

Abstract
Despite the high transduction efficiency in vivo, the application of recombinant E1-deleted adenoviral vectors for in vivo gene therapy has been limited by the attendant innate and adaptive immune responses to adenoviral vectors. Natural killer (NK) cells have been shown to play an important role in innate immune elimination of adenoviral vectors in vivo. However, the mechanisms underlying NK cell activation and function in response to adenoviral vectors remain largely undefined. In this study, we showed that NK cell activation upon adenoviral infection was dependent on accessory cells such as dendritic cells (DCs) and macrophages, and that cell-contact dependent signals from the accessory cells are necessary for NK cell activation. We further demonstrated that ligands of the NK activating receptor, NKG2D, were upregulated in accessory cells upon adenoviral infection and that blockade of NKG2D inhibited NK cell activation upon adenoviral infection, leading to a delay in adenoviral clearance in vivo. In addition, NKG2D was required for NK cell-mediated cytolysis on adenovirus-infected targets. Taken together, these results suggest that efficient NK cell activation and function in response to adenoviral infection is critically dependent on the NKG2D pathway and may help design effective strategies to improve the outcome of adenovirus-mediated gene therapy.
Introduction
Recombinant E1-deleted adenoviruses have been developed for gene therapy due to their high transduction efficiency in vivo. However, significant problems of attendant innate and adaptive immune responses to adenoviral vectors have limited their applications for in vivo gene therapy (1). While the CD8+ and CD4+ T cell responses against both the viral antigens and the transgene contribute significantly to the adaptive immune elimination of adenoviral vectors (2-4), emerging evidence has suggested a critical role for NK cells in innate immune elimination of adenoviral vectors (5-7), leading to the loss of adenoviral genome and the reduction of transgene expression (7). Although our recent work shows that type I interferons (IFNs) are important for the regulation of NK cell activation (7), how NK cells are activated in response to adenoviral infection remains largely unknown. Delineation of this process may help design effective strategies to improve the outcome of adenovirus-mediated gene therapy.
NK cells represent an important component of the innate immune system and play a critical role in innate immune defense against various viral infections in vivo (8, 9). The activation of NK cells is tightly controlled by both inhibitory and activating receptors (10). During murine CMV (MCMV) infection, NK cell activation is mediated by the NK cell activating receptor, Ly49H, which specifically recognizes the m157 gene product of MCMV expressed on the surface of infected cells (11, 12). Similarly, recognition of influenza virus hemagglutinin on virus-infected cells by another activating receptor, NKp46, activates lysis by human NK cells (13), and the murine NKp46 equivalent, NCR1, is required for protection against lethal influenza infection (14). In addition to these pathogen-specific NK cell activation pathways, the NKG2D activating receptor has been shown to recognize host stress proteins induced upon viral infections and play an important role in the control of human CMV and MCMV infections (15, 16).
In this study, we showed that NK cell activation upon adenoviral infection was also dependent on accessory cells such as dendritic cells (DCs) and macrophages, and that the NK cell-accessory cell contact is necessary for NK cell activation in vitro. We further demonstrated that accessory cells upregulated NKG2D ligands upon adenoviral infection and that NKG2D blockade inhibited NK cell activation both in vitro and in vivo, leading to a delay in the clearance of adenoviral vectors. In addition, NKG2D was critical for NK cell-mediated cytolysis on adenovirus-infected targets. Collectively, these results suggest that efficient NK cell activation and function in response to adenoviral infection depends on the NKG2D pathway.
Materials and Methods
Mice
C57BL/6 mice were purchased from the Jackson Laboratories. RAG-2 deficient (RAG-2−/−) mice were purchased from Taconic. Mice used were between 6 and 10 wk of age. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Duke University.
Recombinant E1-deleted adenovirus
Recombinant E1- and E3-deleted adenovirus encoding LacZ (Ad-LacZ) under the control of the cytomegalovirus promoter was generated and purified as described previously (17). For in vivo studies, mice were infected by intravenous injection of 2 ×109 pfu Ad-LacZ in 100 μl of phosphate-buffered saline. After 24 h, splenocytes or intrahepatic lymphocytes were harvested for further analyses. For in vitro stimulation, cells were infected with Ad-LacZ at multiplicity of infection (MOI) of 400 at 37 °C for 18 h.
