

Abstract
BACKGROUND AND AIMS
Immunological disorders of the gastrointestinal tract such as inflammatory bowel disease (IBD) often result in recurrent and persistently elevated levels of pro-inflammatory cytokines. Kinase suppressor of Ras 1 (KSR1) is involved in TNF-mediated colon epithelial cell survival, yet its role in chronic inflammation has not been defined. In this study, we tested the hypothesis that KSR1 is protective against spontaneous experimental colitis.
METHODS
KSR1−/−Interleukin-10 (Il10)−/− mice were generated and histolopathologic parameters of intestinal inflammation were scored. Bone marrow transplants performed on wild-type (WT) and KSR1−/−Il10−/− mice determined the contribution of KSR1 in hematopoietic lineages. Mucosal T helper (Th)1 and Th17 cytokine were also examined. In vitro Th1 and Th17 polarization assays were conducted and interleukin-17A (IL-17A) and interferon-γ (IFN-γ) production analyzed by flow cytometry. Neutralizing antibodies against IgG, IL-17A, or IFN-γ were administered to 3-week old KSR1−/−Il10−/− mice for 3 weeks and scored for colitis.
RESULTS
KSR1−/−Il10−/− mice developed accelerated and severe spontaneous colitis by 4 weeks of age. KSR1 expression in hematopoietic lineages was protective against colitis. Both IFN-γ and IL-17A transcripts were elevated in colons of KSR1−/− and KSR1−/−Il10−/− mice. IFN-γ production was increased in lamina propria T cells isolated from KSR1−/− and KSR1−/−Il10−/− mice. Additionally, in vitro Th1 polarization was increased while Th17 polarization was impaired in KSR1 deficient naïve T cells. Finally, administration of IFN-γ neutralizing antibodies attenuated colitis in KSR1−/−Il10−/− mice.
CONCLUSIONS
Mice lacking both KSR1 and IL-10 develop exacerbated colitis due to dysregulated IFN-γ production in T lymphocytes.
Keywords: KSR1, IL-10, IFN-γ, IL-17A
Introduction
Pathogen defense mechanisms, such as innate and adaptive immunological responses, have greatly contributed to the success of vertebrate evolution. While acute inflammatory responses are beneficial to the host, chronic inflammation can be deleterious. Thus, identification of the molecules involved in regulating these responses is critical for understanding the mechanisms that can contribute to immunological disorders. For patients suffering from inflammatory bowel disease (IBD), dysregulated immune responses result in sustained immune cell activation and elevated cytokine production, which can lead to an increased risk for developing colon cancer over their lifetime.1, 2 Therefore, it is imperative to develop strategies and identify novel targets that modulate immune responses to complement current therapeutic options for IBD patients.
Much of our current understanding of the molecular mechanisms involved in IBD has come from knockout, transgenic, and chemically-induced mouse models. Although mouse models of IBD do not fully recapitulate the human disease, interleukin-10 deficient (Il10−/−) mice display similar characteristics to that of human Crohn’s disease.3, 4 Since the anti-inflammatory effects of IL-10 are required to regulate Th1 cytokine production and promote immune homeostasis,5, 6 loss of IL-10 in mice results in spontaneous enterocolitis driven by an aberrant immunological response to enteric antigens.7, 8
Kinase suppressor of Ras-1 (KSR1) functions as a kinase or molecular scaffold of the Raf/MEK/ERK signaling module to regulate proliferation, apoptosis or function in a cell/tissue context-dependent manor.9–11 For example, KSR1 is required for colon epithelial cell survival downstream of TNF signaling both in vitro and in vivo.12, 13 Given that pro-inflammatory cytokines, including TNF, are elevated in Il10−/− mice, we hypothesized that loss of KSR1 in Il10−/− mice would exacerbate colitis. Therefore, we crossed KSR1−/− mice with Il10−/− mice to generate KSR1−/−Il10−/− mice to examine the role of KSR1 as a protective mediator during chronic inflammation.
In the present study, we found that KSR1−/−Il10−/− mice developed an early onset form of severe colitis resulting from loss of KSR1 expression in hematopoietic lineages. Specifically, KSR1 deficient T cells produced more IFN-γ, had a greater propensity to polarize along the Th1 axis in vitro, and neutralizing IFN-γ attenuated disease in KSR1−/−Il10−/− mice. The data presented here, along with recent reports, reveal an emerging role for KSR1 in immune function.
Materials and Methods
Animals
BALB/c (WT), KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice were bred and maintained on a BALB/c background. Experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Colonoscopy
10-week old mice were sedated with an intraperitoneal injection of ketamine (5 mg/ml)/xylazine (0.5 mg/ml) solution (100 µl/10g body weight). Mouse colons were visualized using a KARL STORZ Veterinary Endoscope (KARL STORZ Imaging, Inc., Goleta, CA) according to procedures previously described.14
Histopathology
Colon sections were stained with Hematoxylin and Eosin (H&E) and analyzed by light microscopy. A pathologist (M.K.W.) blinded to the genotypes and conditions used a scoring method previously described to evaluate colitis.15
Reagents
Fluorochrome coupled anti-CD4, -TCRβ, -IFN-γ, and anti-IL-17 as well as unconjugated anti-CD3, -CD28, -IFN-γ, -IL-4 and rmIL-6 were all purchased from BD Pharmingen (San Jose, CA). rhTGF-β1 was purchased from R&D Systems (Minneapolis, MN).
Lymphocyte isolation
Splenic were isolated using conventional procedures. Lamina propria T cells were obtained as previously described.16
Intracellular cytokine staining
Stimulation, surface and intracellular staining was performed as described.16 Cells were acquired in a FACSCalibur instrument and data analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
In vivo cytokine neutralization
3-week old KSR1−/−Il10−/− mice were given intraperitoneal injections of 100 µg/mouse anti-IgG (eBioscience, cat#16-4301, San Diego, CA), anti-IFN-γ (eBioscience, clone XMG 1.2), or anti-IL-17A (eBioscience, clone: eBioTC11-18H10.1) neutralizing antibodies twice per week for 3 weeks. Mouse colons were harvested and scored as before.
Statistics
Histopathology, immunohistochemistry, and qPCR statistical analysis were compared using one-way ANOVA followed by Dunnett’s post-test. Bone marrow transplant statistical analysis was conducted using one-way ANOVA followed by Bonferroni’s multiple comparison test. Th1/Th17 polarization, intracellular cytokine staining, and cytokine neutralization statistical analysis were performed using an unpaired Student’s t test. P-values of < 0.05 were considered significant. All data was analyzed using Prism 5 (GraphPad Software, Inc.).
Bone marrow transplantation
Please see detailed Supplementary Materials and Methods.
