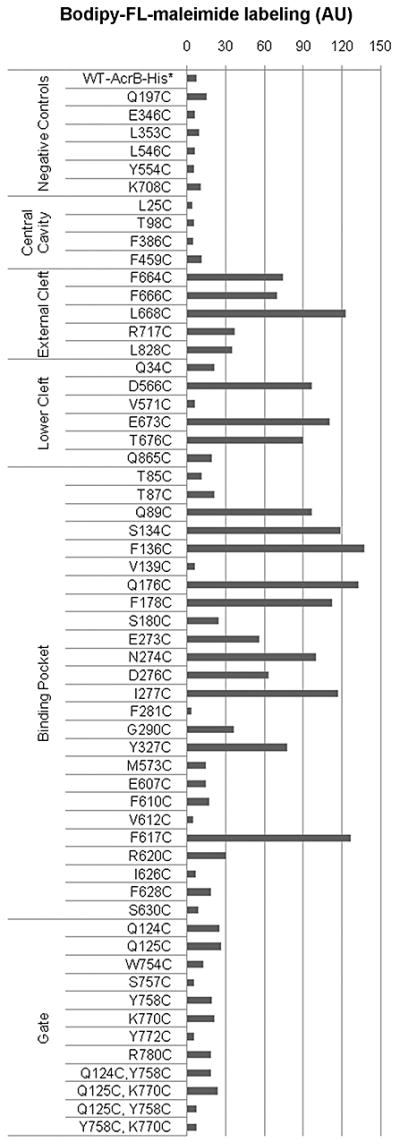
CL-AcrBHis (with no native cysteines) was used to alter the selected residues to cysteines. The extent of labeling was calculated by correcting for the variations in the protein expression level by dividing the fluorescence with the amount of protein, revealed by the Coomassie staining (see Experimental Procedures). The extent of labeling was normalized by using that of N274C as 100. We used 30 as the arbitrary cut-off point to distinguish labeled and unlabeled mutant proteins. WT-AcrBHis has two native cysteines Cys493 and Cys887 outside the presumed substrate path, and thus served as one negative control. AU: arbitrary unit.
AcrB-mediated efflux of Nile Red
Although the Bodipy assay suggested strongly that the identified residues line the substrate path, we wanted to obtain a more conclusive piece of evidence for this interpretation. We reasoned that the covalent modification of these Cys residues with a bulky maleimide reagent might block the normal efflux function of AcrB. For this assay, we needed a direct dye efflux assay that can be monitored right at the time of the modification. This requires a dye that is an AcrB substrate and with a higher fluorescence yield in the cell or cell membrane compared to when in the aqueous medium. Among the dyes easily available, we found that Nile Red and N-phenyl-1-naphthylamine (NPN) showed a clear AcrB-mediated efflux. Nile Red was better suited for our studies because of its very reproducible efflux behavior, as was already shown in our laboratory (Bohnert et al., 2010).
A real-time assay was developed to identify the immediate effect of cysteine-reactive agents on AcrB containing a single cysteine side-chain. The deenergized cells preloaded with Nile Red are fluorescent because Nile Red fluorescence yield is very high when it is partitioned within the membrane interior compared to when in the aqueous environment (Greenspan and Fowler, 1985). This property of Nile Red makes it possible to follow its efflux by observing decreases in the fluorescence intensity (Fig. 4A). In the absence of glucose there was no efflux of Nile Red observed within 140 sec, regardless of the presence or absence of AcrB. When 0.4% glucose was added to cells lacking AcrB, there was a slow quasi-linear efflux of Nile Red, presumably catalyzed by other efflux pumps that are energized by glucose metabolism. In the presence of both 0.4% glucose and AcrB there was a rapid Nile Red efflux that eventually died out as the process approaches its final point (Fig. 4A). We also tested several densities of cells for the reproducibility of efflux assay, and found that cell density at final OD660 0.85 or higher was preferable.
Figure 4. AcrB-mediated Nile Red efflux.