Preparation of dendritic cells (DCs), macrophages, and B cells
Plasmacytoid DCs (pDCs) were generated as described (17). Briefly, bone marrow cells were harvested from femurs and tibiae of C57BL/6 mice and cultured in the presence of 100 ng/ml of Flt-3 ligand (R & D Systems) for 9 days. For generation of cDCs, bone marrow cells were cultured in the presence of mouse GM-CSF (1,000 U/ml) and IL-4 (500 U/ml) (R & D Systems) for 5 days as described (18). pDCs and cDCs were stained with anti-B220-FITC and anti-CD11c-PE and purified by FACS sorting. The purity of cells was 95%-97%. Macrophages were isolated from the peritoneal cavity of mice 2 days after intraperitoneal injection of 1.5 ml of 3 % thioglycollate as described (19). B220+ B cells were purified from the spleen by double selection with B220-microbeads.
NK cell and accessory cell co-culture system
NK cells were enriched from spleens of C57BL/6 mice by positive selection with phycoerythrin-conjugated anti-DX5 and anti-phycoerythrin microbeads (Miltenyi Biotec). The cells were stained with phycoerythrin-Cy5-conjugated anti-CD3ε and then sorted for NK cells (DX5+CD3−). The purity is more than 95%. 1 × 105 sorted NK cells were cultured in 96-well U-bottom tissue culture plates in 100 μl RPMI 1640 medium supplemented with 10% FCS, 2 mM L-glutamine, 10 mM HEPES, 10 μM β-mercaptoethanol and 100 IU/ml penicillin/streptomycin. 2 × 105 of accessory cells (pDCs, cDCs or macrophages) in 100 μl were added to the same wells in the presence or absence of Ad-LacZ at MOI of 400 for 18 h.
For blocking experiments, 10 μg/ml of anti-mouse NKG2D mAb (CX5, eBioscience) or anti-mouse CD226 (10E5, eBioscience), or 30 μg/ml of recombinant mouse NKp46/NCR1/Fc Chimera (R & D systems) was added to the culture as described (20-22).
Intracellular IFN-γ and Granzyme B staining
For ex vivo experiments, splenocytes were restimulated for 4 h in the presence of 5 μg/ml Brefeldin A (Sigma-Aldrich), 40 ng/ml PMA (Sigma-Aldrich) and 100 ng/ml ionomycin (Sigma-Aldrich). After staining of NK cell surface markers (DX5+CD3−), the cells were fixed with CytopermCytofix (BD Biosciences) for 20 min and incubated for 30 min with APC-conjugated rat anti–mouse IFN-γ and FITC-conjugated anti-granzyme B. The cells were then subject to flow cytometry analysis.
For NK co-culture experiments in vitro, only Brefeldin A (5 μg/ml) was added 4 h before the harvest. The cells were harvested and stained for intracellular IFN-γ and granzyme B as above.
Transwell experiments
Transwell experiments were performed in 24-well plates (Corning, Acton, MA). Peritoneal macrophages were plated in the lower wells (2× 106) and NK cells (1× 106) were added to the upper wells in the presence or absence of Ad-LacZ (MOI of 400). After culturing for 18 hours, cells were harvested for intracellular cytokine or cell surface marker staining.
Antibodies and Flow cytometry
The following antibodies were used for flow cytometry: Phycoerythrin (PE)-conjugated anti-Rae-1 (CX1), MULT1 (5D10), and anti-CD49b (DX5); Phycoerythrin-Cy5 (PE-Cy5)-conjugated anti-CD3ε (145-2C11); Fluorescein isothiocyanate (FITC)-conjugated anti-granzyme B (16G6), anti-CD11c (HL3), anti-CD11b (M1/70), and anti-CD69 (H1.2F3); Allophycocyanin (APC)-conjugated rat anti–mouse IFN-γ (XMG1.2), anti-CD11b (M1/70). These antibodies were purchased from BD Biosciences or eBioscience. APC-conjugated anti-F4/80 (CI:A3-1) was purchased from AbD Serotec. FACS Canto (Becton Dickinson, Franklin Lakes, NJ) was used for flow cytometry event collection, which was analyzed using FACS Diva software (Becton Dickinson).
Semiquantitative RT-PCR analysis for NKG2D ligand expression
Total RNA samples were prepared from spleens using Trizol reagent and treated with DNase I (Roche). RNA samples (1 μg) were then converted to first-strand cDNAs using oligo(dT)15 primers and Superscript III reverse transcriptase (Promega). PCR amplification was set up on serial 3-fold dilutions of cDNA using the primer pairs specific for nine NKG2D ligand genes shown in Table 1. RT-PCR products were electrophoresed through a 2% agarose gel.
Table 1.