Generation of Th17 and Th1 cells
Please see detailed Supplementary Materials and Methods.
Results
KSR1−/−Il10−/− mice develop accelerated spontaneous colitis
Although KSR1 inhibits epithelial cell apoptosis downstream of TNF signaling,13 the role of KSR1 during chronic inflammation has not been examined. Since Il10−/− mice on the BALB/c (WT) strain under SPF conditions develop spontaneous colitis between 12–16 weeks of age, we examined the effect of KSR1 deficiency on colitis in Il10−/− mice. The clinical onset of disease was assessed for each group of mice by examination of fecal occult blood and monitoring whole body weight from 3 to 10 weeks. KSR1−/−Il10−/− mice frequently suffered from chronic diarrhea and failed to thrive beginning at 5 weeks of age (Figure 1A). By colonoscopic examination, the colons of 10-week old KSR1−/−Il10−/− mice appeared thickened with a loss of translucency compared to the colons of WT, KSR1−/−, and Il10−/− mice (Figure 1B). Since colon weight per unit length is often indicative of inflammation, the colon length-to-weight (l/w) ratio was quantified for each group. KSR1−/−Il10−/− mice had an average l/w ratio of 15.03 ± 1.5 whereas WT, KSR1−/−, and Il10−/− mice had ratios of 44.36 ± 2.96, 41.64 ± 3.24, and 38.19 ± 2.34 respectively (Figure 1C). Paraffin-embedded colon sections from 10-week old mice were stained with Hematoxylin and Eosin (H&E) and a blinded pathologist scored each section for inflammation and injury. While 10-week old WT, KSR1−/−, and Il10−/− were relatively free of inflammation and injury, KSR1−/−Il10−/− mice suffer from severe colitis (score of 9.83 ± 0.6) (Figures 1D–1E). Barrier permeability was assed by administering FITC-dextran enemas to 10-week old mice, peripheral blood serum collected one hour later, and quantified for the presence of translocated FITC. KSR1−/−Il10−/− mice had increased barrier permeability, possibly due to the increased epithelial cell turnover as determined by apoptotic and proliferative markers (Supplemental Figure 1). We then evaluated the developmental time course of colitis and found that by 4 weeks of age, KSR1−/−Il10−/− mice already have histological signs of disease (Figure 1F). These data indicate that loss of KSR1 expression in Il10−/− mice results in accelerated and severe spontaneous colitis with retarded growth similar to findings in children with Crohn’s disease.17
Figure 1. KSR1−/−Il10−/− mice develop accelerated spontaneous colitis.
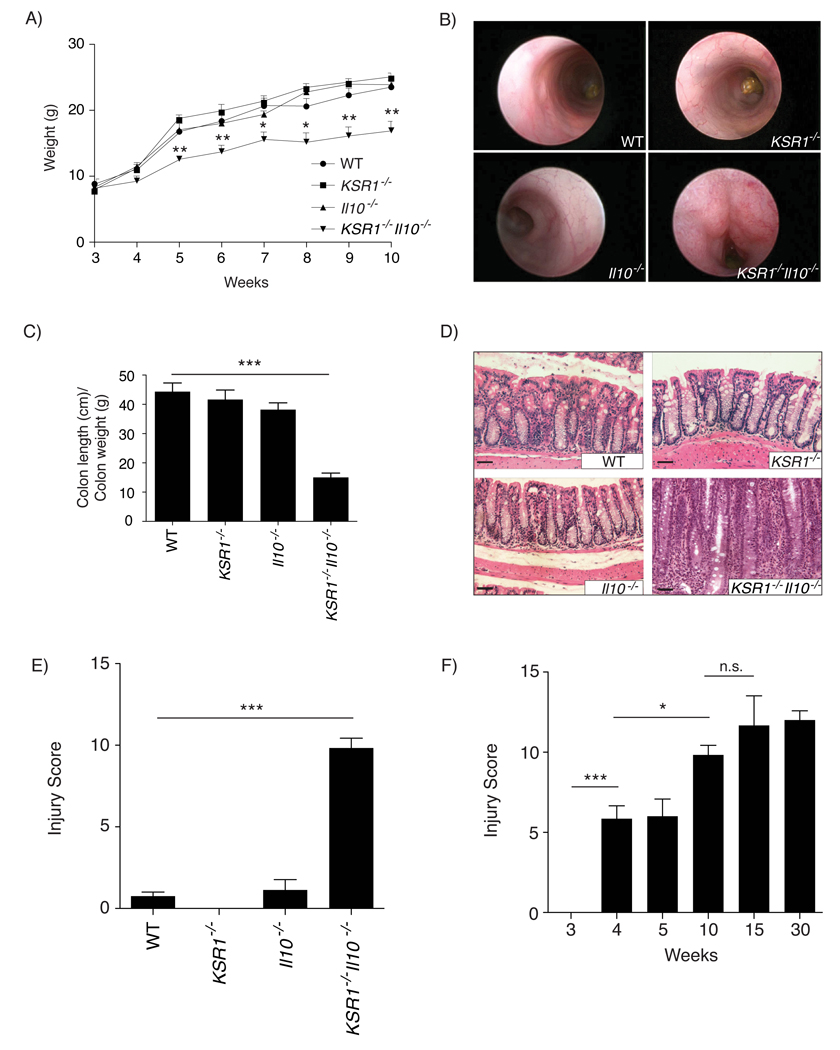
A) WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice were weighed weekly from 3 weeks of age to 10 weeks. Plotted data are the mean weight for each group (n ≥ 5). Error bars represent the SEM. B) Mouse colonoscopic images taken on 10-week old mice. C) Colons were removed from 10-week old mice and flushed, weighed, and measured from the cecum to anus. Solid bars represent the mean (n ≥ 3) of the length/weight ratios. Error bars are the SEM. D) Paraffin embedded colon sections from 10-week old mice stained with Hematoxylin and Eosin (H&E). Images were taken at 20X magnification (scale bars, 50 µm). E) H&E stained 10-week old mouse colon sections were scored by a pathologist blinded to the genotype. Solid bars represent the mean injury and inflammation score for each group (n ≥ 6) and error bars are the SEM. F) KSR1−/−Il10−/− mice were sacrificed at each time point indicated and scored for inflammation and injury as before. * P < 0.05, ** P < 0.01, *** P < 0.001
Expression of KSR1 in hematopoietic lineages is protective against colitis
The disease that develops in Il10−/− mice is attributed to immune hypersensitivity to enteric microflora.7, 8 Therefore, we investigated whether KSR1 expression in hematopoietic lineages mediated protection from disease in Il10−/− mice. Since the KSR1−/−, Il10−/−, and KSR1−/− Il10−/− mice used in this study were engineered as global deletions, we performed bone marrow transplantation on irradiated 4-week old WT or KSR1−/−Il10−/− recipient mice. Following bone marrow transplantation, recipient mice were sacrificed at 10 weeks of age and colitis was assessed as before. Irradiated KSR1−/−Il10−/− mice reconstituted with WT bone marrow (BM) developed mild colitis scores (2.8 ± 1.12) while reconstitution with KSR1−/−Il10−/− bone marrow resulted in severe colitis as expected (11 ± 1) (Figures 2A and 2B panels 1 & 4). Interestingly, restoring IL-10 to the immune system in KSR1−/−Il10−/− recipient mice did not ameliorate colitis (8.1 ± 1.72) (Figures 2A and 2B panel 3). However, restoring KSR1 to the immune system in KSR1−/−Il10−/− recipient mice (5.5 ± 1.25) attenuated the disease (Figures 2A and 2B panel 2). Interestingly, reconstitution of irradiated WT mice with Il10−/− BM was insufficient to cause disease, while reconstitution with KSR1−/−Il10−/− BM was sufficient to drive colitis (5.25 ± 0.84) (Figure 2). Though suppression of colitis in KSR1−/−Il10−/− mice required KSR1 expression in cells of the immune system, KSR1 was not required for leukocyte differentiation or stimulated T cell proliferation in vitro (Supplemental Figures 2A & 2B). Taken together, our data suggest that KSR1 expression in hematopoietic lineages plays a significant role in suppressing colitis in Il10−/− mice.
