

Abstract
OBJECTIVE
The purpose of this article is to present current clinical and research issues in MRI evaluation of nonischemic cardiomyopathy, a diverse set of diseases, many of which have a genetic basis.
CONCLUSION
Cardiac cine MRI along with delayed myocardial enhancement MRI and other MRI techniques can provide information beyond echocardiography for tissue characterization. MRI is increasingly being used for evaluation of genetically positive, phenotypically negative patients as well as for risk stratification.
Keywords: arrhythmogenic right ventricular dysplasia, cardiomyopathy, hypertrophic cardiomyopathy, MRI, myocardium
MRI is one of the most powerful tools currently in use to noninvasively assess the extent and type of cardiovascular disease. Most cardiac disease is due to atherosclerosis in both men and women. Disease of the myocardium associated with atherosclerosis is termed “ischemic” cardiomyopathy. Besides ischemic cardiomyopathy, a wide range of diseases of the myocardium may occur and are unrelated to atherosclerosis. These diseases are classified as nonischemic cardiomyopathy. Whereas ischemic cardiomyopathy is usually well evaluated noninvasively using echocardiography, echocardiography is often deficient for characterization of nonischemic cardiomyopathy. Although the first line of therapy for ischemic cardiomyopathy is directed at improving myocardial blood flow in narrowed coronary arteries (e.g., using coronary stents or bypass surgery), such therapy usually has no role in patients with nonischemic cardiomyopathy. Instead, therapeutic techniques vary widely and may include a combination of medical therapy, implantable cardiodefibrilators or pacemakers, or even cardiac transplantation.
Nonischemic cardiomyopathy is a vague term that does not describe underlying pathology. Nonischemic cardiomyopathy of the heart is reminiscent of the term “interstitial infiltrate” on a chest radiograph. Similar to that of interstitial lung disease, the differential diagnosis of nonischemic cardiomyopathy is lengthy. Historically, nonischemic cardiomyopathies have been described by changes in the morphology or function of the heart [1]. An enlarged, thickened heart was termed “hypertrophic” cardiomyopathy and a thin-walled enlarged heart was termed “dilated” cardiomyopathy. The heart could also be stiff and unable to relax normally due to infiltration—termed “restrictive” cardiomyopathy as in amyloidosis (infiltration by amyloid protein) or sarcoidosis (granulomatous involvement of the heart).
Today, rather than simplistic descriptions of the size or shape of the heart, a more comprehensive definition of nonischemic cardiomyopathy has emerged [1]. Nonischemic cardiomyopathy is defined as disease of the myocardium associated with mechanical or electrical dysfunction exhibiting inappropriate ventricular hypertrophy or dilatation. The causes are numerous, but an increasing number of nonischemic disorders are being recognized as genetic in cause. Nonischemic cardiomyopathy may be either primary (confined to the heart) or secondary to systemic diseases (Table 1), and many of these conditions recently have been extensive reviewed [2–4].
TABLE 1.
Categorization of Nonischemic Cardiomyopathy
| Primary Cardiomyopathy |
Secondary Cardiomyopathy (Result of Systemic Disease) | ||
|---|---|---|---|
| Genetic | Mixed | Acquired | |
| Hypertrophic cardiomyopathy | Dilated cardiomyopathy | Stress cardiomyopathy | Amyloidosis |
| Arrhythmogenic right ventricular dysplasia | Tachycardia-induced | Gaucher disease | |
| Glycogen storage disease | Infants of insulin-dependent mothers | Hemochromatosis | |
| Mitochondrial abnormality | Heavy metal toxicity | ||
| Ion channel diseases | Sarcoidosis | ||
| Hemochromatosis | Etc. | ||
Note—Only selected conditions are mentioned. A more complete listing can be found in [1].
MRI of the heart has been found to have a unique and special role in the evaluation of these diverse and sometimes rare diseases, as will be discussed. The purpose of this article is to examine current key clinical and research issues regarding the role of MRI in the evaluation of nonischemic cardiomyopathy. Comprehensive reviews of the multiple imaging and clinical manifestations have recently been published, and the interested reader is referred to those publications for additional detail regarding diseases that cannot be described in this brief introduction to the field [2–9].
What Is the Unique Value of MRI in Nonischemic Cardiomyopathy?