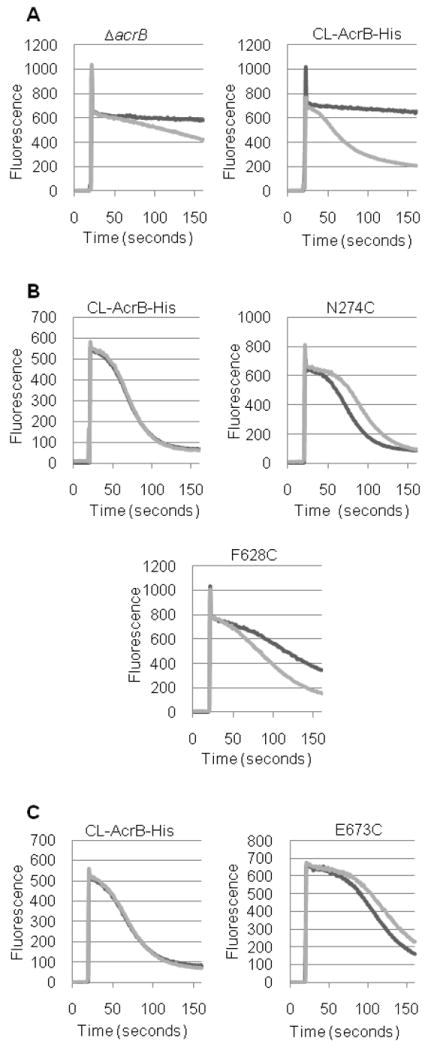
Nile red efflux was followed as described in Experimental Procedures. Intensity of fluorescence emission is shown in arbitrary units. A. Efflux in strains expressing no AcrB (ΔacrB) or expressing CL-AcrBHis, with (dark line) or without (light line) 0.4 % glucose. B. Effect of pyrene-maleimide on Nile Red efflux. Efflux time course in the presence of 0.4% glucose is shown, as examples, in strains expressing CL-AcrBHis and its mutants (N274C and F628C). Experiments were without pyrene-maleimide (dark line) or with 30 μM pyrene-maleimide (light line). C. Effect of CPM on Nile Red efflux. Time course of Nile Red efflux is shown in CL-AcrBHis and the E673C mutant, as typical examples. All samples contained 0.4% glucose. Experiments were without CPM (dark line) or with 12 μM CPM (light line).
Blocking of Nile Red efflux
We examined if covalent modification of cysteine residues of AcrB with cysteine-reactive compounds blocked the AcrB-mediated Nile Red efflux. We tested a few AcrB mutants with methyl methanethiosulfonate, pentyl methanthiosulfonate, 7-diethylamino-3-(4’-maleimidylphenyl)-4-methylcoumarin (CPM), Alexa Fluor 488 C5 maleimide, 2-(4’-(iodoacetamido)anilino)naphthalene-6-sulfonic acid, 2-(4’-maleimidylanilino)naphthalene-6-sulfonic acid (also called MIANS), and N-(1-pyrene) maleimide (pyrene-maleimide). CPM and pyrene-maleimide blocked the Nile Red efflux in some of the AcrB mutants tested. Thus we tested these reagents using our entire array of the single cysteine mutants.
It should be emphasized that we are not testing competition between substrates, because pyrene-maleimide and CPM did not alter the rate of Nile Red efflux in cells expressing CL-AcrBHis (Figs. 4B and 4C). Because both reagents are lipophilic and aromatic, it seems likely that they behave initially as substrates, and go through the substrate path of AcrB in order to reach the target. Nevertheless, this process apparently does not produce visible competition with Nile Red as seen with the experiments with CL-AcrBHis, and we conclude that the inhibition, when seen, is due to the covalent modification of the Cys side-chain that then produces blockage of the pathway (Figs. 4B and 4C).
The time courses of Nile Red efflux by N274C AcrB, in the absence and presence of pyrene-maleimide, are shown in Fig. 4B. The comparison is made somewhat difficult because the time course of fluorescence is not linear. However, in the example shown, at every time point during the early phase of efflux, the slope was steeper in the presence of pyrene-maleimide (thus it is not a decrease in the initial delay in efflux). Furthermore, each of these experiments was repeated at least three times, and the reproducibility of efflux curves was ascertained. Interestingly, AcrBF628C, which is defective in Nile Red efflux, gained efflux competency in the presence of pyrene-maleimide (Fig. 4B), presumably pyrene substitutes for the benzene group in Phe628, which is apparently needed for the aromatic interaction with an aromatic substrate, Nile Red. (In fact, the pyrene-maleimide-mediated activation of essentially non-functional AcrBF628C may be used as an ‘activation switch’ for studying the cellular effects of AcrB efflux in real-time.)
When this assay was performed with all 48 mutants, we found that the addition of pyrene-maleimide inhibited Nile Red efflux in AcrB mutants with following alterations: N274C, E673C, and T676C (Fig. 5). M573C mutant showed a marginal inhibition. CPM inhibited AcrB mutants with following alterations: Q89C, N274C, D276C, I277C, and E673C (Fig. 5).
Figure 5. Extent of Blockage of Nile Red efflux in various AcrB mutants by 30 μM pyrene-maleimide or 12 μM CPM.