Figure 2. KSR1 in hematopoietic lineages suppresses colitis in Il10−/− mice.
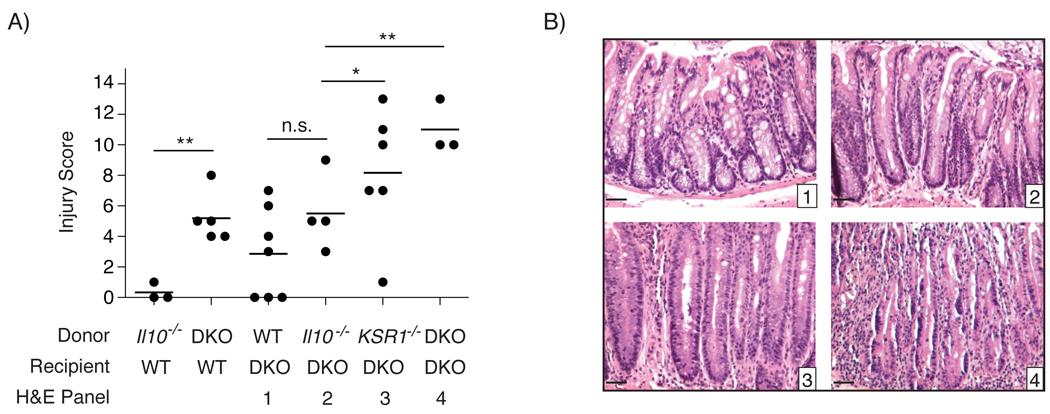
A) 4-week old WT and KSR1−/−Il10−/− (DKO) recipient mice were irradiated with 9 Gy 137Cesium. Bone marrow transplants using the indicated donor mice were performed and mice were sacrificed at 10-weeks of age. Each individual colonic injury and inflammation score is plotted with a solid line indicating the mean score for each group from three independent experiments and error bars are the SEM. B) Representative H&E stained paraffin embedded colon sections from recipient DKO mice transplanted with WT (1), Il10−/− (2), KSR−/− (3), or KSR1−/−Il10−/− (4) bone marrow as indicated. * P < 0.05, ** P < 0.01
Lamina propria T cells isolated from KSR1−/− and KSR1−/−Il10−/− mice have increased IFN-γ production
Pro-inflammatory cytokines including TNF, IFN-γ, and IL-17 are increased in the intestinal mucosa of Crohn’s disease patients and in mouse models of IBD.18–22 Therefore, we determined relative transcript levels of Th1 and Th17 cytokines in the colonic mucosa of WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice by quantitative real-time PCR (qRT-PCR). Both IFN-γ and IL-17A transcripts were increased KSR1 deficient mice (Figure 3A). In addition, colon mucosal lysates were screened and cytokine protein quantified using a Milliplex cytokine bead array. Consistent with the qRT-PCR data, IFN-γ was also increased in colons of KSR1−/− and KSR1−/−Il10−/− mice (8.79 ± 0.6 pg/mg and 93.7 ± 52.6 pg/mg) compared to WT (0.02 ± 0.0 pg/mg) and Il10−/− mice (Figure 3B). Neither message nor protein levels of IL-1β, IL-21, IL-22, or IL-23 were altered by KSR1 expression status (data not shown). We then examined IFN-γ and IL-17A production in T cells isolated from the lamina propria of WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice and stimulated with phorbol 12-myristate 13-acetate (PMA)/ionomycin. The percentage of TCRβ+ lamina propria T cells producing IFN-γ was significantly increased in KSR1−/− mice (10.58% ± 1.21%) and elevated in KSR1−/−Il10−/− mice (5.24% ± 1.22%) compared to Il10−/− mice (1.4% ± 0.12%) and WT mice (2.95% ± 0.52%) (Figure 4). Interestingly, IL-17A production was decreased in KSR1 deficient lamina propria T cells. Consistent with these data, isolated CD4+TCRβ+ splenocytes from KSR1−/− and KSR1−/−Il10−/− mice also exhibited elevated IFN-γ production (Supplemental Figure 3). Thus, KSR1 expression modulates IFN-γ production in TCRβ+ T lymphocytes.
Figure 3. Colons of KSR1−/− and KSR1−/−Il10−/− mice have elevated IFN-γ.
