

Abstract
In the present study, we examined whether activation of p-38α MAPK modulates mechanical allodynia and neuronal hyperexcitability, and if propentofylline (PPF, a glial modulator) modulates specifically localized activated p-38α MAPK expression in caudal regions remote from a low thoracic hemisection injury in rats. T13 spinal hemisection produces bilateral mechanical allodynia in hindpaws with evoked (in response to mechanical stimuli) neuronal hyperexcitability in lumbar spinal wide dynamic range (WDR) neurons compared to sham controls. The mechanical allodynia and the evoked activity of WDR neurons is attenuated by intrathecal and topical administration of SB203580, an inhibitor of p-38α MAPK activation, dose dependently (*p<0.05); however, the spontaneous activity showed no significant differences compared to sham controls. After T13 spinal hemisection, significantly increased phosphorylated (activated form) p-38α MAPK expression was present in both superficial and deep dorsal horn neurons as well as in microglia, but not in astrocytes, in the lumbar spinal cord compared to sham controls (*p<0.05). Intrathecal application of PPF significantly attenuated the expression of phosphorylated p-38α MAPK in superficial dorsal horn neurons (10 mM) and in microglia (1 and 10 mM) in the lumbar spinal cord compared to the hemisection group (*p<0.05). In conclusion, our present data demonstrate that activated neuronal and microglial, but not astrocytic, p-38α MAPK contributes to the maintenance of neuronal hyperexcitability in caudal regions following spinal cord injury.
Keywords: Glia, Hyperexcitability, p38 MAPK, Propentofylline, Spinal cord injury
Introduction
Traumatic thoracic spinal cord injuries (SCI) induce neuronal hyperexcitability in lumbar spinal dorsal horn neurons, which results in abnormally enhanced somatosensory sensation in regions caudal to the injury (Gwak et al., 2008; Hains and Waxman, 2006) that is manifested as mechanical allodynia (non-noxious stimuli become noxious) and thermal hyperalgesia (noxious stimuli become more noxious) (Gwak et al., 2004). The abnormal sensations persist for life and are termed central (since the lesion is in the central nervous system) neuropathic pain syndromes. Neuronal hyperexcitability or central sensitization of spinal dorsal horn neurons following spinal cord injury is the key substrate for central neuropathic pain (CNP) syndromes, however, the mechanisms that contribute to persistent central sensitization are unclear.
Somatosensory transmission, including nociceptive, is regulated and influenced by intra- and extracellular events in the spinal dorsal horn (Ji and Woolf, 2001). Immediately after spinal cord injury, the extracellular concentrations of glutamate dramatically increase and trigger activation of excitatory amino acid receptors and ion channels (Vera-Portocarrero et al., 2002; McAdoo et al., 1999), followed by activation of intracellular downstream pathways, such as p-38 MAPK and CREB, that result in altered gene expression (Crown et al., 2006; Wu et al., 2005). Those events are sufficient to produce persistent neuronal hyperexcitability of spinal dorsal horn neurons following spinal cord injury (Crown et al., 2008, Gwak et al., 2008).
The mitogen-activated protein kinases (MAPK) are responsible factors in survival, proliferation and other intracellular biochemical events in neurons (Widmann et al., 1999). Recent literature supports the important role of activated p-38α MAPK in at-level central neuropathic pain following thoracic contusion spinal cord injury (Crown et al., 2006; 2008). However, the cellular location of activated p-38α MAPK in remote regions after SCI is controversial, with one study reporting that activated p-38 MAPK is predominantly in neurons (Detloff, et al., 2008) and another study reporting that asctivated p-38 MAPK is located in spinal microglia (Hains and Waxman, 2006). Because of the purported role of MAP kinase activity in central neuropathic pain (Hulsebosch, 2008; Corwn et al., 2008) understanding cellular location and pathways involved in remote regions after spinal cord injury is particularly important. In the present study, we tested whether activation of p-38α MAPK modulates mechanical allodynia and neuronal hyperexcitability of spinal neurons in the somatosensory pain pathway. We also tested if propentofylline (PPF), a glial modulator that inhibits astrocytic and microglial activation, modulates specifically localized activated p-38α MAPK expression in remote caudal regions of the spinal cord following spinal hemisection injury.
Materials and Methods
Animal Preparation
Male Sprague-Dawley (225-250 g) rats were obtained from Harlan Sprague-Dawley, Inc., and housed with a reverse light/dark cycle of 12/12h, and fed ad libitium. Animal protocols were reviewed by the UTMB Institutional Animal Care and Use Committee (IACUC) and were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animal. Spinal hemisection injury was done as previously described (Gwak et al., 2004, 2008). Briefly, rats were anesthesized via masked-inhalation with isoflurane (induction 2% and maintenance 1.5%) and unilateral transverse cut of the spinal segment T13 with a # 11 scalpel blade was done after low thoracic laminectomy of the T11-12 vertebral segment with the aid of a surgical microscope (KAPS, Germany). To ensure hemisection, a 28 gauge needle was inserted from dorsal to ventral at the midline of the cord and pulled laterally and the blood aspirated by plunge, which avoided overhemisection and spinal root damage. Sham surgery was produced by only laminectomy of the T11-T12 vertebra in rats of corresponding body weight.