Oligonucleotide primer pairs for semiquantitative PCR analysis.
| Gene name | Forward primer | Reverse primer |
|---|---|---|
| Rae-1α | GGCCGCTGTAGTCAGTTACC | GAGTGCTCCCAGCTGTTTTC |
| Rae-1β | GCAAATGCCACTGAAGTGAA | CCAGGTGGCACTAGGAGTTC |
| Rae-1γ | CTCCTACCCCAGCAGATCCT | TTCAATCGTTGCACAAGGTC |
| Rae-1δ | CTCCTACCCCAGCAGATGAA | TTGCGGATAAATCATGGTGA |
| Rae-1ε | GCTATTGCGGAGGATTTCAG | TCCACTGAGCACTTCACGTC |
| Mult-1 | CATGCCATTGGTGCTCATAG | TGCTTGTGTCAACACGGAAT |
| H60a | ACCAGGGTCTGAGTGTCACC | CGTTTCCTTTGCTGGAGAAG |
| H60b | GGGTCTGAGTTTCACCTGGA | TGAAGATGCACAGGCAATGT |
| H60c | CTCCAGTCATCGAAGGCAAT | ACAGCATAGTGGCATTTCCC |
| β-actin | CTCTCAGCTGTGGTGGTGAA | AGCCATGTACGTAGCCATCC |
Note: All sequences are presented in the 5′-3′ direction.
In vivo antibody blocking
Mice were injected twice with 100 μg of anti-NKG2D mAb (CX5, eBioscience) or a control antibody (Rat IgG1) 24 h and 6 h before adenoviral infection.
NK cell cytotoxicity assay
NK cell cytotoxicity assay was performed as described (7). Briefly, peritoneal macrophages infected with Ad-LacZ at an MOI of 100 for 18 hours were used as target cells. 1×106 target cells were labeled with 100 μCi of Na2[51Cr]O4(51Cr) for 1 h at 37 °C. NK cells were enriched from spleens by positive selection twice with PE-conjugated anti-DX5 and anti-PE microbeads. NK cells were incubated with target cells (1×104/well) labeled with 51Cr for 4 h at various effector-target (E:T) ratios as indicated in triplicate wells. The supernatants were obtained after the incubation and subjected to gamma counting. The maximum or spontaneous release was defined as counts from samples incubated with 1% SDS or medium alone, respectively. Cytolytic activity was calculated with the following formula: % specific lysis = (experimental - spontaneous) release×100/(maximum - spontaneous) release.
Detection of adenoviral genomic DNA by real-time PCR
Total genomic DNA was isolated from the liver tissue as described previously (7). Real-time quantitative PCR was used to measure the amount of adenoviral genomic DNA in liver tissues using primers located in the fiber gene from adenovirus and mouse β-actin gene. Amounts of adenoviral DNA were normalized to β-actin gene within each sample. The sequences of the forward and reverse primers for adenoviral fiber gene were 5′-CCACCGATAGCAGTACCCTT-3′ and 5′-GACCAGTTGCTACGGTCAAA-3′, respectively. Normalized value for adenoviral DNA in each sample was calculated as the relative quantity of adenoviral DNA divided by the relative quantity of β-actin gene.
X-gal staining
The sections of fresh-frozen liver tissue (5 μm) were fixed in 0.5% glutaraldehyde for 10 min, rinsed twice for 10 min in PBS containing 1 mM MgCl2 and incubated in 1 mg/ml of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, and 1 mM MgCl2 in PBS for 2 h at 37°C. Sections were counterstained in hematoxylin and examined microscopically.
β-Galactosidase activity in liver tissues
This assay was performed as described (7). Briefly, liver tissues were homogenized in reporter lysis buffer on ice. Homogenates were then clarified by centrifugation and supernatant was recovered. Total protein concentration was measured in liver homogenate using a standard Bradford protein assay (Bio-Rad). β-galactosidase activity was evaluated, using β-galactosidase enzyme assay (Promega), under conditions recommended by the manufacturer. The absorbance was read at 420 nm in a plate and the results were expressed as units per gram of liver protein.
Statistical analysis
Results were expressed as mean ± SD. Differences between groups were examined for statistical significance using the Student t-test.