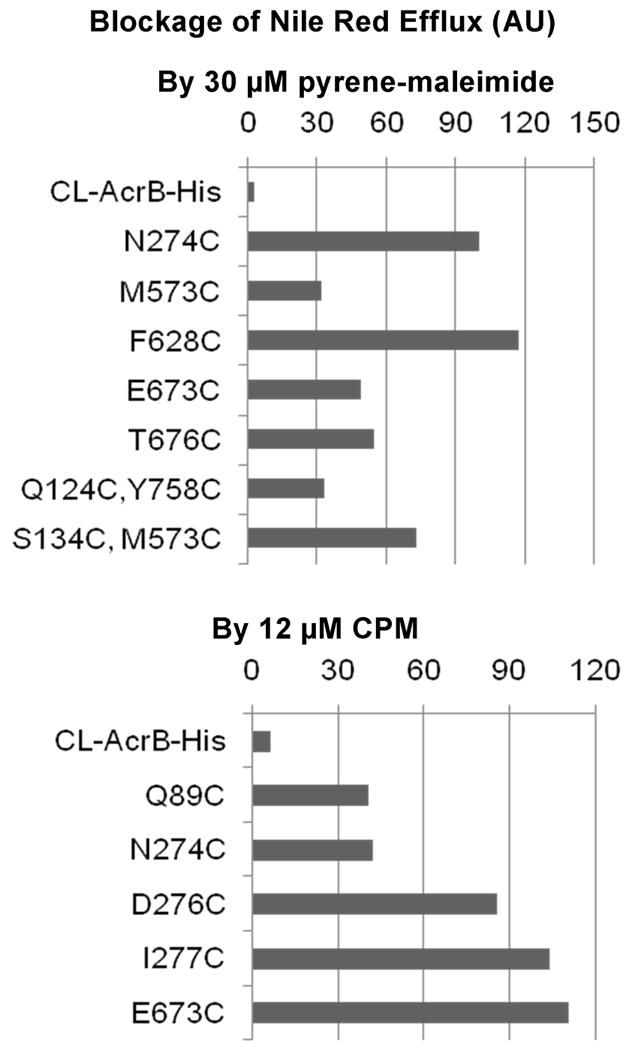
The extent of blockage, calculated as described in Experimental Procedures, is shown as a percentage of blockage seen in reference mutants (N274C for pyrene-maleimide and E673C for CPM). We set the arbitrary cut-off point at 30%. In order to save space, we show only those mutants that showed significant blockage, although we tested all mutants described earlier. The efflux activity of mutant F628C, which was minimal, was significantly stimulated by pyrene-maleimide (Fig. 4B); although this is an inverse of blockage, corresponding to a negative number, it is shown as a positive value here to save space.
Importantly, there is a significant overlap between the positive results in this Nile Red assay and the residues labeled by Bodipy-FL-maleimide (Fig. 3): thus all of the residues that were strongly positive in the pyrene-maleimide assay and all of the residues that were positive in the CPM assay were also positive in the Bodipy assay. This reinforces our interpretation that the Bodipy assay correctly identifies the residues lining the path of the drugs. However, there were residues that were labeled by Bodipy-FL-maleimide but did not block Nile Red efflux in presence of pyrene-maleimide or CPM. Since AcrB has an especially large binding pocket and presumably a broad path for substrates (Neyfakh, 2002; Schumacher et al., 2004; Takatsuka et al., 2010), we speculated that the binding of phenylcoumarin or pyrene to a single cysteine side-chain may not effectively block the path of Nile Red. Indeed some of the residues responded only to one of the reagents. Thus we tested a few double Cys mutants, and pyrene-maleimide blocked the Q124C/Y758C, M573C/S134C, and S134C/L828C mutants, whose single mutation components blocked either insignificantly or only mildly (Fig. 5). In contrast, the use of double mutants did not expand the range of CPM-blocked residues (Fig. 5).
Maleimide inhibitors bind to some Cys residues without blocking Nile Red efflux
As mentioned above, we believe that pyrene-maleimide and CPM covalently modify the Cys side-chains in some AcrB mutants but do not block the Nile Red efflux because of the flexibility and/or the width of the pathway. Direct quantitation of the extent of modification of AcrB mutants (as was done with Bodipy-FL [Fig. 2]) was difficult because pyrene-maleimide and CPM have excitation maxima in the near-UV range (339 and 384 nm). Thus we used a successive two-step labeling approach. We used AcrBN274C, AcrBD276C, AcrBF666C, and AcrBT676C and examined the fate of Cys residues by treating the cells first with pyrene-malemide and CPM, and then with Bodipy-FL-maleimide. We found that pyrene-maleimide (at 15 μM, a lower concentration than that used in the Nile Red efflux blocking assay, 30 μM) strongly protected AcrBN274C, AcrBD276C, and AcrBT676C from subsequent Bodipy-FL-maleimide modification, so that the extent of Bodipy-FL modification fell to 32, 49, and 47% of the control, a data showing that pyrene-maleimide modified these mutant proteins efficiently when applied alone. Thus the fact that pyrene-maleimide did not block the Nile Red efflux in AcrBD276C is not due to the absence of modification by pyrene-maleimide. Similarly, CPM (6 μM) strongly protected AcrBN274C, AcrBD276C, and AcrBT676C from subsequent modification by Bodipy-FL-malemide (the extent of modification fell to 23, 23, and 68% of the control), although CPM did not block Nile Red efflux in AcrBT676C. We conclude therefore that the maleimide reagents may react with some cysteine side-chains but the substitution may not lead to the blocking of Nile Red efflux.