Behaviors
A total of 19 rats were used in behavioral experiments (Hemi+vehicle = 4, Hemi+1 μg SB203580 = 5, Hemi+10 μg SB203580 = 5, Hemi+100 μg SB203580 = 5). To test paw withdrawal behaviors, rats were individually housed in clear plastic boxes (8 ×8 ×24cm) above a metal mesh (0.5 ×0.5 cm) and acclimated for 15 min to avoid the stress of environmental change. To administer three doses of SB203580 (an inhibitor of p38 MAPK activation; 1, 10 and 100 μg/15 μl and flushed by 10 μl saline) onto spinal surface directly, intrathecal implantation was done by insertion of an intrathecal catheter prior to 5 days of paw withdrawal tests. Briefly, under isoflurane anesthesia, a pre-measured length of CS-1 Intrathecal Catheter 32G (RecathCo), was passed caudally from the T8 to the L3 level of the spinal cord and 2 cm of the free end was left exposed in the upper thoracic region. Saline was injected daily to prevent clogging of the tubing. To protect against slipping or loss of the inserted tubing by the rat, the tube was loosely tied using an over hand knot (0.5 cm diameter), fixed by Glas Ionomer Base Cement (Shofu Inc, Japan) and sutured to the surrounding paravertebral musculature just proximal to the intrathecal entry site. The exposed intrathecal tubing was sealed by sterile stainless steel wire to prevent infection. The wire was removed for saline and/or SB203580 administration and immediately replaced after injection.
To test thresholds of mechanical withdrawal responses of both hindpaws on post operative day (POD) 30, calibrated von Frey filaments [a series of von Frey filament log unit; 3.61 (0.45g), 3.84 (0.74g), 4.08 (1.26g), 4.31 (2.04g), 4.56 (3.31g), 4.74 (5.50g), 4.93 (8.32g) and 5.18 (14.45g)] were applied to the center of glaborous surface of hindpaws, not on the keratinized foot pads, in 6 applications (beginning with the 4.31 log unit) with 10 secs intervals between stimuli. The final calculation of 50% withdrawal mechanical threshold was determined by the formula, log (50% threshold) = Xf + κδ. Xf = value of the final von Frey filament (log unit), κ = correction factors (from calibration table), and δ = mean differences of log units between stimuli; the 18 g pressure of 50% threshold measurement (the maximum score according to up-down method) was selected as the cut-off value (Chaplan et al., 1994). The mechanical thresholds were measured before hemisection, after hemisection and 30, 60, 120 and 180 mins after SB203580 administration.
Electrophysiology
A total of 35 rats were used in the electrophysiology experiments (Sham = 8, Hemi+vehicle = 5, Hemi+1 μg SB203580 = 6, Hemi+10 μg SB203580 = 8, Hemi+100 μg SB203580 = 8). To test neuronal hyperexcitability of wide dynamic range (WDR) neurons in the lumbar (L4/5) dorsal horn on post operative day (POD) 30 after spinal hemisection, in vivo extracellular single-unit recordings were performed. Briefly, rats were anesthetized by sodium pentobarbital (60 mg/kg, i.p.) and then a laminectomy of vertebral segments T12-L2 was performed to expose the lumbar enlargement (L4-L5). Tracheal and jugular vein cannulae were inserted for easy breathing and infusion of sodium pentobarbital (5mg/hr/300g) to maintain the stabilization of the rat condition during the single unit recording, respectively. The rats were held in place by a stereotaxic apparatus and rectal temperature was maintained at 37 °C. A single-unit (wide dynamic range, WDR) recording was performed by using a carbon filament-filled single glass microelectrode (0.4-0.8 MΩ, Kation Scientific, USA) in the lumbar L4/5 dorsal horn. The criterion for the identification of WDR neurons is a dorsal horn neuron that displays graded activities in response to increased intensities of mechanical stimuli (Chung et al., 1986; Gwak et al., 2008). After the unit activity was identified, the spontaneous activity was recorded for 20 seconds without stimuli, followed by three graded mechanical stimuli applied to that unit's peripheral receptive field. These were 1) brush stimulation of the skin with a hairy brush, 2) pressure stimulation by applying a large arterial clip (Bulldog Clamps, Tiemann) with weak grip to a fold of the skin (firm pressure) and 3) pinch stimulation by applying a small arterial clip (Serrefines, Tiemann) with a strong grip to a fold of the skin (painful pressure). Three mechanical stimuli were applied successively for 10 sec each with an inter-stimulus interval of 20 sec. The unit activity was amplified and filtered (DAM80; World Precision Instruments, USA), fed either directly or via an oscilloscope (World Precision Instruments, USA) into the data acquisition unit (CED-1401; Cambridge Electronic Design, UK), and stored on computer in order to construct the waveforms or plot peristimulus time histograms (spikes/ 1 second bin width, Spike2 software). To test changes of WDR neuronal activities, SB203580 was given directly onto the spinal surface near the recording electrode (topical administration). Three doses of SB203580 (1, 10 and 100 μg/30 μl) were delivered by Hamilton syringe after characterizing the WDR activity, which was recorded for 10, 30, 60 and 120 min after administration. As a control to ensure that a single and the same WDR unit were held for the duration of the recording experiment, we used the Spike2 program to maintain the same action potential shape and amplitude.