Results
NK cell activation upon adenoviral infection is dependent on accessory cells
Accessory cells such as DCs and macrophages have been implicated in NK cell activation (23). To study whether accessory cells are required in the activation of NK cells in response to adenoviral infection, freshly sorted DX5+CD3− NK cells were stimulated with Ad-lacZ in the presence or absence of macrophages, plasmacytoid DCs (pDCs), conventional DCs (cDCs) or B cells. NK cells were then harvested 18 h later and assayed for the expression of the activation marker CD69, as well as the production of effector molecules, granzyme B and IFN-γ. NK cells stimulated with Ad-lacZ alone did not significantly upregulate CD69 (Fig. 1A), or produce granzyme B (Fig. 1B, C) and IFN-γ (Fig. C), compared to the medium control (Figure 1A, B). However, addition of macrophages, pDCs and cDCs, but not B cells, stimulated NK cells to produce significantly (p <0.001) higher levels of CD69 (Fig. 1A), granzyme B (Fig. 1B, C) and IFN-γ (Fig. 1C). These results indicate that accessory cells such as macrophages and DCs are required for the activation of NK cells in response to adenoviral infection.
FIGURE 1.

Accessory cells are required for NK cell activation in response to adenoviral infection. Purified splenic NK cells (DX5+CD3−) were either cultured alone (NK alone), or co-cultured with peritoneal macrophages (+Mφ), pDCs (+pDCs), cDCs (+cDCs) or B cells (+B cells) in the presence (Ad) or absence (Medium) of Ad-LacZ. 18 h later, brefeldin A was added into each well and incubated for additional 4 h. (A) NK cells were analyzed for the expression of the early activation marker, CD69, by flow cytometry. The mean fluorescent intensity (MFI) of CD69 staining is indicated. Events were gated on DX5+CD3− NK cells. (B-C) NK cells were stained intracellularly for IFN-γ and granzyme B and analyzed by flow cytometry. FACS plots of granzyme B production by NK cells with the percentage of granzyme B positive cells among DX5+CD3− NK cells indicated (B). The mean percentage ± SD of granzyme B or IFN-γ positive cells among DX5+CD3− cells is indicated (n = 5 per group) (C). The data shown are representative of four independent experiments.
NKG2D is required for efficient NK cell activation in response to adenovirus
What are the underlying mechanisms responsible for the accessory cell-dependent NK cell activation to E1-deleted adenovirus? Accessory cells may provide indirect (soluble) and/or direct (contact-dependent) signals to NK cells necessary for their activation. Indeed, we have shown that adenovirus can induce pDCs, cDCs and macrophages to produce type I IFNs (17), and that type I IFNs are critical for NK cell activation and function in response to adenoviral infection (7). To examine whether accessory cells also provided direct contact-dependent signals for NK cell activation upon adenoviral infection, purified NK cells were co-cultured with macrophages either mixed together or separated by a transwell, followed by stimulation with Ad-LacZ for 18 h. Our data showed that the expression of CD69 (Fig. 2A) and the production of IFN-γ and granzyme B (Fig. 2B, C) by NK cells were significantly (p <0.001) reduced in the transwell culture, compared to the mixed culture control. These results suggest that NK cell activation in response to adenovirus also requires contact-dependent signals from the accessory cells.
FIGURE 2.

Accessory cell-NK cell contact is necessary for NK cell activation in vitro. Purified NK cells (DX5+CD3−) were co-cultured with macrophages either mixed together (Mixed) or separated by a transwell system (Transwell), followed by stimulation with Ad-LacZ for 18 h (Ad), or left unstimulated (Medium). Brefeldin A was added into each well and incubated for additional 4 h. (A) DX5+CD3− NK cells were analyzed for the expression of CD69. MFI of CD69 staining is indicated. (B-C) DX5+CD3− NK cells were analyzed for intracellular IFN-γ and granzyme B production. FACS plots of IFN-γ and granzyme B production with the percentage of IFN-γ and granzyme B positive cells indicated (B). The mean percentage ± SD of IFN-γ or granzyme B positive cells is indicated (n = 5 per group) (C). The data shown are representative of three independent experiments.
We next determined what contact-dependent signal(s) was required for adenovirus-induced NK cell activation. The NK cell receptors such as NKG2D, NKp46, and DNAM-1 (CD226), have been shown to play an important role in NK cell activation and function (14-16, 24). We thus investigated the role of NKG2D, NKp46 and DNAM-1 in NK cell activation upon adenoviral infection using a blocking anti-NKG2D or anti-CD226 mAb, or a blocking NKp46-Fc fusion protein. We showed that addition of anti-NKG2D mAb, but not anti-CD226 mAb or NKp46-Fc, to the NK cell-macrophage co-culture markedly inhibited the expression of CD69 (Fig. 3A) and the production of IFN-γ and granzyme B (Fig. 3B, C) by NK cells compared to the control rat IgG-treated group. These results suggest that NKG2D signaling is critical for NK cell activation upon adenoviral infection in vitro.