DISCUSSION
Elucidating the path of substrate travel through the giant transporter AcrB is obviously important not only for understanding the basis of the substrate specificity of this multidrug efflux pump (Nikaido, 1996), but also for designing effective inhibitors of this pump (Lomovskaya et al., 2001; Bohnert et al., 2005). Currently, the point of departure for this discussion could be the periplasmic binding pocket for minocycline and doxorubicin elucidated crystallographically in the binding protomer in the asymmetric trimer structure by Murakami et al. (2006), because this pocket collapses in the extrusion protomer, an observation indicating its functional essentiality. When the possible paths leading to this pocket are explored, we can see a prominent direct path from the external large cleft, as well as a somewhat more tortuous path apparently from the vestibule located between the protomers and close to the membrane surface (Murakami et al., 2002) and a third pathway leading to the central cavity (Fig. 1), as was noted earlier (Murakami et al., 2006; Sennhauser et al., 2007).
In this study, we mutated into cysteine each of the 48 residues that we thought might be on the path of substrates (see Results) to examine experimentally the precise path of the substrates. Such an approach was earlier used for the study of AcrB and its homologs. Thus Mao et al. (2002) mutated critical residues in the periplasmic domain of Pseudomonas aeruginosa MexD, and showed that their modification by a maleimide reagent, MIANS, inhibited its activity. Several residues of Hemophilus influenzae AcrB were mutated to cysteine and with at least one residue the extent of its labeling by fluoresceine maleimide was shown to be affected by the presence of substrate drugs (Dastidar et al., 2007). Our study, however, is much more extensive and our assays are quite different. We used as the main approach the labeling of the Cys residues with a low concentration of Bodipy-FL-maleimide. Because Bodipy-FL-maleimide is quite lipophilic, we expect it to behave as an AcrB substrate and to follow the export path within the AcrB protein; in fact a computer docking simulation using Autodock Vina (Trott and Olson, 2010) predicted that it would bind to the binding pocket as strongly as one of the best substrates, minocycline (unpublished results). This approach showed the potential involvement of many residues (Fig. 3). Importantly, Bodipy-FL-maleimide did not label the residues far away from the predicted substrate path, used as negative controls. Furthermore, our inference that Bodipy-FL-maleimide labels the residues on the correct substrate path was reinforced by the second approach, in which we examined the blockage of Nile Red efflux by covalently modifying such residues with bulky substituents. This approach indeed showed that among the Bodipy-labeled residues, the efflux blockage occurred with N274 and E673 with both CPM and pyrene-maleimide, T676 with pyrene-maleimide, and Q89, D276, and I277 with CPM (Fig. 5). In addition, pyrene-maleimide modification of F628C reactivated the inactive mutant transporter. All residues that were not stained by Bodipy-FL-maleimide showed no blockage of Nile Red efflux by pyrene-maleimide or CPM, except for the marginal blockage at M573 with pyrene, and except when double mutants were used in the blocking assay. These results confirm that at least the residues producing blockage, among the Bodipy-labeled residues, are on the direct path for substrate extrusion. Not all the Bodipy-labeled residues, however, showed blockage, presumably due to the wide and flexible nature of the substrate path. This idea is supported by several observations: (a) In the Nile Red blockage assay, some residues show blockage only with one of the two reagents used (Fig. 5), (b) Some of the residues were shown to be modified by CPM and pyrene-maleimide (Results) presumably because these reagents are concentrated in the substrate path, yet no blockage resulted, (c) Modification of two neighboring residues sometimes produced blockage when that of single residues did not (Fig. 5).