Immunocytochemistry
To test whether propentofylline (PPF, 3,7-dihydro-3-methyl-1-(5-oxohexyl)-7-proplyl-1H-purine-2,6-dione, M.W. 306.4, Sigma) modulates expression of activated p-38α MAPK in neurons and glial cells, double immunofluorescence staining was performed. In double immunofluorescence staining, 16 rats (Sham = 4, Hemi+vehicle = 4, Hemi+0.1 mM PPF = 4, Hemi+10 mM PPF = 4) were used to test changes of phosphorylated p-38α MAPK (p-p-38α MAPK) expression in neurons and glia cells (astrocytes and microglia), respectively. In addition, spinal dorsal horn neurons were divided into two different regional neurons: superficial dorsal horn (predominantly distributed by neurons, < 25 μm in soma diameter) and deep dorsal horn (predominantly distributed by neurons, > 35 μm in soma diameter) neurons (Gwak and Hulsebosch, 2005, Rexed, 1952; Willis and Coggeshall, 1991). Two doses of PPF, 1 and 10 mM (15 μl volume, flushed by 10 μl saline), were administrated intrathecally immediately after spinal hemisection and continued on post operative day (POD) 1, 2, 3, 4, 5, 6 and 7. This regimen effectively prevents activation of astrocytes and microglia up to POD 30 after hemisection (Gwak et al., 2008, 2009a). Intrathecal applications were performed at the same time every morning.
To test expression of p-38α MAPK activation in neurons, astrocytes and microglia on POD 30, rats were deeply anesthetized with sodium pentobarbital (80mg/kg, i.p.) and perfused intracardially with heparinized physiological saline followed by 4% cold buffered paraformaldehyde solution. After perfusion, the lumbar spinal cord (L4/5) was removed immediately and post-fixed overnight in 4% paraformaldehyde, followed by cryoprotection in 30% sucrose in 4% paraformaldehyde over several days. Prior to sectioning, spinal cords were embedded in OCT compound, and then sectioned at 20 μm. Primary rabbit antibody for p-p-38α MAPK (R&D Systems, 1:100, recognizes rat p-38α MAPK), mouse antibody for glial fibrillary acidic protein (GFAP, Chemicon, recongizes astrocytes, 1:500), mouse antibody for CD11b (OX-42, Serotec, recognizes microglia and activated macrophages, 1:200) and NeuN (Chemiscon, 1:5000, recognizes neurons) were incubated with a cocktail solution (0.05M PBS, 0.15% Triton X-100, 1% NGS and 0.3% BSA) at room temperature (overnight). After 3 times washes with 0.05M TBS, sections were incubated with secondary antibodies (2 hours, 1:200, Molecular Probes). Sections were collected by free-floating methods and mounted on gel-coated slides with mounting media (DAPI, Vectashield). Images were captured by Confocal microscope (Bio-Rad) with Lasersharp imaging software and were evaluated by measuring intensity using a computer-assisted image analysis program (Metamorph 6.1).
Statistical Analysis
Statistical analyses were performed using repeated Two way ANOVA with the Student-Newman-Keuls Method for comparisons using the SigmaStat program (Ver 3.1). An alpha level of significance was set at 0.05 for all statistical tests. Data were combined with ipsilateral and contralateral sides because there was no significant differences in side to side comparisons. Data are expressed as means ± S.E. * p<0.05
Results
Inhibition of p38 MAPK activation attenuates mechanical allodynia
Before hemisection, mean paw withdrawal thresholds of the SB203580 (SB) treatment group was 17.4 ± 0.4 g and were not significantly different compared to the vehicle control group (17.7 ± 0.3 g). After hemisection, the paw withdrawal threshold of the vehicle group (4 ± 0.1 g) and SB treatment groups (4.3 ± 0.3 g) significantly decreased when compared to thresholds before hemisection (BH, Figure 1), respectively. On post operative day (POD) 30, however, the mean paw withdrawal threshold after intrathecal treatment (arrow) of 10 and 100 μg SB203580 was 6.1 ± 0.4 g (#p<0.05) and 9.4 ± 0.6 g (*p<0.05) which were significantly increased; whereas the 1 μg SB203580 (3.9 ± 0.4 g) did not show significant differences when compared to after hemisection (AH) but before treatment values (Figure 1), respectively. The vehicle treatment group did not show significant differences when compared to values before treatment (Figure 1).
Figure 1.