Like echocardiography, MRI excels at obtaining high-temporal-resolution cine images of the heart. Using steady-state free precession imaging, a movie loop of a single slice of the heart is easily obtained in 5–6 seconds. Both 1.5- and 3-T MRI scanning produce good image quality on modern scanners. Typically, a stack of parallel short-axis cine loops from the base of the heart to the apex is acquired in about six breath-holds of 10–12 seconds (imaging two slices per breath-hold). The technique depends on a regular R-R interval on the ECG. If that is not the case, real-time cine images are obtained, although the spatial and temporal resolution is lower. Unlike echocardiography, the right ventricle is readily observed at MRI. This is critical for patients with suspected arrhythmogenic right ventricular dysplasia, an increasingly common rule-out diagnosis for MRI. Disease of both the right and left sides of the heart is relatively uncommon in ischemic cardiomyopathy and more likely to be associated with nonischemic cardiomyopathy. For patients with suspected hypertrophic cardiomyopathy, MRI may detect abnormal wall thickness that is otherwise missed by echocardiography (discussed later).
MRI also possesses the unique ability to add information regarding tissue composition. Frequently, nonischemic cardiomyopathy is associated with myocardial fibrosis. The detection of myocardial fibrosis (or scar) is well known from the evaluation of myocardial infarction with delayed enhanced MRI of the heart. After administration of 0.1–0.2 mmol/kg of gadolinium contrast agent, 10- to 20-minute delayed imaging is obtained with cardiac MRI. An inversion pulse is used to suppress normal myocardial tissue, thus highlighting the enhancing region of the heart. For patients with a prior myocardial infarction, myocardial scar is readily identified as transmural or subendocardial focal regions of delayed enhancement. The reason for delayed enhancement is that the wash-out rate of gadolinium is reduced in areas of myocardial fibrosis or collagen deposition. Increased gadolinium concentration causes T1 shortening or enhancement on delayed gadolinium enhancement MRI. Myocardial infarction as small as 1 g of tissue can be identified at MRI. It thus follows that a myocardial scar should be identified by MRI if the patient’s myocardial dysfunction is due to prior myocardial infarction. If scar is absent, a further explanation for the patient’s dysfunction must be sought.
The ability to detect subtle myocardial fibrosis is a major rationale for using MRI for myocardial disease. The pattern of delayed enhancement at MRI often associated with nonischemic cardiomyopathy is distinctly different (Fig. 1) from ischemic cardiomyopathy. Delayed enhancement associated with nonischemic cardiomyopathy often has the following two characteristics. The first is noncoronary artery distribution. In nonischemic cardiomyopathy, myocardial scar may be present in multiple coronary territories. For example, in myocarditis, scar may be present in the septum (left anterior descending coronary artery) as well as the inferior wall (right coronary artery). The second is midwall or epicardial location. In idiopathic dilated cardiomyopathy, for example, midwall septal enhancement is common. These patterns contrast with those of myocardial infarction. Because the most vulnerable zone of the myocardium is the subendocardial layer, the subendocardial layer is always involved by a myocardial infarction. Given that there are more than 100 different types of nonischemic cardiomyopathy, one might question how the above two different features of delayed enhancement are helpful (Fig. 2). Delayed gadolinium enhancement in nonischemic cardiomyopathy is usually not useful for a specific disease but rather to exclude an ischemic cause as the culprit.
Fig. 1. Gadolinium enhancement patterns in ischemic versus nonischemic cardiomyopathy. Top row indicates carton of enhancement pattern, and bottom row shows patient examples from short-axis delayed enhancement MRI. (Adapted from [26]).
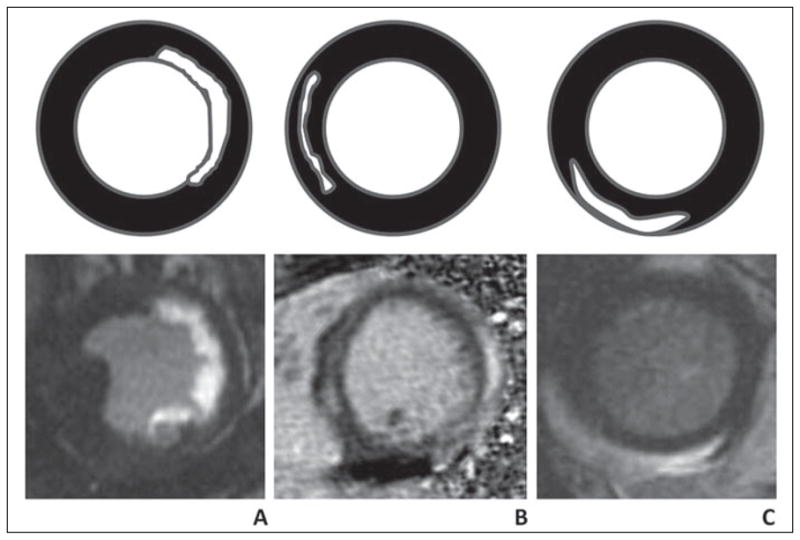
A, Subendocardial enhancement or transmural enhancement is associated with infarction from ischemic disease. In this case, 75-year-old man has had infarction in circumflex artery territory.