We now discuss the location of Bodipy-modified residues, assuming that they are likely to be on the substrate path (Figs. 3 and 6). These residues included, as expected, the residues on the walls of the binding pocket, such as S134, F136, Q176, F178, S180, E273, N274, D276, I277, Y327, and F617 (Figs. 3 and 6). We also believe that T87, F178 and F628 are critical residues as their conversion to Cys significantly reduced Nile Red efflux. Substitution of some of these residues located in the relatively narrow part of the pocket (Q89, N274, D276, I277) with bulky groups also blocked the efflux of Nile Red (Fig. 5), and very interestingly the modification of the inactive F628C mutant protein with pyrene-maleimide produced a functional pump. Our results are also consistent with the site-directed mutagenesis data (Bohnert et al., 2008), which showed that conversion of F136, F178, F610, F617, and F628 into Ala decreased the pumping activity for antimicrobial drugs.
Figure 6. Positions of residues involved in substrate transport through AcrB.
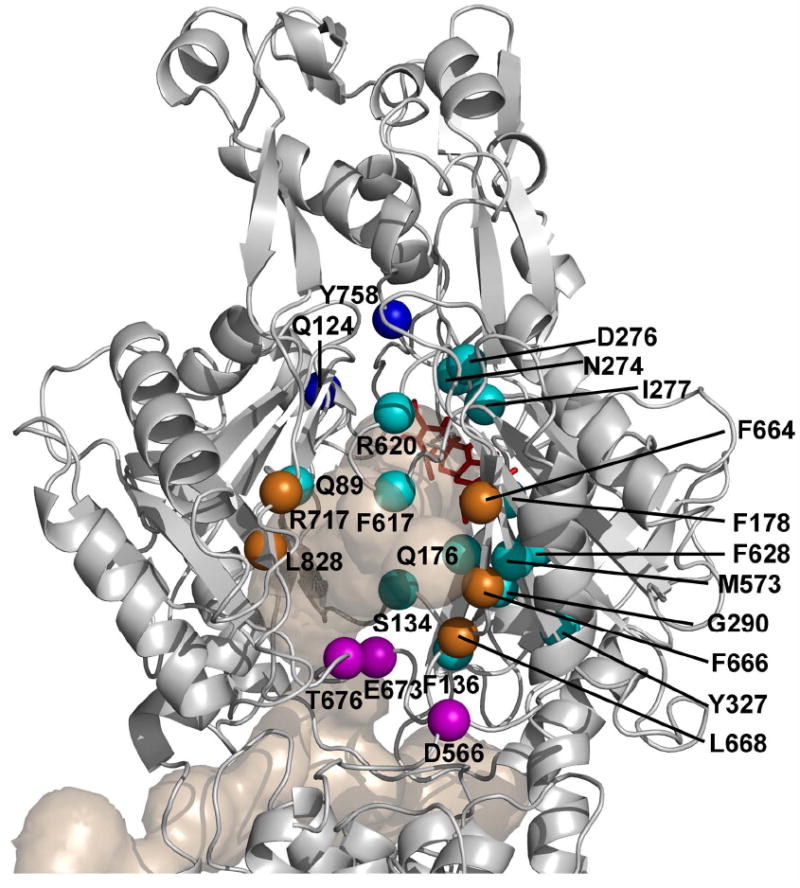
Those residues that gave positive response in Bodipy-FL-maleimide assay or in CPM/pyrene maleimide assay or both are identified and shown as spheres, colored magenta for those in the bottom of cleft, orange for those in cleft, light blue for those in the binding pocket, and deep blue for those in the gate, as in Fig. 1. E273 is hidden behind the cluster of D276, N274, and I277. The Binding Protomer of AcrB (based on the PDB file 2DRD) is shown as a gray ribbon model, oriented as in Fig. 1, so that its TolC-binding region is on top and we look into the protein from the outside of the trimer structure. Tunnels leading to the binding pocket, as well as the bound minocycline found in the pocket, are also shown as in Fig. 1.
Our major objective was to identify residues that are presumably on the path of substrates, outside the binding pocket. Here again we obtained many interesting “hits” (Fig. 6). These contain the residues at the entrance of the cleft, which were shown to bind substrates in crystallographic studies of symmetric AcrB trimers (Yu et al., 2005; Drew et al, 2008). Because this site is quite open, there was some doubt whether the binding represented a part of the normal path for substrate travel. However, we saw several residues in this area (F664, F666, and L668) all prominently labeled with Bodipy-FL-maleimide, and also R717 and L828 labeled somewhat less, yet significantly (Fig. 3). It was shown earlier that F664A, F666A, and R717A mutations decrease the pumping activity of AcrB (Yu et al., 2005), and thus we believe that the interaction of the substrates with this group of residues is one of the earliest steps in transport.