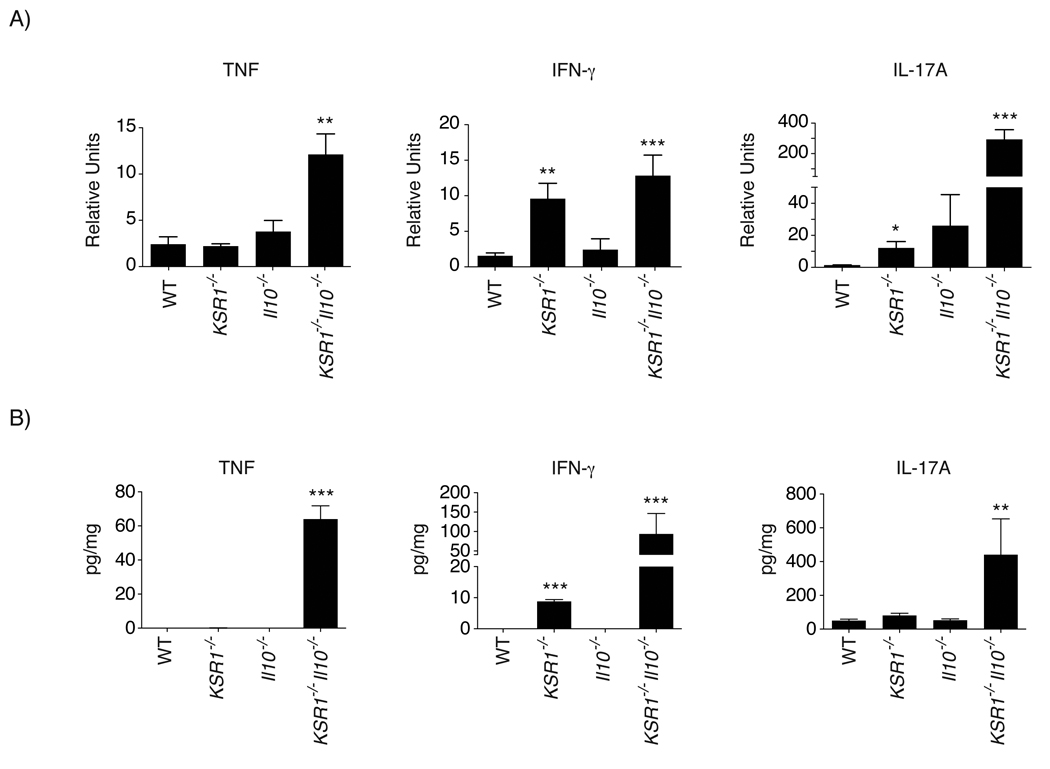
A) Total RNA was isolated from homogenized whole colon tissue from 10 week-old WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice. TNF, IFN-γ, and IL-17A cytokine transcript levels were analyzed by quantitative real-time PCR. Solid bars represent the mean for each genotype and error bars are the SEM. B) Total protein from homogenized mouse colons was analyzed for the detection and quantitation of TNF, IFN-γ, and IL-17A. Solid bars represent the mean quantity of each cytokine per mg of protein. Error bars are the SEM. * P < 0.05, ** P < 0.01, *** P < 0.001
Figure 4. Lamina propria T cells isolated from KSR1−/− mice have increased IFN-γ production.
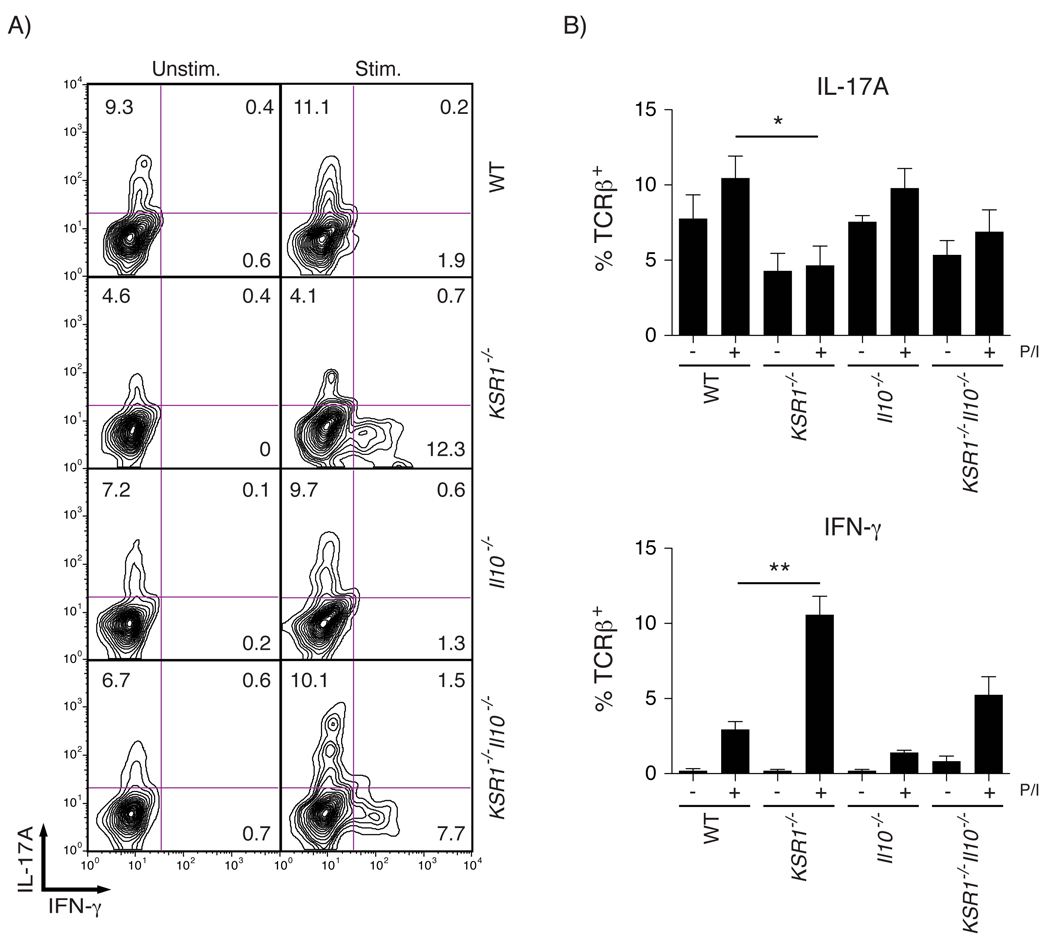
Lamina propria T cells were isolated from WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice and cultured for 5 hours in the presence of the protein transport inhibitor GolgiPlug and treated with or without PMA/Ionomycin. Cells were stained for cell surface TCRβ and intracellular IFN-γ and IL-17A. Samples were analyzed by flow cytometry gated on lymphocyte geometry and TCRβ+ staining. A) Representative flow cytometry contour plots of unstimulated and stimulated WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− lamina propria TCRβ+ T cells stained for intracellular IFN-γ and IL-17A. B) The percent of TCRβ+ T cells positive for intracellular IFN-γ or IL-17A was determined and solid bars represent the mean while error bars are the SEM. * P < 0.05, ** P < 0. 01
In vitro Th1 polarization is enhanced while Th17 polarization is impaired in KSR1 deficient T cells
IL-17A production by Th17 effector cells has been implicated in the pathogenesis of diseases including rheumatoid arthritis, multiple sclerosis, and Crohn’s disease.23–25 Our previous observations that IL-17A transcripts and protein levels were altered in mice lacking KSR1 suggested that KSR1 may be involved in T cell development along the Th1/Th17 axis. To test this, we isolated splenocytes from WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice and cultured them under Th1 or Th17 polarizing conditions. We then stimulated with PMA/ionomycin for 5 hours in the presence of GolgiPlug and measured intracellular IL-17A and IFN-γ production by flow cytometry. We found that under Th17 polarizing conditions, the number of IL-17A expressing cells was reduced in both KSR1−/− (3.20% ± 1.5%) and KSR1−/−Il10−/− (5.9% ± 0.48%) CD4+TCRβ+ T cells compared to WT (13.48% ± 1.34%) and Il10−/− (37.43% ± 8.3%) CD4+TCRβ+ T cells (Figures 5A–B). In fact, the number of CD4+TCRβ+ T cells producing IFN-γ was increased for KSR1−/− (10.32% ± 3.0%) and KSR1−/−Il10−/− (6.8% ± 1.7%) T cells compared to WT (0.89% ± 0.4%) and Il10−/− (2.84% ± 0.7%) T cells even when cultured under Th17 polarizing conditions (Figures 5A–B). We then determined if loss of KSR1 enhanced T cell development along the Th1 axis in vitro. Splenocytes were isolated and cultured under Th1 polarizing conditions and analyzed as before. We found that KSR1−/− T cells expressing IFN-γ under Th1 polarization was increased (56.1% ± 1.9%) compared to WT (44.8% ± 3.4%) (Figures 5C–D). Interestingly, culturing splenocytes from KSR1−/−Il10−/− mice under Th1 polarizing conditions resulted in reduced T cell viability. These findings suggest that KSR1−/− T cells have a greater propensity to develop along the Th1 axis in vitro.