B, Nonischemic cardiomyopathy with midwall stripe pattern in 46-year-old woman. This pattern is common in idiopathic dilated cardiomyopathy.
C, Nonischemic cardiomyopathy with epicardial enhancement pattern in 35-year-old woman. This patient had viral myocarditis.
Fig. 2. Gadolinium enhancement patterns in nonischemic cardiomyopathy.
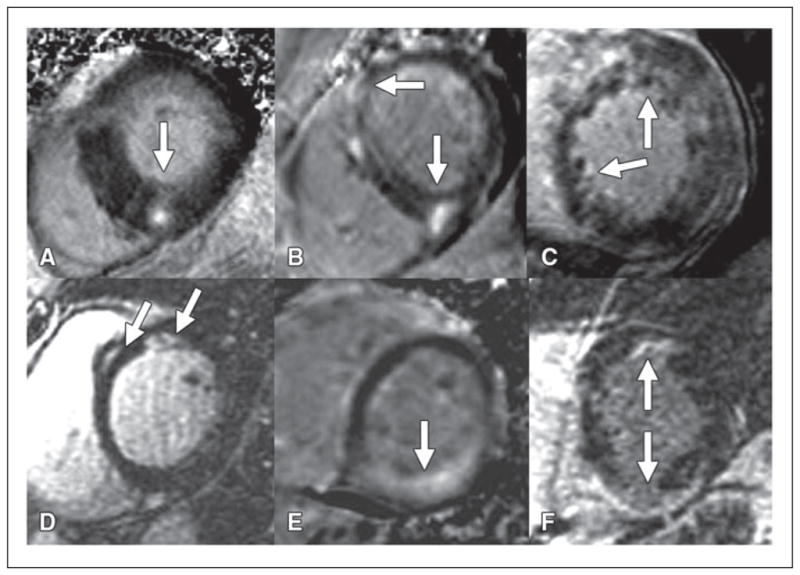
A–F, MRI short-axis views of midwall of left ventricle in six different patients indicate overlap and nonspecific appearance of delayed enhancement. Arrows indicate foci of enhancement in hypertrophic cardiomyopathy in 45-year-old man (A), idiopathic dilated cardiomyopathy in 56-year-old man (B), noncompaction cardiomyopathy in 24-year-old woman (C), sarcoidosis in 47-year-old woman (D), amyloidosis in 34-year-old man (E), and arrhythmogenic right ventricular dysplasia in 25-year-old woman (F).
Finally, there are other tissue characteristics that can be detected by MRI for nonischemic cardiomyopathy. In primary hemochromatosis, the heart may be variably affected by increased iron deposition. Using T2*-weighted imaging, the presence of significant amounts of iron in the heart can be determined. In some nonischemic cardiomyopathies (e.g., myocarditis), edema may be a prominent feature. Using T2-weighted imaging, the presence of edema can be readily assessed.
In summary, cardiac MRI readily visualizes all portions of the myocardium with high spatial resolution with the use of steady-state free precession cine imaging. In addition, patients with nonischemic cardiomyopathy often show a distinct pattern of delayed gadolinium enhancement different from that of patients with ischemic cardiomyopathy. Identification of a nonischemic pattern of delayed gadolinium enhancement is extremely powerful to lead toward the correct diagnosis for the patient. MRI can also provide unique information regarding other tissue characteristics, such as the presence of iron or edema. For patients with a known nonischemic cardiomyopathy, MRI is often used to characterize the extent of the abnormality. When the cause of the myocardial dysfunction is unknown, a major benefit of MRI is to help separate an ischemic (atherosclerotic) cause from a nonischemic cause. Patients with ischemic cardiomyopathy are managed with β blockers, myocardial stenting, risk factor modification, or coronary artery bypass. Patients with nonischemic cardiomyopathy may be managed medically, but myocardial transplantation or defibrillator implantation may be the only options in certain severe cardiomyopathies. For nonischemic cardiomyopathy with a known genetic cause, screening of family members also becomes of paramount importance.