Attenuation of mechanical allodynia by intrathecal treatment of p-38 MAPK inhibitor. After hemisection (AH), the mean paw withdrawal thresholds of all groups significantly decreased compared to before hemisection (BH). Post operative day (POD) 30 (arrow), inthrathecal administration of 10 (#) and 100 μg (*) SB203580 significantly increased paw withdrawal thresholds whereas 1 μg and vehicle treatments did not show significant differences compared to after hemisection but before treatment (AH) (#, *p<0.05). Data are expressed as means ± S.E. * p<0.05.
Inhibition of p38 MAPK attenuates neuronal hyperexcitability
The mean spontaneous activity of WDR neurons in the sham control group was 0.9 ± 0.5 spikes/sec. After hemisection, the means of spontaneous activity of WDR neurons in all groups (Hemi-vehicle, Hemi+1, Hemi+10 and Hemi+100 μg SB203580) were 1.1 ± 0.6, 0.9 ± 0.4, 0.8 ± 0.2 and 1.1 ± 0.3 spikes/sec, respectively and were not significantly different compared to the sham control (Figure 2).
Figure 2.

The comparison of spontaneous activity in lumbar WDR dorsal horn neurons following low thoracic hemisection. After hemisection, the spontaneous activity (recorded for 20 seconds) in all hemisection groups did not show significant differences compared to the sham control. Thus, increased spontaneous activity, which is thought to contribute to neuropathic pain, cannot account for the behavioral observations. Data are expressed as means ± S.E..
Figure 3A shows the typical response patterns of WDR neurons to graded intensity of mechanical stimuli applied on the receptive fields of the hindpaw. The means of mechanically evoked activity of WDR neurons in the sham control group were 5.6 ± 1.3 (brush), 10.7 ± 1.5 (pressure) and 11.2 ± 1.9 spkes/sec (pinch), respectively. However, spinal hemisection produced hyperexcitability to values of 15.2 ± 2.6 (brush), 23.1 ± 1.9 (pressure) and 26.9 ± 5.9 spikes/sec (pinch) in the lumbar spinal dorsal horn WDR neurons. Moreover, topical administration of SB203580 dose dependently attenuated the hyperexcitability. After 30 min of 100 μg SB203580 administration, the neuronal activity was significantly attenuated to 9.3 ± 1.3 (brush), 13.9 ± 2.8 (pressure) and 11.9 ± 2.2 spikes/sec (pinch) compared to SB203580 administration, respectively (Figure 3B, p<0.05). The attenuation of hyperexcitability lasted over 120 mins after SB203580 administration.
Figure 3.

The comparison of evoked activity in lumbar WDR dorsal horn neurons following low thoracic hemisection. (A) Spinal hemisection produced increased single WDR neuronal activity (upper) and topical administration of 100 μg SB203580 attenuated the increased activity (bottom). The typical (peristimulus spikes activity) histograms and lines of waveforms (below histograms) are represented 30 mins after SB203580 administration. (B) Topical administration of SB203580 dose dependently attenuated lumbar WDR neuronal hyperexcitability following T13 spinal hemisection for all tested stimuli (*: 100 μg, #: 10 μg). Br - brush, Pr – pressure, Pi - pinch stimulation with 10 seconds, SCI – after hemisection and before SB203580 administration. Data are expressed as means ± S.E. * p<0.05.
Propentofylline attenuates neuronal p38αMAPK activation
In superficial dorsal horn neurons (Figure 4A), the expression level (intensity) of phosphorylated p-38α MAPK in the sham control group was 17.3 ± 3.3. After hemisection, the expression level of phosphorylated p-38α MAPK was 35.3 ± 2.8, a significant difference compared to the sham control group (*p<0.05). However, 10 mM PPF treatment significantly inhibited activation of phosphorylated p-38α MAPK expression (21.5 ± 2.8) compared to the hemisection group (#p<0.05). In deep dorsal horn neurons (Figure 4B), the expression level of phosphorylated p-38α MAPK in the sham control group was 14.6 ± 2. After hemisection, the expression level of phosphorylated p-38α MAPK was 22.1 ± 2.3, a significant difference compared to the sham control group (*p<0.05). However, the level of phosphorylated p-38α MAPK expression in neurons > 35 μm after 10 mM PPF (21.6 ± 1.6) treatment was not significantly different compared to the hemisection group.
Figure 4.