Figure 5. In vitro Th17 polarization is impaired while in vitro Th1 polarization is increased in KSR1−/− T cells.
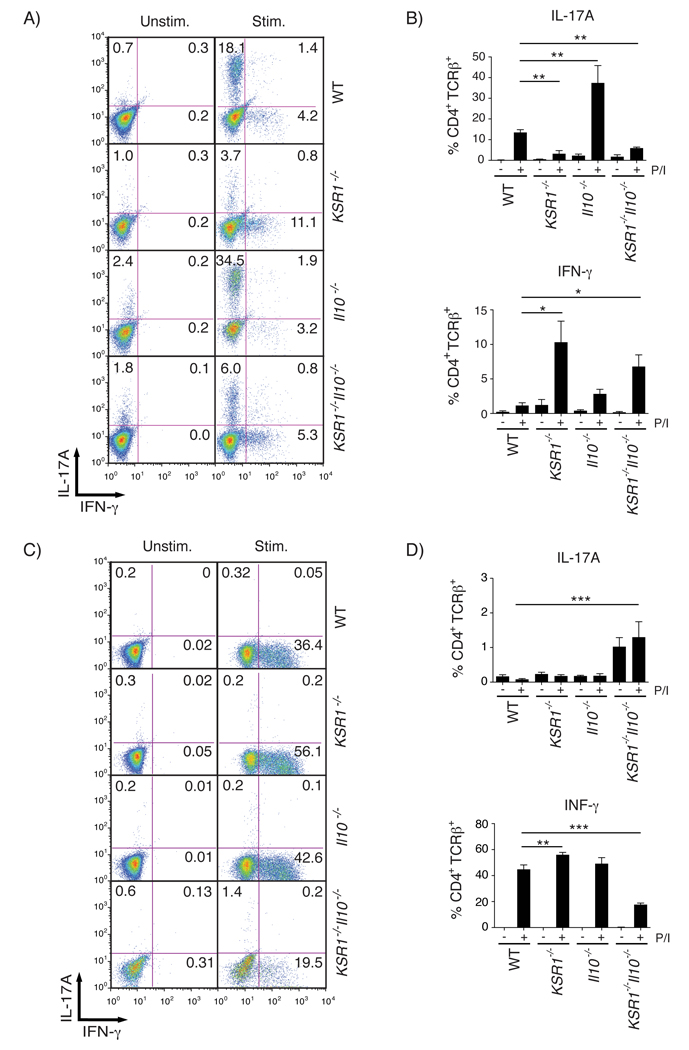
Splenocytes isolated from WT, KSR1−/−, Il10−/−, and KSR1−/−Il10−/− mice were cultured under Th17 or Th1 polarizing conditions. Lymphocytes were stained for cell surface CD4 and TCRβ and intracellular IFN-γ and IL-17A. Samples were analyzed by flow cytometry gated on lymphocyte geometry and CD4+TCRβ+ cell surface staining. A) Representative flow cytometry dot plots of unstimulated and stimulated T cells cultured under Th17 polarizing conditions. B) Intracellular cytokine staining on Th17 polarized cells was quantified and reported as the percent CD4+TCRβ+ cells staining positive for IL-17A or IFN-γ. Bar graphs represent the mean for each cultured T cell population from two independent experiments. Error bars are the SEM. C) Representative flow cytometry dot plots of unstimulated and stimulated T cells cultured under Th1 polarizing conditions. D) Intracellular cytokine staining on Th1 polarized cells was quantified and reported as the percent CD4+TCRβ+ cells staining positive for IL-17A or IFN-γ. Bar graphs represent the mean for each cultured T cell population. Error bars are the SEM. * P < 0.05, ** P < 0.01, *** P < 0.001
Neutralization of IFN-γ attenuates severity of disease in KSR1−/−Il10−/− mice
Our data showing that colons of KSR1−/− and KSR1−/−Il10−/− mice had increased in IFN-γ transcript and protein levels, together with the data indicating that KSR1−/− and KSR1−/−Il10−/− CD4+TCRβ+ T cells have enhanced IFN-γ production, suggest a potential mechanism driving the pathogenesis of colitis in KSR1−/−Il10−/− mice. Since colitis in KSR1−/−Il10−/− mice was detected by 4 weeks of age, we administered intraperitoneal injections of anti-IgG (αIgG), anti-IFN-γ (αIFN-γ), or anti-IL-17A (αIL-17A) neutralizing antibodies in 3-week old KSR1−/−Il10−/− mice twice per week for 3 weeks to determine the role of IFN-γ in disease KSR1−/−Il10−/− pathogenesis. Three days following the final antibody injection mice were sacrificed and colons were flushed, measured, and weighed and then paraffin embedded. The colon length to weight ratio for mice receiving αIFN-γ was increased (26.92 ± 1.86) compared to mice receiving αIgG or αIL-17A (18.70 ± 0.90 and 21.60 ± 1.29 respectively), indicative of reduced inflammation (Figure 6A). H&E stained mouse colon sections were obtained and scored for inflammation and injury as before. While neutralization of IL-17A caused no statistical decrease in colitis severity over isotype control (8.75 ± 1.5 vs. 9.75 ± 1.0), treatment with αIFN-γ significantly reduced the severity of colitis (6.3 ± 1.1) in KSR1−/−Il10−/− mice (Figures 6B & 6C). In addition, cytokine transcript levels were measured for TNF, IFN-γ, IL-1β, IL-17A, IL-22, and KC for all three groups of mice. While neutralizing IFN-γ resulted in diminished TNF and IFN-γ mRNA levels, neutralizing IL-17A significantly reduced KC transcription, indicating IL-17A inhibition (Supplemental Figure 4). Collectively, these data indicate that the severity of colitis in KSR1−/− Il10−/− mice is mediated, at least in part, by the increase in IFN-γ that is associated with loss of KSR1.