Examples of Nonischemic Cardiomyopathy
To illustrate the importance of MRI in the management and diagnosis of patients with nonischemic cardiomyopathy, I will discuss two very distinct conditions and the current role of MRI in each disease. In hypertrophic cardiomyopathy (HCM), the wall of the left ventricle is abnormally thickened. In arrhythmogenic right ventricular dysplasia (ARVD), the wall of the right ventricle is abnormally thinned. Both conditions have been identified as having a genetic basis and both are associated with sudden cardiac death.
Hypertrophic Cardiomyopathy
HCM is a genetic cardiomyopathy that is autosomal dominant. The incidence in the United States is approximately one in 500, or 0.2%, of the population [2]. HCM is the leading cause of sudden cardiac death in competitive athletes, adolescents, and young adults in the United States. The reason for sudden cardiac death is due to arrhythmia. Treatment may consist of medical therapy, surgery for partial resection of the septum in patients with obstruction of the outflow tract, implanted defibrillator, or a combination of these treatments.
The underlying abnormality in HCM is a mutation involving sarcomere contractile proteins. More than 800 different mutations have been identified in at least 11 contraction-encoding genes in HCM. Approximately 65% of patients with familial HCM have a genetic abnormality that can be recognized. Genetic testing for HCM may be considered a clinical test and is routinely used in research protocols. As a result, some patients are diagnosed as genotype positive but may be phenotype negative if they have not yet expressed the disease.
Histologically, HCM shows myocardial disarray and collagen deposition, eventually resulting in a thickened, poorly contracting myocardium. Echocardiography (or MRI) is used to diagnose HCM when there is unexplained focal or diffuse thickening of the myocardium greater than 15 mm in left ventricular wall thickness. Systolic function of the heart is normal or even increased early in the disease, but diastolic function is reduced, detected by echocardiography [10]. After gadolinium administration, delayed enhancement images most often show a diffuse speckled pattern in the midwall of the myocardium, frequently in the area of myocardial thickening (Fig. 3).
Fig. 3. MRI of hypertrophic cardiomyopathy in 46-year-old woman.
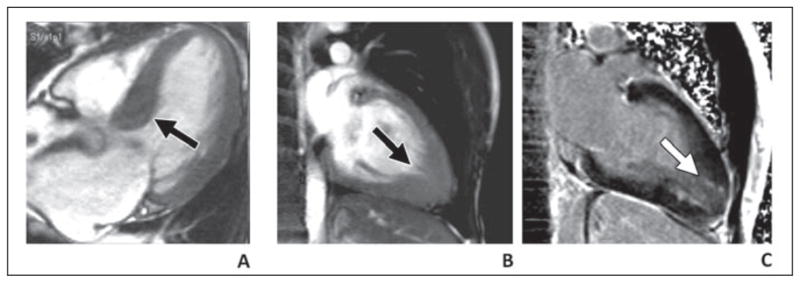
A, Septal thickening (arrow) of left ventricular outflow tract with hypertrophic obstructive cardiomyopathy. B and C, Thickening of apex (B) with myocardial delayed enhancement (C) with apical variant hypertrophic cardiomyopathy.
All patients with suspected hypertrophic cardiomyopathy undergo echocardiographic evaluation of the myocardium. Until recently, MRI was not thought to be necessary because the diagnosis relied strongly on wall thickness measurements. Increasing use of MRI has shown that HCM is undetected in 6–12% of patients by echocardiography [11, 12].
An emerging application of MRI in HCM is to characterize the presence of myocardial fibrosis in HCM. I conducted an informal meta-analysis of papers from 2002 to 2009 that described delayed enhancement by MRI in 675 patients. Of these, delayed enhancement was reported in 62% of patients with HCM. Delayed enhancement in HCM is related to collagen deposition, and collagen deposition interspersed with normal myocardium represents an arrhythmogenic substrate. A large series of 177 patients by Adabag et al. [13] showed that the presence or absence of delayed enhancement at MRI was the primary predictor of nonsustained ventricular tachycardia. Thus, risk stratification in HCM may potentially be significantly improved by identifying the presence of myocardial delayed enhancement at MRI [14].