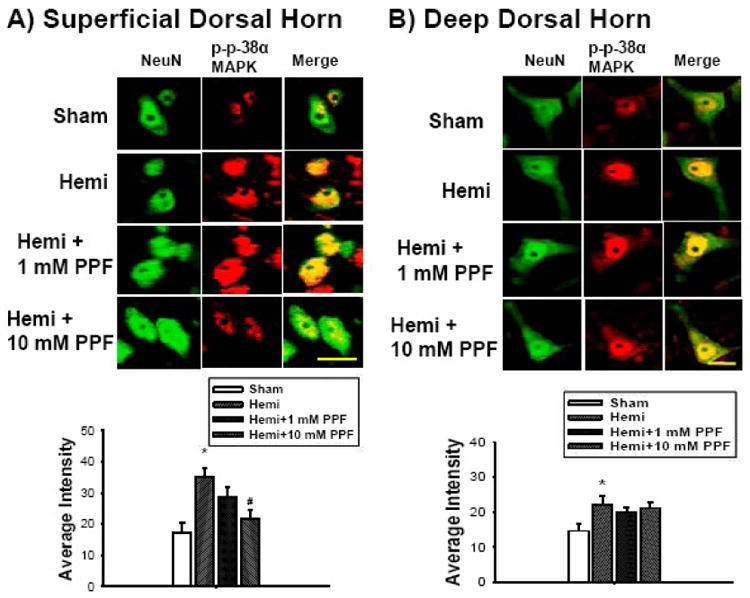
The comparison of phosphorylated p-38α MAPK expression levels (red) in lumbar dorsal horn neurons (NeuN-green) following low thoracic hemisection. The expression of phosphorylated p-38α MAPK in both superficial (laminae I and II, A) and deep dorsal horn neurons (laminae III-V, B) significantly increased in the lumbar dorsal horn following hemisection compared to the sham control (*p<0.05). Note that ten mM intrathecal treatment of PPF significantly attenuated expression of the phosphorylated p-38α MAPK in superficial dorsal horn neurons (#p<0.05, A) compared to the hemisection group whereas deep dorsal horn neurons (B) did not show significant differences. Scale bar : 30 μm. Data are expressed as means ± S.E.
Propentofylline attenuates microglia p38 MAPK activation
In astrocytes, the expression of astrocytic phosphorylated p-38α MAPK after hemisection was not significantly different compared to the sham control group (Figure 5A). By contrast, in microglia, the expression level of phosphorylated p-38α MAPK in the sham control was 15.5 ± 1; after hemisection, the expression level of phosphorylated p-38α MAPK was 47.9 ± 8.7, a significant difference compared to the sham control group (Figure 5B. right, *p<0.05). However, the level of phosphorylated p-38α MAPK expression in microglia after intrathecal applications 1 mM (21.2 ± 1.2) and 10 mM (11.7 ± 0.5) PPF was decreased significantly compared to the hemisection group, respectively (#p<0.05, Figure 5B).
Figure 5.

The comparison of phosphorylated p-38α MAPK expression levels in lumbar astrocytes and microglia following low thoracic hemisection. The expression level of phosphorylated p-38α MAPK (red) in astrocytes (GFAP-green, A) did not show significant change in the lumbar dorsal horn. However, the expression level of phosphorylated p-38α MAPK in microglia (OX-42-green, B) significantly increased in the lumbar dorsal horn following hemisection compared to the sham control (*p<0.05). Quantitative intensity measurements are graphed as histograms for the four groups studied. Note that one and ten mM intrathecal treatments of PPF significantly attenuated the expression level of phosphorylated p-38α MAPK in the lumbar dorsal horn compared to the hemisection group (#p<0.05). Scale bar : 30 μm. Data are expressed as means ± S.E.
Discussion
This is the first report that activated p-38 MAPK is located in both neurons and microglia, but not astrocytes (however, see Crown et al., 2008), in caudal regions remote from a low thoracic spinal cord injury (SCI); and that propentofylline modulates both neuronal and microglial p-38α MAPK activation in caudal regions and attenuates below-level central neuropathic pain via MAPK pathways in a rodent SCI model. Previously, we determined that activation of p-38α MAPK contributed to at-level central neuropathic pain following spinal contusion injury (Crown et al., 2008) and was localized to neurons, microglia and astrocytes in regions just rostral to the contusion site. The current study extends previous studies to caudal regions remote from a low thoracic SCI and differs from those that propose that p-38 MAPK pathways are involved in below-level pain and located only in microglial cells (Hains and Waxman, 2006) or only in neuronal populations (Detloff et al., 2008) in that the current study demonstrates that p-38α MAPK is localized to both neurons and microglia. Moreover, we show that propentofylline modulates the expression of both neuronal and microglial p-38α MAPK and attenuates below-level central neuropathic pain following low thoracic SCI (Gwak et al., 2008).
It is note worthy that the cellular location of p-38α MAPK activation after SCI is not unique. In spinal hemisection injury, we observed activation of p-38α MAPK in neurons and microglia in the dorsal horn in the lumbar enlargement, caudal and remote to the injury, which would be the substrate for below-level neuropathic pain syndromes. But our previous study showed that activation of p-38α MAPK developed in neurons, astrocytes and microglia as well as astrocytic and microglial activation in the region near the injury (at-level) following moderate spinal contusion injury using Infinite Horizon Impactor, which is a computer software controlled machine (150 Kdyne Force and 1 second dwell time, Crown et al., 2008). This parameter causes moderate spinal injury with robust neuropathic pain behaviors (Carter et al., 2004). Hains and Waxman reported that spinal contusion injury using the NYU Impactor produced p-38 MAPK activation in microglia in caudal regions (Hains and Waxman, 2006), but did not comment on neuronal or astrocytic location. In addition, other authors reported that neurons show activation of p-38α MAPK or only activation of microglia at “below-level” regions after moderate spinal cord injury using the OSU electromagnetic device for moderate spinal injury (Detloff et al., 2008) using the same antibodies as in Hains and Waxman, 2006.