Another interesting observation is the role of residues close to the membrane surface, but that are located somewhat farther from the external surface of the cleft (Fig. 6). These residues (D566, E673, and T676) were stained very strongly with Bodipy-FL-maleimide (Fig. 3), and the modification of E673 or T676 blocked the Nile Red efflux (Fig. 5). Ciprofloxacin binding to cleft surface appears to involve also D566 and E673, and E673A mutation weakens the pumping activity (Yu et al, 2005). A MexD residue corresponding to T676 (N673) was shown earlier to be involved in substrate recognition (Mao et al., 2002). These observations suggest that the residues are on the integral pathway of drug uptake. Interestingly T676C reacts with Bodipy-FL-maleimide regardless of whether the AcrB is active or not (Results). Possibly the binding of the substrates to T676 (and perhaps other residues in this group) may be the first step of substrate translocation within AcrB.
Early in the crystallographic study of AcrB, cocrystallized substrates were found at the ceiling of the central cavity, near L25, T98, F386 and F459 (Yu et al, 2003). In this study however, we found no evidence that these residues are on the normal path of drug export. The conversion of F386 and F459 into alanine also does not appear to affect activity (Yu et al., 2005).
Bodipy-FL-maleimide did not label the residues in the gate region between the binding pocket and the funnel, such as Q124, Q125, W754, S757, Y758, K770, Y772, and R780. We could not show the blockage of Nile Red efflux in single mutants of this group, but a Q124C/Y758C double mutant inhibited Nile Red efflux upon pyrene-maleimide treatment (Fig. 5). Possibly the presence of bulky substituents on both sides of the path is needed to block efflux.
In any case we note that this study did not include many residues lining the alternative tunnels leading to the vestibule or to the central cavity, and that these and other residues remain to be examined. We also emphasize that negative results in our study do not disprove the functional importance of the residues studied.
Experimental Procedures
Bacterial strains, plasmids, chemicals, and media
Strains used in this study were plasmid-containing derivatives of RAM-1334 (MC4100 Δara ΔacrB), a kind gift from Rajeev Misra. The plasmids were derivatives of pSportI-acrB expressing cysteine-less AcrB with His-tag at C-terminus (henceforth referred to as CL-AcrBHis)(Takatsuka and Nikaido, 2007). LB medium with 25 μg/ml ampicillin was used as the growth medium. Nile red, BODIPY FL N-(2-aminoethyl)maleimide (Bodipy-FL-maleimide), 7-diethylamino-3-(4’-maleimidylphenyl)-4-methylcoumarin (CPM), and N-(1-pyrene) maleimide (pyrene-maleimide) were acquired from Invitrogen. DDM (n-dodecyl β-D-maltoside) was obtained from Anatrace. Dimethyl sulfoxide was used as solvent to produce stock solutions of Nile Red, Bodipy-FL-maleimide, and CPM. N,N-Dimethyl formamide was used as solvent for the stock solution of pyrene-maleimide. Solvents, His-select cobalt affinity gel, and other chemicals were purchased from Sigma.
Site-directed-mutagenesis
The specific codons were mutated using the standard protocol using the QuikChange kit from Stratagene. The plasmid pSport1, plasmids expressing CL-AcrBHis and some of its mutants (Q89C, F673C, F666C, and Q124C/Y758C), the plasmid expressing WT-AcrBHis (with native cysteines) and primers for altering the acrB codon Q34C and D407A were kind gifts of Yumiko Takatsuka. The sequence for primers is available on request.
Bodipy-FL-maleimide labeling assay
Cells (5 ml) were grown overnight at 37° C with vigorous shaking. They were washed twice and resuspended with 5 ml buffer (50 mM potassium phosphate, 0.5 mM MgCl2, pH 7.0, hereafter called “phosphate buffer”) and the cell density was adjusted to OD660 of 3.5 using Shimadzu 1240 spectrophotometer. To 5 ml cells, 0.4% glucose and 6 μM Bodipy-FL-maleimide were added and the mixture was left standing for 1 h (with vortexing at 30 min) at room temperature. The cells were washed with 5 ml phosphate buffer containing 0.4% glucose and again washed with 5 ml phosphate buffer. The cells were lysed using a sonicator and the membrane fraction was isolated. The membrane fraction was solubilized and the His-tagged AcrB was isolated using an equivalent of 200 μl His-select cobalt affinity gel in a total of 800 μl phosphate buffer containing 1% DDM, kept at 4° C overnight with slow shaking. His-tagged AcrB was then isolated by using Micro Bio-spin Chromatography Columns (from Bio-Rad), first by washing with 500 μl buffer containing 0.02% DDM, and then eluting using 100 μl phosphate buffer containing 500 mM imadazole and 0.02% DDM.
The eluted fraction (7.5 μl in sample buffer) was used to resolve the AcrB band with SDS-PAGE (0.2% SDS and 11.5% acrylamide). Fluorescence in the AcrB band was observed using Typhoon Phosphorimager, with an excitation filter (488 nm) and an emission filter (510 nm) at normal sensitivity, to detect labeling by Bodipy-FL-maleimide. The protein bands in the gel were then stained using Coomassie Blue.