Finally, genetically positive HCM patients who do not have myocardial hypertrophy (phenotype negative) have emerged as an important clinical issue [15]. Should young, competitive athletes who come from families with HCM be allowed to participate in sports if they have a “normal” echocardiogram? As discussed, it appears that 6–12% of these individuals will have abnormal morphology at MRI. Early in their disease course, tissue Doppler echocardiography has shown that diastolic function is abnormal despite normal wall thickness. Delayed-enhancement MRI is expected to be normal in this situation because a positive MRI for fibrosis is usually only present when wall thickness is abnormal. A sensitive MRI method to assess wall motion during diastole is myocardial tissue tagging. Myocardial tagging in either a grid or line pattern is performed by applying pulses that eliminate the MRI signal along the tag lines. The tag lines deform with the myocardium as it contracts or relaxes, allowing precise determination of myocardial contraction patterns Because several experimental medical treatments are now being studied for HCM, MRI will be used in future clinical trials to determine if subtle improvements in regional contraction or relaxation may occur after therapy.
Arrhythmogenic Right Ventricular Dysplasia
ARVD is a complex topic because of the difficulty in providing a definitive imaging diagnosis early in the course of the disease. Whereas HCM is the most common genetic cardiac disease, ARVD is relatively rare, occurring in one in 10,000 individuals in the United States. The condition is more common in Italy and in individuals who live in the region surrounding the Mediterranean Sea. The diagnosis is made by a combination of clinical features, imaging abnormalities (aneurysm or enlargement of the right ventricle), electrical abnormalities, and family history [16] (Table 2).
TABLE 2.
Diagnosis of Arrhythmogenic Right Ventricular Dysplasia (ARVD) [25]
| Diagnosis | Criteria | Finding |
|---|---|---|
| Global or regional dysfunction and structural alterationsa | Major | Severe dilatation and reduction of right ventricular ejection fraction with no (or only mild) left ventricle impairment, localized right ventricular aneurysms (akinetic or dyskinetic areas with diastolic bulging), severe segmental dilatation of the right ventricle |
| Minor | Mild global right ventricular dilatation or ejection fraction reduction with normal left ventricle, mild segmental dilatation of the right ventricle, regional right ventricular hypokinesia | |
| Tissue characterization of wall | Major | Fibrofatty replacement of myocardium on endomyocardial biopsy |
| Repolarization abnormalities | Minor | Inverted T waves in right precordial leads (V2 and V3) in people aged > 12 years, in absence of right bundle branch block |
| Depolarization–conduction abnormalities | Major | ε waves or localized prolongation (> 110 milliseconds) of the QRS complex in right precordial leads (V1–V3) |
| Minor | Late potentials (signal-averaged ECG) | |
| Arrhythmias | Minor | Left bundle branch block type ventricular tachycardia (sustained and nonsustained) by ECG, Holter, or exercise testing, frequent ventricular extrasystoles (> 1,000/24 h) (Holter) |
| Family history | Major | Familial disease confirmed at necropsy or surgery. |
| Minor | Family history of premature sudden death (< 35 years) due to suspected right ventricular dysplasia, family history (clinical diagnosis based on present criteria) | |
Note—The diagnosis of ARVD would be fulfilled by the presence of two major, one major plus two minor, or four minor criteria from different groups.
Detected by echocardiography, angiography, MRI, or radionuclide scintigraphy.
The most common age range for ARVD is 20–40 years and the clinical presentation is quite nonspecific, including syncope or ventricular tachycardia. In the United States, ARVD accounts for 5% of sudden cardiac deaths in individuals younger than 35 years [16]. Because of the adverse risk of the condition, cardiologists may aggressively pursue the diagnosis of ARVD when other tests, including ECG, Holter monitor, and echocardiography, are normal. Thus, despite being somewhat rare, ARVD is a common reason for referral for MRI. It is not unusual for 50% of nonischemic cardiomyopathy patient referrals to be for the purpose of excluding ARVD.