Specifically, p-38 MAP kinases are serine threonine kinases that are activated by several upstream kinases in response to inflammation. In turn, p-p38 MAPK plays a key role in monocyte/macrophage inflammatory responses and inhibition of p-p38 MAPK is associated with reduction in iNOS, TNF α, IL-1β, COX-2 and iNOS, all proinflammatory mediators known to be involved in neuropathic pain. Cellular localization of activated p-38 MAPK is involved in neuronal and glial cell death, days after SCI (Crown et al., 2006), and is involved in persistent activation of both astrocytes and microglia (Zhuang et al., 2005; Svensson et al., 2005; Hua et al., 2005) and in dorsal horn hyperexcitability (Crown et al., 2008 and the present study), presumably by different pathways but possibly may share the same pathways in which the results are concentration dependent. In the latter case, for example, p-p38 downstream activation of the transcription factor CREB can lead to feed forward phosphorylation of the NMDA receptors leading either to maintained hyperexcitability or neuronal death if sufficient numbers of NMDA are activated. It should be noted that selection of antibodies may determine cellular localization and may provide the reason for differential findings between laboratories.
Taken collectively, our study in combination with previous studies suggest that differential mechanical injury (surgical hemisection vs. contusion injury) on the spinal cord influences common pathways of intracellular downstream molecular events, such as p-38 MAPK activation, which is involved in the development and maintenance of below-level neuropathic pain.
It is now well documented that traumatic neural injury causes glial activation, characterized by somatic hypertrophy of astrocytes and microglia (Watkins et al., 2001; Sweitzer et al., 2001, Gwak et al., 2008). In addition, our data suggest that activated astrocytes and/or microglia play important roles in modulating neuronal p-38α MAPK activation in caudal regions remote from low thoracic spinal injury. To determine this, we used a putative glial modulator agent, propentofylline (PPF), to inhibit astrocytic and microglial activation following SCI (Gwak et al., 2008; Raghavendra et al., 2003). We previously reported that spinal treatment of PPF attenuated neuronal hyperexcitability, mechanical allodynic behaviors and activation of astrocytes and microglia at regions caudal after spinal hemisection (Gwak and Hulsebosch, 2009a). Propentofylline is thought to regulate the synthesis and release of proinflammatory cytokines and cAMP signaling (Si et al., 1998), which trigger intracellular downstream pathways in neurons via activation of mitogen activated protein kinase (MAPKs) followed by activation of transcription factors, such as pCREB (Crown et al., 2006). The p-38 MAPK→ pCREB pathways modulate expression of membrane-bound receptors and inflammatory cytokine production, which results in persistent hyperexcitability of spinal dorsal horn neurons at the level of injury (at-level, Crown et al., 2008) and several segments remote from the injury (below-level, present study). Furthermore, we reported that changes in astrocytic and microglial morphology, increased GFAP (a useful marker of astrocytes activation) and increased OX-42 (a useful marker of microglial/macrophage activation) were produced after spinal hemisection and inhibited by intrathecal treatment of PPF (Gwak et al., 2008, Gwak and Hulsebosch, 2009a). We interpreted these observations to be consistent with astrocytic and microglial activation that resulted as a consequence of SCI. Furthermore, glial activations, neuronal hyperexcitability and mechanical allodynia were inhibited by intrathecal treatment of PPF (Gwak et al., 2008, Gwak and Hulsebosch, 2009a).
Spinal cord injury results in increased extracellular glutamate concentrations (McAdoo et al., 1999) and cytokines (Nesic et al., 2001), invasion of granulocytes (Kigerl et al., 2006) and release of blood serum and products (Noble et al., 2002), all factors that can directly activate neurons and glial cells and contribute to dysfunctional responses. Subsequent prolonged changes in synaptic efficacy produce continued activation of receptors and ion channels in both neurons and glial cells, followed by prolonged and abnormal activation of intracellular downstream events that result in enhanced neurotransmitter release and altered protein expression. In addition, activated glial cells actively participate in synaptic reorganization and remodeling synaptic circuits in the nervous system (Lee et al., 2005). These anatomical and functional changes in glial cells also result in enhanced release of gliotransmitters (released neurotransmitter by glial cells), proinflammatory cytokines, reactive oxygen species, and chemokines (Johnstone et al., 1999). These substances continue to provide changes in the activation state of receptors, for example all three general groups of glutamate receptors (NMDA, kainate/AMPA, metabotropic) are involved in CNP after SCI (Bennett et al., 2000; Mills et al., 2002; Hulsebosch, 2003), and in the activation state of ion channels, for example Nav1.3 (Hains et al., 2003) in neuronal membranes. The alterations in neuronal receptors and ion channels, coupled with increased production of putative ligands from activated glia cells, provide for continued production of reactants that contribute to intracellular downstream biochemical pathways that provide an intracellular feed forward mechanism for continued phosphorylation/activation of receptors and ion channels that ensures persistent neuronal hyperexcitability. Additionally, it is important to note that chronically activated astrocytes lead to permanent blood spinal cord barrier breakdown that ensure continued immune cell infiltration and feed forward continued activation of both astrocytes and microglia (Nesic et al., 2005).