Figure 6. Neutralizing IFN-γ attenuates colitis in KSR1−/−Il10−/− mice.
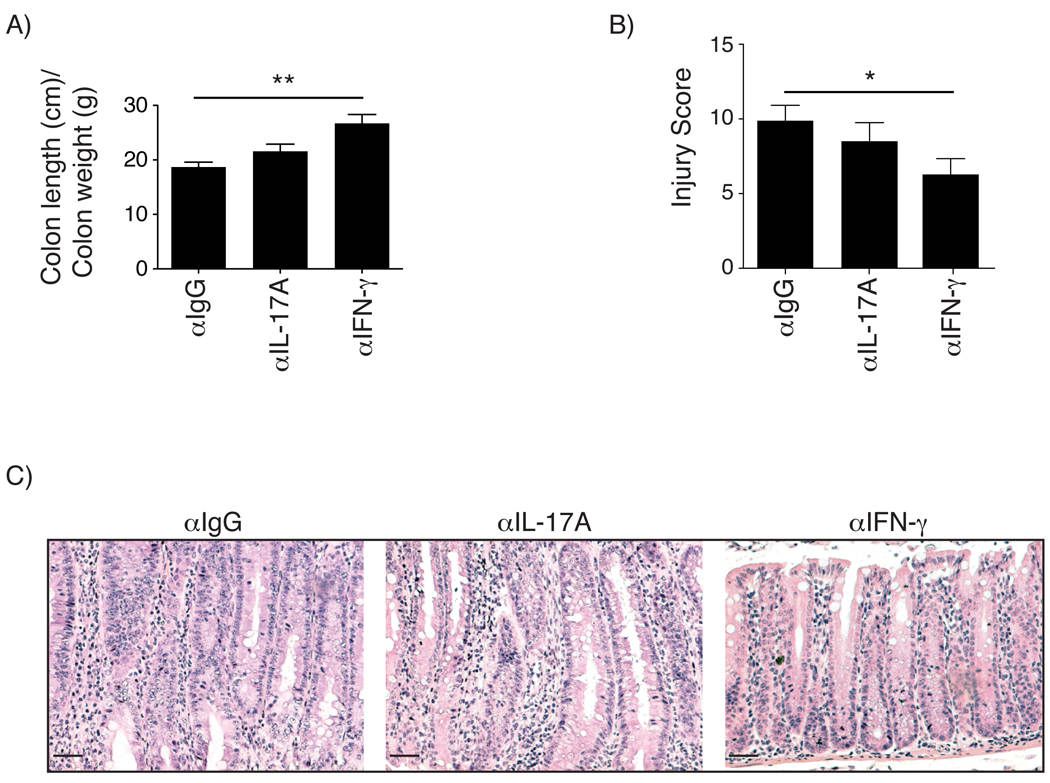
3-week old KSR1−/−Il10−/− mice were administered 100 µg/mouse neutralizing antibodies against αIgG, αIFN-γ, or αIL-17A intraperitoneally twice per week for a period of 3 weeks and assessed for colitis. A) Mouse colons were removed, flushed, weighed, and measured from the cecum to anus. Solid bars represent the mean length/weight ratio. Error bars are the SEM. B) Colon sections were scored as before for inflammation and injury. Solid bars represent the mean and error bars represent the SEM. C) Representative H&E stained colon sections from KSR1−/−Il10−/− mice administered αIgG, αIFN-γ, or αIL-17A neutralizing antibody. * P < 0.05, ** P < 0.01
Discussion
While distinct roles for KSR1 have been reported for intestinal epithelial cells,12, 13 T cell proliferation,26 T cell differentiation,27, 28 and recently NK cell-mediated cytolysis,29 the role of KSR1 in inflammatory diseases has not been defined. In this study, we utilized the IL-10 deficiency-induced mouse model of spontaneous experimental colitis to investigate the role of KSR1 during chronic inflammation. We found that KSR1−/−Il10−/− mice developed accelerated severe spontaneous colitis with 100% penetrance by 4 weeks of age. The pathogenesis of the disease was predominantly attributed to loss of KSR1 in hematopoietic lineages. In addition, IFN-γ transcript and protein levels were elevated in colons of KSR1−/− and KSR1−/−Il10−/− mice. The percent of IFN-γ producing TCRβ+ T cells isolated from the lamina propria of KSR1−/− and KSR1−/−Il10−/− mice were increased compared to WT and Il10−/− mice. KSR1 deficient naïve T cells also exhibited a greater propensity to develop along the Th1 axis in vitro while in vitro Th17 development was impaired. Finally, administration of αIFN-γ neutralizing antibody attenuated colitis severity in KSR1−/−Il10−/− mice. Collectively, the data presented here implicate KSR1 as a regulatory molecule that functions to suppress IFN-γ production in T cells and promotes Th1/Th17 developmental homeostasis.
Early reports characterizing the spontaneous colitis that develops in Il10−/− mice established the requirement for intestinal bacterial flora in mediating disease.7, 8 Since KSR1−/− Il10−/− mice develop colitis by 4 weeks of age and enteric microbial populations are established just post-weaning,30 we initially suspected that loss of KSR1 might contribute to epithelial barrier defects. Though epithelial barrier permeability was increased in diseased 10-week old KSR1−/−Il10−/− mice, barrier permeability in 10-week old KSR1−/− and Il10−/− mice were similar to WT mice (Supplemental Figure 1A). Although KSR1 does not appear to be required for epithelial barrier function in the absence of challenging conditions, we cannot rule out the effect of KSR1 deficiency in the presence of inflammatory conditions since KSR1 protects colon epithelial cells from TNF-induced apoptosis.12, 13
We were surprised, however, to find that the protective role of KSR1 was attributed to expression in cells of the immune system (Figure 2). Interestingly, Il10−/− BM was insufficient to cause disease in irradiated WT mice, and is consistent with a previous report.31 The finding that KSR1−/−Il10−/− BM was sufficient to cause disease in irradiated recipient WT mice established a role for KSR1 in disease pathogenesis and immune cell function. Since IFN-γ and IL-17A transcripts were elevated in the colons of disease-free KSR1−/− mice and diseased KSR1−/−Il10−/− mice (Figure 3A), it seemed plausible that KSR1 was involved in T cell development or function. Though current data on the pathogenesis of IL-17A in inflammatory diseases remains unclear,32–35 the colitis in KSR1−/−Il10−/− mice appears to be exacerbated by IFN-γ and not IL-17A (Figure 6). The observation that IFN-γ production and in vitro Th1 polarization was elevated in KSR1 deficient T cells suggests a potential pathway by which KSR1 deficiency might exacerbate colitis in KSR1−/−Il10−/− mice (Figures 4B & 5D). KSR1 expression modifies signal transduction pathways through association with multiple protein kinases and protein phosphatases including protein phosphatase 2A (PP2A) and 2B (PP2B).36,37, 38 Suppression of PP2A activity increases IFN-γ production in NK cells.39 It is attractive to speculate that antigenic stimulus in the absence of KSR1 perturbs signaling pathways normally regulated by protein phosphatases. It will be interesting to see if KSR1-associated phosphatases in T lymphocytes are involved in this process.