The most common genetic abnormality in ARVD affects the desmosome. The desmosome is a structure between adjacent myocytes that is involved in mechanical cell coupling. The PKP2 gene encodes for the protein plakophilin-2, which is a component of the desmosome. Deficient cell-to-cell coupling may result in contraction abnormalities, cell death, or disruption in gap junctions, which otherwise facilitate electrical coupling of cells. Histologically, there is apoptosis of myocytes and replacement by fibrofatty tissue. Inflammation has also been reported in affected regions of the heart.
The predominant expression of ARVD is in the right ventricle. The reason for this is not known although the theory is that the right ventricle is the weak chamber. Using tagged MRI, we have been able to detect contraction abnormalities in both the right and left ventricles. However, more obvious morphologic changes of dilatation and aneurysm formation are much more common in the right heart than the left.
The treatment of ARVD is a permanently implanted automatic cardiac defibrillator (AICD) to disrupt an otherwise lethal arrhythmia. Unfortunately, after AICD placement, MRI is typically not performed for safety reasons. Thus, it is difficult to determine the accuracy of MRI for the diagnosis.
The complexity of using MRI for the diagnosis of ARVD is to distinguish normal from abnormal size and contraction of the right ventricle [17]. In a sense, this becomes a paradox for the cardiac imager: one cardiac MRI article indicated that the majority of right ventricles in healthy volunteers showed “abnormal” contraction patterns. The authors assumed that our knowledge of left ventricle contraction patterns could be applied to the right ventricle. Unfortunately, this is not the case. The right ventricle is deformed anteriorly by retrosternal ligaments attached to the pericardium, and small bulges in the anterior wall are relatively common. These structural details are not seen using echocardiography. Hence, the cardiac imager who pays little attention to the right ventricle for most cases is suddenly able to discover a host of potential “abnormalities” in patients referred for suspected ARVD [18]. Nevertheless, regional wall motion abnormalities in the right ventricle remain the most reliable feature of early ARVD [19].
Other MRI characteristics of importance are the imaging correlates of histologically identified fibrofatty infiltration. In our experience, fatty infiltration of the right ventricle is detected only late in the course of the disease. At that point, frequently dilatation and aneurysm formation are seen in the right ventricle. Fatty infiltration of the left ventricle may also be present. It is problematic that most MRI scanners cannot adequately image the right ventricle using T1-weighted turbo or fast spin-echo imaging [20, 21].
Delayed enhancement of the right ventricle is present in about 60–70% of patients with confirmed ARVD [22] (Fig. 4). Together with fatty infiltration, the identification of myocardial fibrosis involving primarily the right heart helps confirm the diagnosis of ARVD. In our experience, functional abnormalities of the right ventricle may occur without either definite fat infiltration or delayed gadolinium enhancement. Our current concept of the disease is that early ARVD is manifested by subtle contraction abnormalities that may involve both the left and right ventricles. More advanced disease is accompanied myriad abnormalities, including right ventricular enlargement, delayed gadolinium enhancement, and fatty infiltration. The diagnosis appears more specific when these morphologic and functional changes have a predominant right-sided involvement. If biventricular disease is present, other causes for myocardial dysfunction should be sought. In the multicenter United States Study of ARVD, the most common differential diagnosis for ARVD was sarcoidosis. A multidisciplinary team that is expert in understanding the manifestations of ARVD is helpful to avoid a false-positive MRI diagnosis [23].
Fig. 4. MRI of arrhythmogenic right ventricular dysplasia in 32-year-old man.
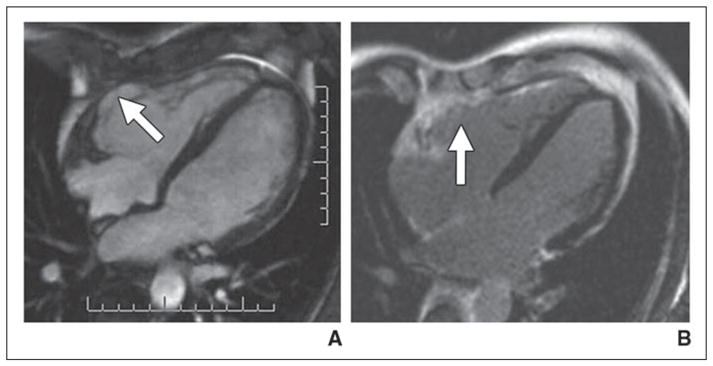
A, Axial cine steady-state free precession image shows aneurysm (arrow) involving anterior wall of right ventricle.
B, Delayed gadolinium-enhanced MRI shows enhancement (arrow) in location of right ventricular aneurysm.