The Coomassie stained gel was dried and attached to a printed copy of fluorescent image (from Typhoon phosphorimager) and both were scanned using HP Officejet 6500. With ImageJ (from NIH) the scanned image was converted to 8-bit digital data, background was removed digitally (using 50 pixels radius) and finally the image was inverted to negative image for quantification. The Bodipy-FL-maleimide fluorescence intensity was normalized with the Coomassie band intensity in order to correlate fluorescence intensity to the amount of the protein. Each set of assay was run with the positive control AcrBN274C, which was used to calculate the relative intensity of other bands. The normalized intensity of AcrBN274C staining is set as 100 in bar graphs (Fig. 3).
Nile Red efflux assay
Cells (5 ml) were grown overnight at 37° C with vigorous shaking. The cells were washed twice and resuspended with 5 ml phosphate buffer and treated with 10 μM CCCP for 30 min at room temperature with gentle shaking. They were washed twice and resuspended with 5 ml phosphate buffer. The cell density was adjusted to an OD660 of 8.5 using Shimadzu 1240 spectrophotometer. The de-energized cells were treated (preloaded) with 10 μM Nile Red, by letting them stand for 3 h at room temperature.
For the assay 1.8 ml phosphate buffer was added to a cuvette, with or without glucose (final concentration 0.4%), and with or without cysteine-reactive compounds (either 30 μM pyrene-maleimide or 12 μM CPM, final concentrations). For control mixtures, pure solvents were added in the same amounts. Fluorescence was recorded every sec using a Shimadzu RF-5301 PC spectrofluorophotometer, set at excitation 545 nm (slit 5 nm) and emission 630 nm (slit 10 nm). Exactly 20 sec after the recording was started, 200 μl of Nile-Red-preloaded cells were added. The fluorescence was recorded for additional 140 sec.
The result was quantified as the average difference between the fluorescent readings with the maleimide reagent and those without maleimide, during 51 to 151 sec. In the comparison of residues with pyrene-maleimide blocking (Fig. 5, top) the results were normalized against the value for N274C (which was 90), set as 100. In the assays with CPM (Fig. 5, bottom) the results were normalized against the value for E673C (which was 60), here set as 100.
Pretreatment of mutant AcrB with pyrene-maleimide or CPM followed by labeling with Bodipy-FL-maleimide
The cells were grown, washed and adjusted for density as described for Bodipy-FL-maleimide labeling assay above. Then 0.4% glucose was added, followed by either 15 μM pyrene-maleimide or 6 μM CPM (pure solvents were added to controls), and the mixture was left standing for 5 min at room-temperature. After 5 min incubation, 6 μM Bodipy-FL-maleimide was added and the same steps were followed as described for Bodipy-FL-maleimide labeling assay.
Presentation and variation of quantitative data
In order to make the paper concise, we have presented the comparative data as bar diagrams (Figs. 3 and 5). All the bars represent average of two independent assays, where the difference between the two values was usually less than 10% when the relative activity was between 20 and 90. When the activity was below 20, the two assays often gave wider variations as expected. In all assays 30% or above of the reference value was interpreted as positive.
Acknowledgments
We thank Etsuko Sugawara for many constructive suggestions, Yumiko Takatsuka for important suggestions, plasmids, and primers, and Rajeev Misra for the strain RAM-1334. This work was supported by U.S. Public Health Service (AI-09644).