The crosstalk between innate immune defenses and acquired immunity is actively being investigated for autoimmune diseases and IBD.40–42 While our data support a role for KSR1 in lymphocyte function in IL-10 deficiency-induced colitis, we cannot exclude a role for KSR1 in innate immune cell function. Consistent with a functional role for KSR1 in innate immune system, we observed decreased nitric oxide production from stimulated bone marrow derived KSR1−/− macrophages (unpublished observations). In a secondary mouse model of colitis using dextran sulfate sodium (DSS), which is not dependent on lymphocyte function, 43 KSR1−/− mice also exhibited greater sensitivity to DSS-induced injury compared to WT mice (Supplemental Figure 5). While emerging data implicate KSR1 in innate immune cell function,29 further investigations are necessary to elucidate if KSR1 is involved in innate immune responses and susceptibility to other models of inflammation-associated diseases.
Finally, there may be a connection between vitamin D and KSR1 in immune regulation. Vitamin D receptor (VDR) signaling regulates T cell development and helps maintain immunological tolerance with implications for multiple sclerosis, type-1 diabetes mellitus, and IBD.44, 45 Recently, polymorphisms at the VDR locus were linked to IBD and other autoimmune disorders.46 Vitamin D supplementation holds promise as a therapeutic agent in the treatment of Crohn’s disease by increasing NOD2 expression that then couples to the expression the antimicrobial peptide defensin β2.47 Interestingly, the KSR1 promoter region contains a vitamin D responsive element and moreover, KSR1 protein is upregulated by 1, α25-dihydroxyvitamin D3.28 In fact, VDR−/−Il10−/− mice develop severe accelerated spontaneous colitis harboring many phenotypic similarities to those observed in KSR1−/−Il10−/− mice including increased levels of IFN-γ.48, 49 It is attractive to speculate that vitamin D-mediated suppression of pathogenic immune responses is, in part, regulated by KSR1 expression and suppression of IFN-γ production in Th1 effector cells.
We conclude that KSR1 expression suppresses IFN-γ production in T lymphocytes and promotes T cell developmental homeostasis along the Th1/Th17 axis. Therefore, induction of KSR1 expression may be an ideal strategy for modulating IFN-γ in Th1-mediated diseases.
Supplementary Material
Acknowledgements
We thank Valerie Hilliard for helpful critique of this manuscript, Amy Major for assistance with bone marrow transplantation, Abudi Nashabi and Shivesh Punit for their excellent technical assistance. We also thank the Vanderbilt Immunohistochemistry and Human Tissue Acquisition Core facilities.
Grant Support:
This work was supported by NIH grants DK066176 (D.B.P.), AI072417 (L.V.K.), R01AT004821 and R01DK053620 (K.T.W), pilot and feasibility project from the Digestive Disease Research Center supported by NIH grant P30DK058404, and by the Medical Research Service of the Department of Veterans Affairs (C.D.A.-2, H.M.S.A.).
Abbreviations
- CD
cluster of differentiation
- DSS
dextran sulfate sodium
- FITC
fluorescein isothiocyanate
- H&E
Hematoxylin and Eosin
- IBD
inflammatory bowel disease
- IFN-γ
interferon-gamma
- IgG
Immunoglobulin G
- IL-10
interleukin-10
- IL-17A
interleukin-17A
- KSR1
kinase suppressor of Ras 1
- NK
natural killer
- qRT-PCR
quantitative real-time polymerase chain reaction
- SPF
specific pathogen free
- TCR
T cell receptor
- Th
T helper
- TNF
tumor necrosis factor
- VDR
Vitamin D receptor
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors do not have any conflicts of interest to disclose
Author contributions:
J.A.G. wrote the paper, J.A.G., H.M.S.A., D.O.V. designed experiments and acquired the data, M.K.W., R.C., K.T.W., and L.V.K. assisted with data interpretation, and D.B.P supervised the study.