Like HCM, ARVD presents the possibility of genetically positive disease but a phenotypically normal myocardium. In this setting, we assume that the risk of sudden cardiac death is much higher than in the average rule-out ARVD patient. If MRI and other tests are negative, patients return yearly for follow-up MRI. After several years of negative MRI, the interval for follow-up can be lengthened to 2–3 years unless symptoms appear or other tests become positive.
Other Nonischemic Cardiomyopathies
The MRI features of other nonischemic cardiomyopathies are myriad but have been recently reviewed [2]. Several other diseases are worthy of distinct mention because of frequent referrals to MRI. For example, patients with sarcoidosis present with conduction abnormalities and frequently have evidence of systemic disease. Gadolinium enhancement is present as patchy enhancement in the myocardium. Idiopathic dilated cardiomyopathy was mentioned previously. In this condition, the left heart is markedly dilated and thinned, and midwall enhancement especially in the septum is present in more than 50% of patients. Cardiac amyloidosis shows enhancement that is frequently subendocardial but in a noncoronary distribution and may involve all four cardiac chambers as well as valvular enhancement. Patients with viral myocarditis show patchy enhancement in the midwall and epicardial portions of the left ventricle.
In most of these conditions, the delayed-enhancement gadolinium images provide unique information that is useful to categorize the disease as other than typical ischemic cardiomyopathy. An unfortunate consequence of myocardial scarring is that it appears to not be reversible. Thus, gadolinium enhancement from acute viral myocarditis may diminish, but the secondary injury of collagen deposition or scar formation appears to be constant over time.
Conclusions and Clinical Implications
Nonischemic cardiomyopathy is a term that encompasses a heterogeneous group of diseases. Increasingly, the disorders are recognized as a having a genetic component. The myocardium has a very limited response to cellular injury and can be replaced by either fibrosis or fat tissue. Thus, whereas nonischemic myocardial disease usually can be distinguished from ischemic disease, determining the particular cause of nonischemic cardiomyopathy requires clinical correlation and often myocardial biopsy.
There are two compelling reasons these somewhat unusual diseases focus our attention for cardiac MRI. First, we are increasingly presented with referrals for patients who are genetically positive for known mutations but who have no clinically apparent disease. Because radiation is not required and the risk of MRI is low, cardiac MRI is often used to supplement evaluation by echocardiography.
Second, many nonischemic cardiomyopathy disorders are associated with the presence of scar tissue. Emerging evidence suggests that myocardial scar is often the genesis for cardiac arrhythmia that can be life threatening [24]. Thus, the use of delayed gadolinium MRI is routine in evaluating these patients. There is currently an extraordinary research effort aimed at risk prediction and localized treatment on the basis of cardiac MRI evaluation of myocardial scar. It appears likely that cardiac MRI will continue to be a major investigational tool for nonischemic cardiomyopathy, both in research and in clinical practice.
Footnotes
The opinions and assertions contained herein are the private views of the author and are not to be construed as official or as representing the views of the National Institutes of Health.
References
- 1.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 2.Shehata ML, Turkbey EB, Vogel-Claussen J, Bluemke DA. Role of cardiac magnetic resonance imaging in assessment of nonischemic cardiomyopathies. Top Magn Reson Imaging. 2008;19:43–57. doi: 10.1097/RMR.0b013e31816fcb22. [DOI] [PubMed] [Google Scholar]
- 3.Ordovas KG, Reddy GP, Higgins CB. MRI in nonischemic acquired heart disease. J Magn Reson Imaging. 2008;27:1195–1213. doi: 10.1002/jmri.21172. [DOI] [PubMed] [Google Scholar]
- 4.Macedo R, Schmidt A, Rochitte CE, Lima JA, Bluemke DA. MRI to assess arrhythmia and cardiomyopathies: relationship to echocardiography. Echocardiography. 2007;24:194–206. doi: 10.1111/j.1540-8175.2007.00376.x. [DOI] [PubMed] [Google Scholar]