FIGURE 3.

NKG2D is critical for NK cell activation in response to adenovirus in vitro. Purified DX5+CD3− NK cells were co-cultured with macrophages, and with stimulated Ad-LacZ for 18 h (Ad), or left unstimulated (Medium). In some cultures, anti-NKG2D (+Anti-NKG2D), anti-CD226 (+Anti-CD226), NKp46-Fc (+NKp46-Fc), or a control rat IgG (+Rat IgG) was added. The cultures were incubated for additional 4 h in the presence of Brefeldin A. (A) CD69 expression on DX5+CD3− NK cells. MFI of CD69 staining is indicated. (B-C) IFN-γ and granzyme B production by DX5+CD3− NK cells. FACS plots of IFN-γ and granzyme B production with the percentage of IFN-γ and granzyme B positive cells indicated (B). The mean percentage ± SD of IFN-γ or granzyme B positive cells is indicated (n = 5 per group) (C). The data shown are representative of three independent experiments.
NKG2D ligands are upregulated in accessory cells upon adenoviral infection
We then investigated whether NKG2D ligands were induced upon adenoviral infections. Murine NKG2D recognizes nine ligands: Rae-1α, Rae-1β, Rae-1γ, Rae-1δ, Rae-1ε, Mult1, H60a, H60b, and H60c (25). We first investigated if adenoviral infection up-regulated the expression of NKG2D ligands. RNA samples from naïve or Ad-lacZ infected mice were analyzed for the expression of NKG2D ligands by semi-quantitative PCR technology. As shown in Fig. 4A, the mRNA levels for Rae-1α, Rae-1γ, Rae-1δ, and Mult1 were markedly increased in Ad-infected splenocytes, whereas mRNAs coding for Rae-1β, Rae-1ε, H60a, H60b, and H60c were not detected. To further determine whether accessory cells upregulate the expression of NKG2D ligands in response to adenovirus in vivo, splenic macrophages (CD11b+F4/80+) and cDCs (CD11c+B220−) from naïve or Ad-lacZ infected mice were analyzed for the expression of Rae-1 and MULT1 by flow cytometry. Indeed, in Ad-lacZ infected mice, the expression of MULT1 on the surface of macrophages and cDCs was uniformly upregulated compared to the uninfected naïve controls (Fig. 4B). Although the expression of Rae-1 was also uniformly upregulated on macrophages of the infected mice, it appears only a subpopulation of cDCs upregulated Rae-1 (Fig. 4B). This Rae-1+ subpopulation increased in size that is proportional to increasing doses of Ad-lacZ infection in vivo (data not shown), suggesting that induction of Rae-1 in cDCs by adenovirus requires a higher viral load. Collectively, these results suggest NKG2D ligands MULT1 and Rae-1 are induced in accessory cells upon adenoviral infection in vivo.
FIGURE 4.

NKG2D ligands are upregulated in accessory cells upon adenoviral infection. C57BL/6 mice were infected intravenously with 2×109 pfu Ad-LacZ (Ad) or left uninfected (Naïve). 24 h after infection, spleens were harvested for analyses. (A) Total RNA was extracted from splenocytes and analyzed for the induction of NKG2D ligands by semi-quantitative RT-PCR using 3-fold serial dilution of the template. Mouse β-actin was used as an internal control. (B) Expression of Rae-1 and MULT-1 on macrophages and DCs from splenocytes. MFI of Rae-1 and MULT-1 staining is indicated. Data shown is representative of two independent experiments.
NKG2D is critical for NK cell activation and function upon adenoviral infection in vivo
We next examined the role of NKG2D signaling in NK cell activation in vivo. To address this question, mice were treated with the blocking NKG2D antibody or a control antibody, followed by infection with 2×109 pfu of Ad-LacZ administered intravenously. 24 h after infection, splenic NK cells were analyzed for their activation. NK cells from the control rat IgG-treated mice produced large amounts of IFN-γ and granzyme B, compared to the naïve mice (Fig. 5A, B). However, the production of IFN-γ and granzyme B by NK cells was significantly (p <0.05) reduced in the anti-NKG2D mAb treated group (Fig. 5A, B). These results indicated that the NKG2D pathway is also important in the activation of NK cells upon adenoviral infection in vivo.
FIGURE 5.