References
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 2.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–G17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 3.Rennick D, Davidson N, Berg D. Interleukin-10 gene knock-out mice: a model of chronic inflammation. Clin Immunol Immunopathol. 1995;76:S174–S178. doi: 10.1016/s0090-1229(95)90144-2. [DOI] [PubMed] [Google Scholar]
- 4.Scheinin T, Butler DM, Salway F, Scallon B, Feldmann M. Validation of the interleukin-10 knockout mouse model of colitis: antitumour necrosis factor-antibodies suppress the progression of colitis. Clin Exp Immunol. 2003;133:38–43. doi: 10.1046/j.1365-2249.2003.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 7.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 8.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Yao B, Delikat S, Bayoumy S, Lin XH, Basu S, McGinley M, Chan-Hui PY, Lichenstein H, Kolesnick R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell. 1997;89:63–72. doi: 10.1016/s0092-8674(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 10.Michaud NR, Therrien M, Cacace A, Edsall LC, Spiegel S, Rubin GM, Morrison DK. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc Natl Acad Sci USA. 1997;94:12792–12796. doi: 10.1073/pnas.94.24.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zafrullah M, Yin X, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Kinase suppressor of Ras transphosphorylates c-Raf-1. Biochemical and Biophysical Research Communications. 2009;390:434–440. doi: 10.1016/j.bbrc.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan F, John SK, Polk DB. Kinase suppressor of Ras determines survival of intestinal epithelial cells exposed to tumor necrosis factor. Cancer Res. 2001;61:8668–8675. [PubMed] [Google Scholar]
- 13.Yan F, John SK, Wilson G, Jones DS, Washington MK, Polk DB. Kinase suppressor of Ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. Journal of Clinical Investigation. 2004;114:1272–1280. doi: 10.1172/JCI21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker C, Fantini MC, Neurath MF. High resolution colonoscopy in live mice. Nat Protoc. 2006;1:2900–2904. doi: 10.1038/nprot.2006.446. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy RJ, Hoper M, Deodhar K, Erwin PJ, Kirk SJ, Gardiner KR. Interleukin 10-deficient colitis: new similarities to human inflammatory bowel disease. The British journal of surgery. 2000;87:1346–1351. doi: 10.1046/j.1365-2168.2000.01615.x. [DOI] [PubMed] [Google Scholar]
- 16.Olivares-Villagómez D, Mendez-Fernandez YV, Parekh VV, Lalani S, Vincent TL, Cheroutre H, Van Kaer L. Thymus leukemia antigen controls intraepithelial lymphocyte function and inflammatory bowel disease. Proc Natl Acad Sci USA. 2008;105:17931–17936. doi: 10.1073/pnas.0808242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heuschkel R, Salvestrini C, Beattie RM, Hildebrand H, Walters T, Griffiths A. Guidelines for the management of growth failure in childhood inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:839–849. doi: 10.1002/ibd.20378. [DOI] [PubMed] [Google Scholar]
- 18.MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–305. doi: 10.1111/j.1365-2249.1990.tb03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2-and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–131. [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 22.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koenders MI, Joosten LA, van den Berg WB. Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann Rheum Dis. 2006;65 Suppl 3:iii29–iii33. doi: 10.1136/ard.2006.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagi Y, Andoh A, Inatomi O, Tsujikawa T, Fujiyama Y. Inflammatory responses induced by interleukin-17 family members in human colonic subepithelial myofibroblasts. J Gastroenterol. 2007;42:746–753. doi: 10.1007/s00535-007-2091-3. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen A, Burack WR, Stock JL, Kortum R, Chaika OV, Afkarian M, Muller WJ, Murphy KM, Morrison DK, Lewis RE, McNeish J, Shaw AS. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002;22:3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Studzinski GP. Kinase suppressor of RAS (KSR) amplifies the differentiation signal provided by low concentrations 1,25-dihydroxyvitamin D3. J. Cell. Physiol. 2004;198:333–342. doi: 10.1002/jcp.10443. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Wang T-T, White JH, Studzinski GP. Induction of kinase suppressor of RAS-1(KSR-1) gene by 1, alpha25-dihydroxyvitamin D3 in human leukemia HL60 cells through a vitamin D response element in the 5'-flanking region. Oncogene. 2006;25:7078–7085. doi: 10.1038/sj.onc.1209697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giurisato E, Lin J, Harding A, Cerutti E, Cella M, Lewis RE, Colonna M, Shaw AS. The mitogen-activated protein kinase scaffold KSR1 is required for recruitment of extracellular signal-regulated kinase to the immunological synapse. Mol Cell Biol. 2009;29:1554–1564. doi: 10.1128/MCB.01421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savage DC, Dubos R, Schaedler RW. The gastrointestinal epithelium and its autochthonous bacterial flora. J Exp Med. 1968;127:67–76. doi: 10.1084/jem.127.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bamba S, Lee C-Y, Brittan M, Preston SL, Direkze NC, Poulsom R, Alison MR, Wright NA, Otto WR. Bone marrow transplantation ameliorates pathology in interleukin-10 knockout colitic mice. J. Pathol. 2006;209:265–273. doi: 10.1002/path.1967. [DOI] [PubMed] [Google Scholar]
- 32.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 34.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 35.Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, Waisman A. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–69. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugimoto T, Stewart S, Han M, Guan KL. The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. EMBO J. 1998;17:1717–1727. doi: 10.1093/emboj/17.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13:1356–1364. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- 38.Stewart S, Sundaram M, Zhang Y, Lee J, Han M, Guan KL. Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell Biol. 1999;19:5523–5534. doi: 10.1128/mcb.19.8.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trotta R, Ciarlariello D, Dal Col J, Allard J, 2nd, Neviani P, Santhanam R, Mao H, Becknell B, Yu J, Ferketich AK, Thomas B, Modi A, Blaser BW, Perrotti D, Caligiuri MA. The PP2A inhibitor SET regulates natural killer cell IFN-gamma production. J Exp Med. 2007;204:2397–2405. doi: 10.1084/jem.20070419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19:353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Kalyan S, Chow AW. Linking innate and adaptive immunity: human Vgamma9Vdelta2 T cells enhance CD40 expression and HMGB-1 secretion. Mediators Inflamm. 2009;2009:819408. doi: 10.1155/2009/819408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pletneva M, Fan H, Park JJ, Radojcic V, Jie C, Yu Y, Chan C, Redwood A, Pardoll D, Housseau F. IFN-producing killer dendritic cells are antigen-presenting cells endowed with T-cell cross-priming capacity. Cancer Res. 2009;69:6607–6614. doi: 10.1158/0008-5472.CAN-09-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krieglstein CF, Cerwinka WH, Sprague AG, Laroux FS, Grisham MB, Koteliansky VE, Senninger N, Granger DN, de Fougerolles AR. Collagen-binding integrin alpha1beta1 regulates intestinal inflammation in experimental colitis. J Clin Invest. 2002;110:1773–1782. doi: 10.1172/JCI200215256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cantorna MT. Vitamin D and its role in immunology: multiple sclerosis, and inflammatory bowel disease. Prog Biophys Mol Biol. 2006;92:60–64. doi: 10.1016/j.pbiomolbio.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 45.Ginanjar E, Sumariyono, Setiati S, Setiyohadi B. Vitamin D and autoimmune disease. Acta Med Indones. 2007;39:133–141. [PubMed] [Google Scholar]
- 46.Naderi N, Farnood A, Habibi M, Derakhshan F, Balaii H, Motahari Z, Agah MR, Firouzi F, Rad MG, Aghazadeh R, Zojaji H, Zali MR. Association of vitamin D receptor gene polymorphisms in Iranian patients with inflammatory bowel disease. J Gastroenterol Hepatol. 2008;23:1816–1822. doi: 10.1111/j.1440-1746.2008.05525.x. [DOI] [PubMed] [Google Scholar]
- 47.Wang T-T, Dabbas B, Laperriere D, Bitton AJ, Soualhine H, Tavera-Mendoza LE, Dionne S, Servant MJ, Bitton A, Seidman EG, Mader S, Behr MA, White JH. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem. 2010;285:2227–2231. doi: 10.1074/jbc.C109.071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol. 2003;17:2386–2392. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 49.Froicu M, Zhu Y, Cantorna MT. Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology. 2006;117:310–318. doi: 10.1111/j.1365-2567.2005.02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.