- 5.Masci PG, Dymarkowski S, Bogaert J. The role of cardiovascular magnetic resonance in the diagnosis and management of cardiomyopathies. J Cardiovasc Med (Hagerstown) 2008;9:435–449. doi: 10.2459/JCM.0b013e32827ab49f. [DOI] [PubMed] [Google Scholar]
- 6.Harris SR, Glockner J, Misselt AJ, Syed IS, Araoz PA. Cardiac MR imaging of nonischemic cardiomyopathies. Magn Reson Imaging Clin N Am. 2008;16:165–183. vii. doi: 10.1016/j.mric.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 7.White JA, Patel MR. The role of cardiovascular MRI in heart failure and the cardiomyopathies. Cardiol Clin. 2007;25:71–95. vi. doi: 10.1016/j.ccl.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Rochitte CE, Tassi EM, Shiozaki AA. The emerging role of MRI in the diagnosis and management of cardiomyopathies. Curr Cardiol Rep. 2006;8:44–52. doi: 10.1007/s11886-006-0010-5. [DOI] [PubMed] [Google Scholar]
- 9.Isbell DC, Kramer CM. The evolving role of cardiovascular magnetic resonance imaging in nonischemic cardiomyopathy. Semin Ultrasound CT MR. 2006;27:20–31. doi: 10.1053/j.sult.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Ho CY, Sweitzer NK, McDonough B, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002;105:2992–2997. doi: 10.1161/01.cir.0000019070.70491.6d. [DOI] [PubMed] [Google Scholar]
- 11.Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220–228. doi: 10.1016/j.jacc.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation. 2005;112:855–861. doi: 10.1161/CIRCULATIONAHA.104.507723. [DOI] [PubMed] [Google Scholar]
- 13.Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51:1369–1374. doi: 10.1016/j.jacc.2007.11.071. [DOI] [PubMed] [Google Scholar]
- 14.Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375–1376. doi: 10.1016/j.jacc.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Maron BJ, Ho CY. Hypertrophic cardiomyopathy without hypertrophy: an emerging pre-clinical subgroup composed of genetically affected family members. JACCl Cardiol Cardiovasc Imaging. 2009;2:65–68. doi: 10.1016/j.jcmg.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Jain A, Tandri H, Calkins H, Bluemke DA. Role of cardiovascular magnetic resonance imaging in arrhythmogenic right ventricular dysplasia. J Cardiovasc Magn Reson. 2008;10:32. doi: 10.1186/1532-429X-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tandri H, Friedrich MG, Calkins H, Bluemke DA. MRI of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Magn Reson. 2004;6:557–563. doi: 10.1081/jcmr-120030583. [DOI] [PubMed] [Google Scholar]
- 18.Sievers B, Addo M, Franken U, Trappe HJ. Right ventricular wall motion abnormalities found in healthy subjects by cardiovascular magnetic resonance imaging and characterized with a new segmental model. J Cardiovasc Magn Reson. 2004;6:601–608. doi: 10.1081/jcmr-120038528. [DOI] [PubMed] [Google Scholar]
- 19.Tandri H, Castillo E, Ferrari VA, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J Am Coll Cardiol. 2006;48:2277–2284. doi: 10.1016/j.jacc.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz SJ, Macedo R, Malayeri AA, et al. Axial black blood turbo spin echo imaging of the right ventricle. Magn Reson Med. 2009;61:307–314. doi: 10.1002/mrm.21864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castillo E, Tandri H, Rodriguez ER, et al. Arrhythmogenic right ventricular dysplasia: ex vivo and in vivo fat detection with black-blood MR imaging. Radiology. 2004;232:38–48. doi: 10.1148/radiol.2321030688. [DOI] [PubMed] [Google Scholar]
- 22.Tandri H, Bomma C, Calkins H, Bluemke DA. Magnetic resonance and computed tomography imaging of arrhythmogenic right ventricular dysplasia. J Magn Reson Imaging. 2004;19:848–858. doi: 10.1002/jmri.20078. [DOI] [PubMed] [Google Scholar]
- 23.Bomma C, Rutberg J, Tandri H, et al. Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Electrophysiol. 2004;15:300–306. doi: 10.1046/j.1540-8167.2004.03429.x. [DOI] [PubMed] [Google Scholar]
- 24.Spector PS. Diagnosis and management of sudden cardiac death. Heart. 2005;91:408–413. doi: 10.1136/hrt.2003.024026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunold P, Schlosser T, Vogt FM, et al. Myocardial late enhancement in contrast-enhanced cardiac MRI: distinction between infarction scar and non-infarction-related disease. AJR. 2005;184:1420–1426. doi: 10.2214/ajr.184.5.01841420. [DOI] [PubMed] [Google Scholar]