References
- Bohnert JA, Karamian B, Nikaido H. Optimized Nile Red efflux assay of AcrAB-TolC multidrug efflux system shows competition between substrates. Antimicrob Agents Chemother. 2010 doi: 10.1128/AAC.00620-10. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert JA, Kern WV. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob Agents Chemother. 2005;49:849–852. doi: 10.1128/AAC.49.2.849-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF) J Antimicrob Chemother. 2007;59:1216–1222. doi: 10.1093/jac/dkl426. [DOI] [PubMed] [Google Scholar]
- Bohnert JA, Schuster S, Seeger MA, Fähnrich E, Pos KM, Kern WV. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J Bacteriol. 2008;190:8225–8229. doi: 10.1128/JB.00912-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastidar V, Mao W, Lomovskaya O, Zgurskaya HI. Drug-induced conformational changes in multidrug efflux transporter AcrB from Haemophilus influenzae. J Bacteriol. 2007;189:5550–5558. doi: 10.1128/JB.00471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh T, Paulsen IT, Saier MH., Jr A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J Bacteriol. 1994;176:3825–3831. doi: 10.1128/jb.176.13.3825-3831.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew D, Klepsch MM, Newstead S, Flaig R, De Gier JW, Iwata S, Beis K. The structure of the efflux pump AcrB in complex with bile acid. Mol Membr Biol. 2008;25:677–682. doi: 10.1080/09687680802552257. [DOI] [PubMed] [Google Scholar]
- Elkins CA, Nikaido H. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominantly by two large periplasmic loops. J Bacteriol. 2002;184:6490–6498. doi: 10.1128/JB.184.23.6490-6498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fralick JA. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J Bacteriol. 1996;178:5803–5805. doi: 10.1128/jb.178.19.5803-5805.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan P, Fowler SD. Spectrofluorometric studies of the lipid probe., nile red. J Lipid Res. 1985;26:781–789. [PubMed] [Google Scholar]
- Hearn EM, Gray MR, Foght JM. Mutations in the central cavity and periplasmic domain affect efflux activity of the resistance-nodulation-division pump EmhB from Pseudomonas fluorescens cLP6a. J Bacteriol. 2006;188:115–123. doi: 10.1128/JB.188.1.115-123.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother. 2001;45:105–116. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J Bacteriol. 1993;175:6299–6313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol. 1995;16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- Mao W, Warren MS, Black DS, Satou T, Murata T, Nishino T, Gotoh N, Lomovskaya O. On the mechanism of substrate specificity by resistance nodulation division (RND)-type multidrug resistance pumps: the large periplasmic loops of MexD from Pseudomonas aeruginosa are involved in substrate recognition. Mol Microbiol. 2002;46:889–901. doi: 10.1046/j.1365-2958.2002.03223.x. [DOI] [PubMed] [Google Scholar]
- Middlemiss JK, Poole K. Differential impact of MexB mutations on substrateselectivity of the MexAB-OprM multidrug efflux pump of Pseudomonas aeruginosa. J Bacteriol. 2004;186:1258–1269. doi: 10.1128/JB.186.5.1258-1269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Murakami S, Tamura N, Saito A, Hirata T, Yamaguchi A. Extramembrane central pore of multidrug exporter AcrB in Escherichia coli plays an important role in drug transport. J Biol Chem. 2004;279:3743–3748. doi: 10.1074/jbc.M308893200. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Neyfakh AA. Mystery of multidrug transporters: the answer can be simple. Mol Microbiol. 2002;44:1123–1130. doi: 10.1046/j.1365-2958.2002.02965.x. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178:5853–5859. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. The role of outer membrane and efflux pumps in the resistance of gram-negative bacteria. Can we improve drug access? Drug Resist Updat. 1998;1:93–98. doi: 10.1016/s1368-7646(98)80023-x. [DOI] [PubMed] [Google Scholar]
- Petrek M, Otyepka M, Banás P, Kosinová P, Koca J, Damborský J. CAVER: a new tool to explore routes from protein clefts., pockets and cavities. BMC Bioinformatics. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Miller MC, Brennan RG. Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. EMBO J. 2004;23:2923–2930. doi: 10.1038/sj.emboj.7600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Nat Acad Sci USA. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y, Nikaido H. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol. 2006;188:7284–7289. doi: 10.1128/JB.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y, Nikaido H. Site-directed cross-linking shows that cleft flexibility in the periplasmic domain is needed for the multidrug efflux pump AcrB of Escherichia coli. J Bacteriol. 2007;189:8677–8684. doi: 10.1128/JB.01127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y, Nikaido H. Covalently linked trimer of the AcrB multidrug efflux pump provides support for the functional rotating mechanism. J Bacteriol. 2009;191:1729–1737. doi: 10.1128/JB.01441-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y, Chen C, Nikaido H. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proc Natl Acad Sci U S A. 2010;107:6559–6565. doi: 10.1073/pnas.1001460107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry. 2005;44:11115–11121. doi: 10.1021/bi050452u. [DOI] [PubMed] [Google Scholar]
- Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukagoshi N, Aono R. Entry into and release of solvents by Escherichia coli in an organic-aqueous two-liquid-phase system and substrate specificity of the AcrAB-TolC solvent-extruding pump. J Bacteriol. 2000;182:4803–4810. doi: 10.1128/jb.182.17.4803-4810.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehmeier C, Schuster S, Fähnrich E, Kern WV, Bohnert JA. Site-directed mutagenesis reveals amino acid residues in the Escherichia coli RND efflux pump AcrB that confer macrolide resistance. Antimicrob Agents Chemother. 2009;53:329–330. doi: 10.1128/AAC.00921-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. AcrA is a highly asymmetric protein capable of spanning the periplasm. J Mol Biol. 1999;285:409–420. doi: 10.1006/jmbi.1998.2313. [DOI] [PubMed] [Google Scholar]