NKG2D is required for NK cell activation upon adenoviral infection in vivo. (A-B) C57BL/6 mice were treated with 100 μg of anti-NKG2D mAb (+Anti-NKG2D) or the control rat IgG (+Rat IgG) 24 h and 6 h before intravenous injection of Ad-lacZ (Ad). Some mice were left uninfected as controls (Naïve). 24 h after infection, splenocytes were harvested and restimulated with PMA/Inomycin in the presence of Brefeldin A for 4 h. Intracellular IFN-γ and granzyme B was determined by flow cytometry gated on DX5+CD3− NK cells. FACS plots of IFN-γ and granzyme B production with the percentage of IFN-γ and granzyme B positive cells indicated (A). The mean percentage ± SD of IFN-γ or granzyme B positive cells is indicated (n = 4 per group) (B). The data shown are representative of three independent experiments.
The critical role of NKG2D in NK cell activation in response to adenoviral infection suggested the blockade of NKG2D may improve the outcome of adenovirus-mediated gene therapy in vivo. To eliminate the potential influence of the adaptive immunity on the clearance of adenovirus, RAG-2−/− mice were used to test this strategy. RAG-2−/− mice were treated with either the control rat IgG or the anti-NKG2D mAb 24 h and 6 h prior to infusion of Ad-LacZ. Mice treated with the NKG2D blocking mAb showed significantly (p <0.05) higher levels of LacZ expression evaluated by X-gal histochemistry (Fig. 6A) and a quantitative β-gal assay (Fig. 6B), in the liver 3 days after infection. Furthermore, the copy numbers of adenoviral genome in the liver were evaluated by real time quantitative PCR. Significantly (p <0.05) larger amounts of adenoviral DNA were detected in mice treated with anti-NKG2D mAb than the control antibody-treated mice (Fig. 6C). These results indicate that the NKG2D pathway is critical for NK cell-mediated elimination of adenoviral infection in vivo.
FIGURE 6.

NKG2D blockade delays adenoviral clearance in vivo. RAG-2−/− mice were treated with 100 μg of anti-NKG2D mAb (Anti-NKG2D) or the control rat IgG (Rat IgG) 24 h and 6 h before intravenous injection of Ad-lacZ. 5 days after infection, liver tissues were harvested for analyses. (A) Cryosections were stained for LacZ expression by X-gal histochemistry. (B) Liver tissues were assayed for β-galactosidase (β-gal) activity. Data represent β-gal units ± SD per gram of liver protein. (C) Total genomic DNA were extracted and analyzed for adenoviral genome copies by quantitative real-time PCR. Data are representative of two independent experiments.
Previous studies have shown that NKG2D also plays a critical role in recognizing NKG2D ligands expressed on target cells and mediating NK cell-mediated cytolysis in the setting of human CMV and MCMV infections (15, 16). To address whether NKG2D is involved in NK cell-mediated cytolysis on adenovirus-infected targets, NK cells isolated from Ad-lacZ-infected or naive control mice were assayed for their lytic activities on Ad-lacZ infected macrophages in vitro in the presence of NKG2D blocking mAb. A significant (p <0.05) reduction in cytolytic activities was observed with the NKG2D blockade (Fig. 7), suggesting that NKG2D is also critical for NK cell-mediated cytolysis on adenovirus-infected target cells.
FIGURE 7.
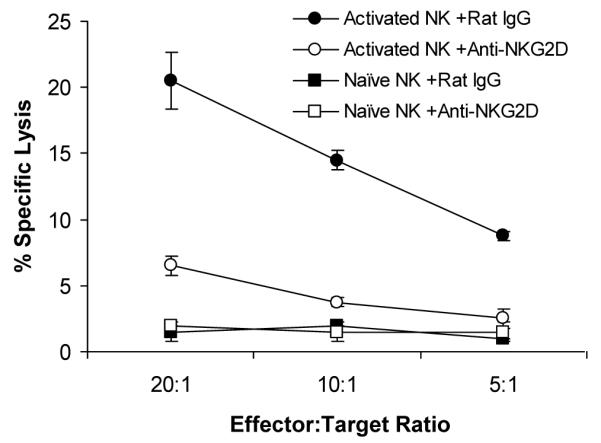
NKG2D is critical for NK cell-mediated cytolysis on adenovirus-infected targets. C57BL/6 mice were injected intravenously with 2×109 pfu Ad-LacZ or left uninfected as the control. 24 h after infection, splenic NK cells were enriched from Ad-lacZ infected (Activated NK) or naïve (Naïve NK) mice, and assayed for NK cell lytic activity on Ad-lacZ infected macrophages in the presence of anti-NKG2D mAb (+Anti-NKG2D) or the rat IgG control (+Rat IgG) for 4 hour at different effector:target ratio. The mean percentage ± SD of specific lysis is indicated (n=3 per group). Data shown is representative of two independent experiments.