In the mammalian system, we propose that normal glial function becomes abnormal and dysfunctional after CNS injury. The dysfunctional glial state contributes to conditions that initiate and ensure persistence of neuropathic pain. While the concept of glia-neuronal and neuronal-glial interactions are not new and were described in invertebrate systems several decades ago (see Lasek et al., 1974; Villegas, 1972); the conceptual basis of dysfunctional glial cells in mammals contributing to neuropathic pain is novel (Crown et al., 2008; Detloff, et al., 2008; Tawfik et al., 2007; Gwak et al., 2008; Nesic et al., 2005; Romero-Sandoval et al., 2008; Milligan et al., 2008). We propose that permanently dysfunctional glial cells, or gliopathy, play key roles in the development and maintenance of central neuropathic pain following SCI (Gwak and Hulsebosch, 2009b; Hulsebosch, 2008).
In conclusion, several pathways are implicated in the maintenance of mechanical allodynia and neuronal hyperexcitability at remote regions following spinal cord injury (Tan et al., 2008; Yu and Yezierski, 2005). However, our current finding suggests a novel pathway in which activation of p-38α MAPK downstream events in neurons and microglia, but not in astrocytes, actively participates in below-level neuropathic pain following SCI. The data presented in this study demonstrate direct evidence for the role “gliopathy” (see Hulsebosch, 2008) plays in modulating one pathway, the p-38 MAP kinase pathway, that alter neuronal hyperexcitability and consequently, nociceptive transmission and mechanical allodynia, in caudal spinal regions remote from the site of spinal cord injury.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett AD, Everhart AW, Hulsebosch CE. Intrathecal administration of an NMDA or 1 non-NMDA receptor antagonist reduces mechanical but not thermal allodynia in a rodent model of central neuropathic pain after spinal cord injury. Brain Res. 2000;859:72–82. doi: 10.1016/s0006-8993(99)02483-x. [DOI] [PubMed] [Google Scholar]
- Carter MW, Tan HY, Johnson KM, Hulsebosch CE. Effects of force and dwell-time in modeling chronic central neuropathic pain after contusive spinal cord injury (SCI) Neurosci Soc 2004 [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chung JM, Surmeier DJ, Lee DJ, Sorkin LS, Honda CN, Tsong Y, Willis WD. Classification of primate spinothalamic and somatosensory thalamic neurons based on cluster analysis. J Neurophysiol. 1986;56:308–327. doi: 10.1152/jn.1986.56.2.308. [DOI] [PubMed] [Google Scholar]
- Crown ED, Gwak YS, Ye Z, Johnson KM, Hulsebosch CE. P38 MAP Kinase inhibition attenuates central neuropathic pain following spinal cord injury in rats. Exp Neurol. 2008;213:257–267. doi: 10.1016/j.expneurol.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown ED, Ye Z, Johnson KM, McAdoo DJ, Hulsebosch CE. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp Neurol. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE. Remote Astrocytic and Microglial Activation Modulate Neuronal Hyperexcitability and Below-Level Neuropathic Pain after Spinal Injury in Rat. Neuroscience. 2009a;161:895–903. doi: 10.1016/j.neuroscience.2009.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE. Horizons of Neuroscience Research. Vol. 1. Nova Publisher; New York: 2009b. Gliopathy maintains persistent hyperexcitability of spinal dorsal horn neurons after spinal cord injury. in press. [Google Scholar]
- Gwak YS, Crown ED, Unabia GC, Hulsebopsch CE. Propentofylline attenuates allodynia, glial activation and modulates GABAergic tone after spinal cord injury in the rat. Pain. 2008;138:410–422. doi: 10.1016/j.pain.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE. Upregulation of Group I Metabotropic Glutamate Receptors in Neurons and Astrocytes in the Dorsal Horn following Spinal Cord Injury. Exp Neurol. 2005;195:236–243. doi: 10.1016/j.expneurol.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Gwak YS, Hains BC, Johnson CE, Hulsebosch CE. Effect of age at time of spinal cord injury on behavioral outcome in rat. J Neurotrauma. 2004;21:983–993. doi: 10.1089/0897715041650999. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Inthrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE. Gliopathy ensures persistent inflammation and chronic pain after spinal cord injury. Exp Neurol. 2008;214:6–9. doi: 10.1016/j.expneurol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsebosch CE. Recent advances in pathophysiology and treatment of spinal cord injury. Adv Physiol Educ. 2003;26:238–255. doi: 10.1152/advan.00039.2002. [DOI] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal Plasticity and Signal Transduction in Nociceptive Neurons: Implications for the Inhibition and Maintenance of Pathological Pain. Neurobiol Disease. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Johnstone M, Gearing AJ, Miller KM. A central role for astrocytes in the inflammatory response to beta-amyloid: chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol. 1999;93:182–193. doi: 10.1016/s0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neuro. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek RJ, Gainer H, Przybyski RJ. Transfer of newly synthesized proteins from Schwann cells to the squid giant axon. Proc Natl Acad Sci. 1974;71:1188–1192. doi: 10.1073/pnas.71.4.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Lindgvist E, Kiehn O, Widenfalk J, Olson L. Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. J Comp Neurol. 2005;489:1–10. doi: 10.1002/cne.20567. [DOI] [PubMed] [Google Scholar]
- McAdoo DJ, Xu GY, Robak G, Hughes MG. Changes in amino acid concentrations over time and space around an impact injury and their diffusion through the rat spinal cord. Exp Neurol. 1999;159:538–544. doi: 10.1006/exnr.1999.7166. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Watkins LR. Glia in pathological pain: a role for fractalkine. J Neuroimmunol. 2008;198:113–120. doi: 10.1016/j.jneuroim.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Johnson KM, Hulsebosch CE. Group I metabotropic glutamate receptors in spinal cord injury: roles in neuroprotection and the development of chronic central pain. J Neurotrauma. 2002;19:23–42. doi: 10.1089/089771502753460213. [DOI] [PubMed] [Google Scholar]
- Nesic O, McAdoo DJ, High KW, Hulsebosch CE, Perez-Polo R. IL-1 receptor antagonis prevent apoptosis and caspase-3 activation after spinal cord injury. J Neurotrauma. 2001;18:947–956. doi: 10.1089/089771501750451857. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, Wood TG, McAdoo DJ, Westlund KN, Hulsebosch CE, Region-Polo J. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J Neurochem. 2005;95:998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Lgarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain. 2003;104:655–664. doi: 10.1016/S0304-3959(03)00138-6. [DOI] [PubMed] [Google Scholar]
- Rexed B. The cytoarchitectonic organization of the spinal cord in the cat. J Comp Neurol. 1952;96:414–495. doi: 10.1002/cne.900960303. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath RJ, DeLeo JA. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs. 2008;9:726–734. [PMC free article] [PubMed] [Google Scholar]
- Si Q, Nakamura Y, Ogata T, Kataoka K, Schubert P. Differential regulation of microglial activation by propentofylline via cAMP signaling. Brain Res. 1998;812:97–104. doi: 10.1016/s0006-8993(98)00954-8. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Powell HC, Lai J, Porreca F, Yaksh TL. Prostaglandin E2 release evoked by intrathecal dynorphin is dependent on spinal p38 mitogen activated protein kinase. Neuropeptides. 2005;39:485–494. doi: 10.1016/j.npep.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Schubert P, Deleo JA. Propentophylline, a glial modulating agent, exbihits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Therapeu. 2001;297:1210–1217. [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, Hains BC. Neuropathic apin memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci. 2008;28(49):13173–13183. doi: 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik VL, Nutile-McMenemy N, Lacroix-Fralish ML, DeLeo JA. Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav Immuno. 2007;21:238–246. doi: 10.1016/j.bbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Mills CD, Ye Z, Fullwood SD, McAdoo DJ, Hulsebosch CE. Rapid changes in expression of glutamate transporters after spinal cord injury. Brain Res. 2002;927:104–110. doi: 10.1016/s0006-8993(01)03329-7. [DOI] [PubMed] [Google Scholar]
- Villegas J. Axon-Schwann cell interaction in the squid nerve fibre. J Physiol. 1972;225:275–296. doi: 10.1113/jphysiol.1972.sp009940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new players in pain. Pain. 2001;93:201–205. doi: 10.1016/S0304-3959(01)00359-1. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-Activated Protein Kinase: Conversation of a Three-Kinase Module from Yeast to Human. Physiol Rev. 1999;79:173–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Willis WD, Coggeshall RE. Structure of the dorsal horn. In: Willis WD, Coggeshall RE, editors. Sensory mechanisms of the spinal cord. PLENUM Press; NY: 1991. pp. 79–132. [Google Scholar]
- Wu X, Yoo S, Wrathall JR. Real-time quantitative PCR analysis of temporal-spatial alterations in gene expression after spinal cord contusion. J Neurochem. 2005;93:943–952. doi: 10.1111/j.1471-4159.2005.03078.x. [DOI] [PubMed] [Google Scholar]
- Yu CG, Yezierski RP. Activation of the ERK1/2 signaling cascade by excitotoxic spinal cord injury. Mol Brain Res. 2005;138:244–255. doi: 10.1016/j.molbrainres.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]