Discussion
NK cells play an important role in innate immune control of adenoviral infection in vivo (5-7). We have previously shown that type I IFN, induced upon adenoviral infection, acts directly on NK cells to regulate their activation and function (7). However, how NK cells are activated upon adenoviral infection remains to be further defined. In this study, we provided evidence that the NKG2D pathway is critical for NK cell activation and function in response to adenoviral infection. This is supported by upregulation of NKG2D ligands in accessory cells upon adenoviral infection and a requirement of the accessory cell-NK cell contact for NK cell activation. Furthermore, NKG2D blockade inhibits NK cell activation and function, and delays adenoviral clearance in vivo.
The NKG2D activating receptor has been shown to play an important role in NK cell activation in response to human and murine CMV infections (15, 16). NKG2D recognizes host stress proteins induced upon viral infections (10). The stress-induced NKG2D ligands are RAE1, MULT1 and H60 classes in mice and the ULBP and MIC classes in humans. Here we showed that both RAE1 and MULT1 are induced upon adenoviral infection and that NK cell activation and NK cell-mediated control of adenoviral infection in vivo are critically dependent on the NKG2D pathway. Furthermore, NKG2D is required for the recognition of adenovirus-infected target cells for NK cell-mediated cytolysis. This is in line with the previous observation that NK cell-mediated cytolysis on adenovirus-infected primary human fibroblasts is also dependent on NKG2D (26). Our results may have important implications for the design of effective strategies to reduce NK cell-mediated early toxicity and improve the outcome of adenovirus-mediated gene therapy. Specifically, the observation that NKG2D blockade significantly blunts innate immune elimination of adenoviral vectors suggests a feasible strategy to reduce early phase of hepatotoxicity and prolong transgene expression.
NKG2D is not only expressed by most NK cells, but also by subsets of T cells including activated CD8+ T cells (27). This could complicate the interpretation of the NKG2D blocking experiments in immunocompetent mice in vivo. To address this question, we have used RAG-2−/− mice. Indeed, our data in RAG-2−/− mice clearly demonstrated a critical role of NKG2D in NK cell-mediated clearance of adenovirus in vivo. We further demonstrated that NKG2D plays an important role both in the activation of NK cells and NK cell recognition of target cells.
What triggers the expression of NKG2D ligands on accessory cells upon adenoviral infection remains to be determined. Previous studies in other models have shown that NKG2D ligands can be induced in response to cellular stress, such as the response to DNA damage (28), infection (29) as well as TLR stimulation (30). In addition, it has been shown that wild-type adenovirus E1A protein can regulate the expression of Rae-1, which sensitizes tumor cells to NKG2D-dependent NK cell lysis and promotes tumor rejection (31). It remains to be determined what component(s) of the E1-deleted adenovirus or which mechanism (s) is responsible for the induction of NKG2D ligands upon adenoviral infection. Understanding this process will help us design effective NK cell-based strategies to improve adenovirus-mediated gene therapy in vivo.
In conclusion, we have shown that the NKG2D pathway is critical for NK cell activation in response to adenoviral infection in vitro and in vivo. We have further demonstrated that NKG2D is required for the recognition of virus-infected targets and that it plays an important role in elimination of adenoviral vectors. These results may suggest potential strategies to improve the outcome of adenovirus-mediated gene therapy in vivo.
Acknowledgments
This work was supported by National Institutes of Health grants AI083000, CA136934, and CA047741 from the National Institutes of Health ( to Y.Y.), and a grant from the Alliance for Cancer Gene Therapy (to Y.Y.).
Footnotes
Publisher's Disclaimer: Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
References
- 1.Huang X, Yang Y. Innate immune recognition of viruses and viral vectors. Hum Gene Ther. 2009;20:293–301. doi: 10.1089/hum.2008.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Y, Ertl HC, Wilson JM. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 3.Dai Y, Schwarz EM, Gu D, Zhang WW, Sarvetnick N, Verma IM. Cellular and humoral immune responses to adenoviral vectors containing factor IX gene: tolerization of factor IX and vector antigens allows for long-term expression. Proc Natl Acad Sci U S A. 1995;92:1401–1405. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Jooss KU, Su Q, Ertl HC, Wilson JM. Immune responses to viral antigens versus transgene product in the elimination of recombinant adenovirus-infected hepatocytes in vivo. Gene Ther. 1996;3:137–144. [PubMed] [Google Scholar]
- 5.Liu ZX, Govindarajan S, Okamoto S, Dennert G. NK cells cause liver injury and facilitate the induction of T cell-mediated immunity to a viral liver infection. J Immunol. 2000;164:6480–6486. doi: 10.4049/jimmunol.164.12.6480. [DOI] [PubMed] [Google Scholar]
- 6.Ruzek MC, Kavanagh BF, Scaria A, Richards SM, Garman RD. Adenoviral vectors stimulate murine natural killer cell responses and demonstrate antitumor activities in the absence of transgene expression. Mol Ther. 2002;5:115–124. doi: 10.1006/mthe.2002.0529. [DOI] [PubMed] [Google Scholar]
- 7.Zhu J, Huang X, Yang Y. A critical role for type I IFN-dependent NK cell activation in innate immune elimination of adenoviral vectors in vivo. Mol Ther. 2008;16:1300–1307. doi: 10.1038/mt.2008.88. [DOI] [PubMed] [Google Scholar]
- 8.French AR, Yokoyama WM. Natural killer cells and viral infections. Curr Opin Immunol. 2003;15:45–51. doi: 10.1016/s095279150200002x. [DOI] [PubMed] [Google Scholar]
- 9.Lee SH, Miyagi T, Biron CA. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 2007;28:252–259. doi: 10.1016/j.it.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 12.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 13.Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, Davis DM, Strominger JL, Yewdell JW, Porgador A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409:1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- 14.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7:517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]
- 15.Guma M, Angulo A, Lopez-Botet M. NK cell receptors involved in the response to human cytomegalovirus infection. Curr Top Microbiol Immunol. 2006;298:207–223. doi: 10.1007/3-540-27743-9_11. [DOI] [PubMed] [Google Scholar]
- 16.Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposti MA. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- 17.Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol. 2007;81:3170–3180. doi: 10.1128/JVI.02192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5:508–515. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 19.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang M, Lanier LL, Sigal LJ. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog. 2008;4:e30. doi: 10.1371/journal.ppat.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnon TI, Achdout H, Lieberman N, Gazit R, Gonen-Gross T, Katz G, Bar-Ilan A, Bloushtain N, Lev M, Joseph A, Kedar E, Porgador A, Mandelboim O. The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood. 2004;103:664–672. doi: 10.1182/blood-2003-05-1716. [DOI] [PubMed] [Google Scholar]
- 22.Dardalhon V, Schubart AS, Reddy J, Meyers JH, Monney L, Sabatos CA, Ahuja R, Nguyen K, Freeman GJ, Greenfield EA, Sobel RA, Kuchroo VK. CD226 is specifically expressed on the surface of Th1 cells and regulates their expansion and effector functions. J Immunol. 2005;175:1558–1565. doi: 10.4049/jimmunol.175.3.1558. [DOI] [PubMed] [Google Scholar]
- 23.Newman KC, Riley EM. Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat Rev Immunol. 2007;7:279–291. doi: 10.1038/nri2057. [DOI] [PubMed] [Google Scholar]
- 24.Chan CJ, Andrews DM, McLaughlin NM, Yagita H, Gilfillan S, Colonna M, Smyth MJ. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol. 184:902–911. doi: 10.4049/jimmunol.0903225. [DOI] [PubMed] [Google Scholar]
- 25.Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol. 2007;7:737–744. doi: 10.1038/nri2144. [DOI] [PubMed] [Google Scholar]
- 26.Tomasec P, Wang EC, Groh V, Spies T, McSharry BP, Aicheler RJ, Stanton RJ, Wilkinson GW. Adenovirus vector delivery stimulates natural killer cell recognition. J Gen Virol. 2007;88:1103–1108. doi: 10.1099/vir.0.82685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 28.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodoen MB, Lanier LL. Natural killer cells as an initial defense against pathogens. Curr Opin Immunol. 2006;18:391–398. doi: 10.1016/j.coi.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamerman JA, Ogasawara K, Lanier LL. Cutting edge: Toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J Immunol. 2004;172:2001–2005. doi: 10.4049/jimmunol.172.4.2001. [DOI] [PubMed] [Google Scholar]
- 31.Routes JM, Ryan S, Morris K, Takaki R, Cerwenka A, Lanier LL. Adenovirus serotype 5 E1A sensitizes tumor cells to NKG2D-dependent NK cell lysis and tumor rejection. J Exp Med. 2005;202:1477–1482. doi: 10.1084/jem.20050240. [DOI] [PMC free article] [PubMed] [Google Scholar]